
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Site-specific recombination systems are widely used as bioengineering tools. However, the traditional site-specific recombination system requires a consensus sequence for the specific site. Such sequence-level constraints limit effective recombination between sites. Therefore, in order to develop an efficient site-specific recombination system, we investigated the attC site of the bacterial integration subsystem and built a predictive model to infer the important features that contribute to recombination. Here, we design an attC site prediction algorithm based on a combination optimisation strategy. Based on the structural features of attC sites, the prediction algorithm realises the high-precision prediction of the recombination frequencies between sites and the screening of the top 20 important features that play a role in recombination, which are effective for improving the design method of attC sites. The algorithm has better portability and higher prediction accuracy compared with the existing advanced algorithms, among which the Pearson correlation coefficient is 0.87, explained variance score is 0.73, root mean square error is 0.006 and mean absolute error is 0.041. This can not only provide ideas for the research of efficient recombination systems but also provide a theoretical basis for developing genetic engineering further.
1. Introduction
Gene recombination is a way that organisms use recombinase to recombine different genes to produce new genotype individuals. It is widely present in prokaryotes and has important meanings such as maintaining biological genetic diversity and promoting biological evolution (Epum & Haber, Citation2022). Common recombination includes: homologous recombination, translocation recombination and site-specific recombination. Currently, with the development of site-specific recombination systems, site-specific recombination technology has been extensively used in various biological genetic engineering operations, especially in higher eukaryotes (Bessen et al., Citation2019; Häcker et al., Citation2017). Site-specific recombination refers to the integration, excision and transformation of DNA fragments between specific sites, which is catalysed by integron integrase. This type of recombination is associated with specific DNA sequences in bacteriophages and bacteria, and the reaction always involves two DNA-specific sites. However, these two specific sites usually have very similar or even exactly the same DNA sequences. Such sequence-level constraints restrict efficient recombination between the two sites (Tian & Zhou, Citation2021). Therefore, in order to solve the problem of sequence constraints, it is necessary to study the structure of specific recombination sites in the recombination system.
In this paper, we study based on the bacterial integration subsystem. The bacterial integration system is an important application of site-specific recombination, which can capture and express foreign gene cassettes and convert them into functional gene expression units by site-specific recombination (Domingues et al., Citation2012). Through the recombination of DNA fragments between sites, bacteria can acquire properties that are beneficial to themselves, such as increasing bacterial resistance. Studies have shown that integron-promoted horizontal gene transfer enables bacteria to acquire foreign drug resistance genes as an important reason for accelerating the spread of clinically relevant Gram-negative pathogens(Mazel et al., Citation2015; Stalder et al., Citation2012). The function of the integron depends on the activity of the integrase (Nivina et al., Citation2016; Weiss et al., Citation2019), which is a site-specific tyrosine recombinase with the special ability to excise, integrate, reverse and translocate DNA fragments in organisms. Gene cassettes are usually mobile elements that carry a single gene and an associated recombination site (attC). Investigation shows that many gene cassettes often carry antibiotic resistance genes. Gene cassettes can exist independently in the form of a free ring, or they can be recognised and catalysed by integrases, and site-specific recombination becomes a part of the integron structure (Ghaly et al., Citation2021; Vit et al., Citation2020).
In the integron system, the excision of the gene cassette occurs between two adjacent attC sites, and the insertion of the gene cassette is mainly achieved through the recombination of the attI site on the integrator platform and the attC site on the free gene cassette (Mukhortava et al., Citation2019). The bottom strand of DNA at the attC site is folded into a hairpin-like structure and recombined as a single strand, and the conserved structure of the folded ensures the specificity of the reaction. Previous research has shown that tyrosine recombinase has a high sequence homology requirements for recombining attI sites, but recombinase can efficiently recombine attC sites with highly variable sequence and structure, and the recombination of attC sites is highly dependent on its structural features (Olorunniji et al., Citation2016). Meanwhile, attC sites appear to have very few sequence-level constraints. The attC site allows insertion into the cassette sequence of the integron-related recombination site (attI), and the recombination mainly occurs between A and C in the consensus sequence 5'-RYYYAAC-3’ of the R box (Larouche & Roy, Citation2011). Investigations revealed that integrase binding and recombination are mainly driven by the structural features of the stem loop at the attC site: outer helical bases (EHBs), unpaired central spacer (UCS) and variable end structure (VTS) (Frumerie et al., Citation2010; Grieb et al., Citation2017). The favourable properties of these attC sites make them key to developing efficient site-specific recombination systems. Therefore, studying the relationship between the function and structure of attC sites and seeking effective features that play a role in recombination can not only expand new ideas for the synthesis of efficient attC sites but also be important for understanding bacterial evolution and the promotion of antibiotic resistance.
This paper proposes an attC site prediction algorithm based on the combination optimisation strategy for the attC site of the bacterial integration subsystem. By analyzing and quantifying the structural characteristics of attC site, three regression algorithms are used to establish prediction models, which can effectively improve the accuracy of prediction and avoid the shortcomings of a single model. Meanwhile, by visualising the correlation between structure and function, we can find unknown important features that contribute to reorganisation, which plays a key role in improving the design method for synthetic sites and improving the recombination rate. Compared with four traditional regression algorithms: Decision Tree Regression (Lewis, Citation2000), Ridge Regression (Hoerl & Kennard, Citation2000), Support Vector Regression (Klopfenstein & Vaiter, Citation2021) and Random Forest Regression (Pellegrino et al., Citation2021), this algorithm has obtained higher credibility. More importantly, the algorithm is highly flexible and extensible.
2. Related works
With the rapid development of gene sequencing, traditional biochemical experiments can not meet the needs of massive gene data. At this time, the application of computer technology in biology has shown its strong performance advantages, and machine learning technology has been applied in various fields of biology (Liu et al., Citation2022; Wang et al., Citation2021; Wei et al., Citation2021). In the field of science, research on the biological significance and value behind the data is also the current research hotspot. Machine learning has the learning ability to acquire knowledge from data and experience. It can not only extract knowledge from massive biological data and realise data-based predictions, but also continuously improve its performance and realise self-learning in the process of learning (Cao et al., Citation2020; Libbrecht & Noble, Citation2015). Therefore, it is very effective to apply machine learning to the field of site prediction to improve testing efficiency. At present, the typical algorithms applied by machine learning in this field include Random forest, hidden Markov and logistic regression.
With the development of synthetic biology, the strategies and tools for rapidly constructing new biochemical pathways will become more and more valuable (Doudna, Citation2020). Institut et al. proposed a synthetic integron for genetic recombination in vivo, providing a way to construct and optimise metabolism using the inherent gene recombination activity of natural bacterial site-specific recombination system integrators. This method demonstrated the ability to use site-specific recombination to efficiently generate a large number of gene combinations and arrangements in vivo for the first time (Bikard et al., Citation2010). This indicates that studying the structure of attC sites and synthesising a recombinant and efficient attC site will effectively improve the integration subsystem.
Pereira et al. presented a new method HattCI based on Hidden Markov Model (HMM) to quickly and accurately identify attC sites in DNA sequence data. This model describes each core component of an attC site separately, that is, the attC site can be directly identified in the fragment data without any additional information about the integrated substructure. The results of cross-validation showed that HattCI achieved a high sensitivity of up to 91.9% while maintaining a satisfactory false positive rate (FPR) (Pereira et al., Citation2016).
Meanwhile, there are many programmes that can recognise attC sites. The XXR programme uses pattern matching technology to recognise attC sites in Vibrio vulgaris integrons (Rowe-Magnus et al., Citation2003), and the programmes ACID (Joss et al., Citation2009) and Attacca (Tsafnat et al., Citation2009) are designed to search for categories 1 to categories 3 mobile integrators. However, these identification programmes based on sequence conservation can only identify attC sites in a limited integration subcategory, and have greater difficulty in identifying distant relative attC sites with highly different sequences.
The identification programme IntegronFinder proposed by Jean et al. aims to accurately identify any integrase and attC sites belonging to integrons. This model uses 291 artificially managed attC sites to establish a covariance model for attC sites, and uses the covariance model to search for attC sites on 2484 complete bacterial genomes. The recognition programme IntegronFinder has achieved 96% sensitivity while ensuring maximum attC site diversity (Cury et al., Citation2016).
Nivina et al. designed a structure-specific recombination system based on attC sites to construct a large-scale mutation library for attCr0 sites with low recombination rates. By analyzing and quantifying the structural features of the sites in the library, a Random forest-based approach was realised and obtained a higher prediction accuracy of attC site recombination (Nivina et al., Citation2020). This model is also applicable to other specific sites and other genetic elements based on sequence characteristics, which have good portability. However, the model used in this method is relatively single, so there may be data deviation.
With the further development of genome research, a growing number of biological genomes have been completely sequenced, and the prediction of DNA recombination sites has also become a significant goal of computational analysis of biological information. Among them, the attC site is an important element in the realisation of site-specific recombination systems. Studies have shown that this site is closely related to gene therapy and drug development. Therefore, accurate prediction of the attC site can provide theoretical support for the treatment of certain diseases. Traditional biochemical experiments have been unable to meet the problem of processing a large amount of biological data. On the contrary, the advantages of combining biological information with computer technology have gotten more and more obvious. The prediction model of attC sites established by biological data can not only effectively help the development of recombinant systems, but also be applied to other research fields, with high flexibility and portability. After learning from previous studies, we found that a large number of studies were used to identify attC sites in DNA sequences, ignoring the prediction of their recombination rates. The high-precision prediction of the recombination rate based on the characteristics of the attC site is of great significance for the establishment of an efficient recombination system.
Previous research has shown that the attC site has highly conserved structural features and can be used to identify highly variable sequences (Bouvier et al., Citation2009; Lacotte et al., Citation2017). Therefore, it is an effective method to establish prediction experiments based on the structural features data (Liu et al., Citation2019; Liu et al., Citation2019). However, the predictions of attC sites defined by traditional research methods are different. Most of them have problems such as single feature, unrepresentative, single model and complex extraction of biological data. At the same time, a large amount of feature data not only wastes time, but some error features also have a great impact on the experimental results. Therefore, it has great development potential to comprehensively use the relevant knowledge of machine learning and deep learning to improve the prediction performance for the above problems.
3. Materials and methods
3.1. Prediction of attC site
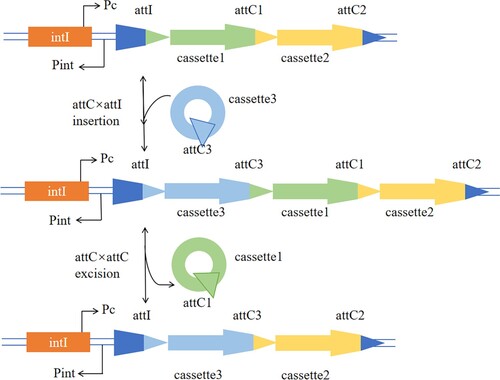
The attC site is the main site for site specific recombination, and its bottom strand is folded into a hairpin-like structure and recombined as a single strand (Figure ). The attC site can mediate the insertion and excision of gene cassettes under the catalysis of integrase. The excision of the gene cassette mainly occurs between two adjacent attC sites on the integrative system, and the insertion of the gene cassette involves the attC site on the gene cassette and the attI site on the integrative system (Figure ).
Figure 1. Schematic diagram of attC recombination site structure.
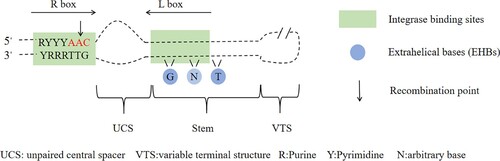
Figure 2. Schematic representation of the integron system. intI, integrase; Pint, integrase promoter; Pc, gene cassette promoter; attI, gene cassette insertion site; attC, gene cassette recombination site.
At present, the research of site prediction is developing rapidly, and there are mainly two technologies at this stage. The first is to predict the site through biochemical experiments, and the second is to quickly predict the site through bioinformatics combined with computer-related techniques. However, the prediction of recombination sites based on bacterial integration subsystems is currently mostly limited to biochemical experiments and a small number of bioinformatics methods, which consume a lot of economic and time costs. Consequently, this paper aims at the attC site of bacterial integration subsystem to study the correlation between its structure and function, and realises the accurate and efficient prediction of attC site, which is a beneficial supplement to the existing experimental methods.
3.2. Data sources for attC sites
In order to determine which structural features can contribute to the high recombination rate of the attC site, we visited the attCr0 mutant library for analysis (Nivina et al., Citation2020). The library included all sequences with a single mutation in the constant region of the attCr0 site and a sequence containing all possible combinations of two mutations. This paper selects the attC site sequence containing two mutations in the library as the initial data set. The data set contains 12879 attCr0 mutants and 292 structural features, including 9 global features and 283 basic features. Meanwhile, we employ five-fold cross-validation to randomly select validation set and training set from the data set at a ratio of 1:4 during each training session.
3.3. Data preprocessing and segmentation
Data preprocessing refers to some processing performed on the data before the main processing. Since the data quality of the data set will directly affect the experimental results, and a good data set will have a positive effect on the results, it is particularly important to perform preprocessing operations on the data before training the model (Zhu et al., Citation2019). First, the invalid features in the database are filtered and removed, including features with all values of 0, features with the variance of 0 and features with low variance, where low variance features refer to more than 80% of the numbers in a single feature with the same value. These features have little influence on the results and are not representative. The variance judgment formula is shown in formula (1). A total of 14 features with zero variance are removed here (Table ).
(1)
(1)
Table 1. Table of 14 zero variance features removed.
Second, standardise and linearly normalise the remaining features, and scale the value of the feature to [0,1]. The standardised formula and the normalised formula are shown in formula (2) and formula (3) respectively:
(2)
(2)
(3)
(3)
Among them, µ is the average value of a single feature, and σ is the standard deviation of a single feature. m is the number of values of a single feature. Xmin is the minimum value among m values of a single feature, and Xmax is the maximum value among m values of a single feature.
Finally, an oversampling operation is performed to construct a balanced data set. By defining the threshold of recombination rate (0.46), positive sites and negative sites were screened out. That is to classify the sites, marking the positive sites greater than the threshold as 1 (positive sample) and the negative sites less than the threshold as 0 (negative sample). A total of 1762 positive samples and 11117 negative samples were collected at last. Then, we performed replacement sampling on the selected positive samples, resulting in a total of 11117 positive samples, which together with the selected 11117 negative samples constituted a balanced data set of 22234 samples. At this point, a list of class features is added to the data set to label the properties of each site. Here, we mark the positive sites as 1 and the negative sites as 0.
The validation set and training set are randomly selected from 22234 data based on the ratio of 1:4, so the numbers of training set and validation set in this experiment are 17787 and 4447, respectively.
3.4. Parameter optimisation of the model
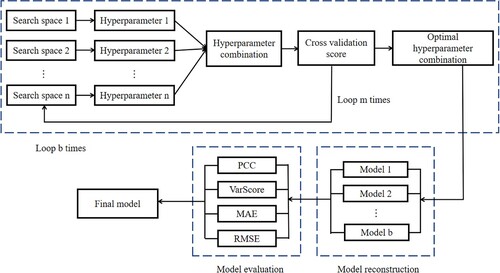
Hyperparameters are parameters that can control the behaviour of machine learning model and affect the performance of model to a great extent. For the most machine learning models, the optimisation training of hyperparameters plays a decisive role in the performance of the model (Chen et al., Citation2022). Therefore, in order to get the optimal hyperparameters of each model, we use Optuna, an efficient hyperparameter optimisation framework. Optuna allows users to dynamically set the search space of hyperparameters according to their own needs, which has the advantages of high efficiency and convenience. Meanwhile, Optuna also provides a visualisation window to visualise the hyperparameter optimisation process, which is very helpful for finding the optimal hyperparameter combination.
The specific process of hyperparameter optimisation is: first, estimate the space of possible values of the hyperparameter, and search for the hyperparameter in this space. When new results appear, update the interval and continue searching, using repeated searches and evaluation updates to find better performing hyperparameters. Here, we perform 4 iterative experiments of 100 rounds for each model, and select the four groups of hyperparameter parameter combinations with the highest score by five-fold cross-validation and then select the optimal hyperparameter combination through model evaluation indicators to establish the final predictive model (Figure ).
Figure 3. Flow chart of model hyperparameter optimisation. b is the number of hyperparameter training; m is the number of hyperparameter optimisation iterations.
3.5. Algorithm description of XRLattCPred
Machine learning can integrate different algorithms for optimisation, which can bring the advantages of each model into full play and achieve higher performance. Random forest is to build a forest in a random way. The forest is composed of many decision trees (CART), and there is no correlation between each decision tree (Nagra et al., Citation2019; Zhang et al., Citation2020). Random forest utilises many decision trees to train and predict samples, which can be used to implement features such as feature selection (Liu et al., Citation2022; Too et al., Citation2021), regression prediction (Xu et al., Citation2021), and classification prediction (Cao et al., Citation2021). The simple average method is usually used in regression, and the regression results obtained by multiple weak learners are arithmetic averaged, which is the final model output (Abdulkareem & Abdulazeez, Citation2021). The XGBoost (Extreme Gradient Boosting) algorithm is improved from the GBDT algorithm. It is an integrated learning algorithm on account of boosting. XGBoost integrates multiple decision trees to form a strong classifier from multiple weak classifiers. On this basis, the objective function adopts the second-order Taylor expansion and adds a regularisation term to find the optimal solution of the model (Chen & Guestrin, Citation2016). LightGBM algorithm uses the histogram algorithm. The basic idea is to discretize continuous floating-point feature values into k integers, and at the same time construct a histogram with a width of k. LightGBM supports parallel learning, so that it has faster training speed and higher efficiency, and has the ability to process big data (Song et al., Citation2021).
Based on the idea of machine learning, this paper adopts the strategy of combination optimisation and parameter optimisation to construct the prediction algorithm XRLattCPred, which is to find the combination with the best performance by combining and reconstructing the above three algorithms together. The algorithm XRLattCPred achieves a significant improvement in prediction efficiency, and the algorithm has high flexibility and portability. Figure shows the prediction process of the algorithm XRLattCPred.
Figure 4. Work flow chart of the algorithm XRLattCPred. The green box indicates the file format required for a given step.
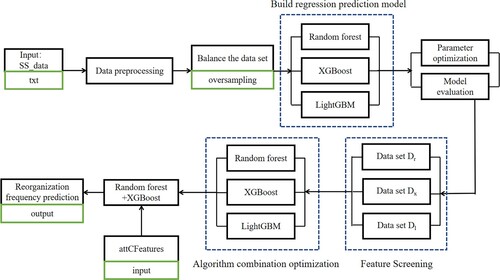
The algorithm XRLattCPred performs site prediction by learning the relationship between the structural characteristics of the recombination site and the recombination rate. The input of the algorithm is the structural feature data set D of the attC site, the attC data set Z to be predicted, and the threshold of the recombination rate, and the output is the recombination rate of each point in the data set Z. The models used in the algorithm XRLattCPred are Random forest, XGBoost and LightGBM. Through parameter optimisation and model evaluation, the study found that the model constructed by Random forest + XGBoost has better performance, so it is used as the final prediction model. The specific description is as follows: First, perform the preprocessing operation on the initial data set D to obtain the data set D’, and then obtain a balanced data set D” after oversampling; Using the algorithm Random forest, the algorithm XGBoost and the algorithm LightGBM to construct the initial prediction model, and perform operations such as parameter optimisation and model reconstruction; Input the data set D” into the above model to obtain the first 20 important features in the feature sequence output by each model, and name them as the data sets Dr, Dx and Dl; The above three data sets are input into Random forest algorithm, XGBoost algorithm and LightGBM algorithm as training sets respectively, and nine algorithm combination models are constructed; The Random forest + XGBoost model is constructed as the model to obtain the optimal score, which is used as the final prediction model; Finally, the data set Z is input to predict the recombination rate. The pseudo code of the algorithm XRLattCPred is as follows:
Table
As shown in the algorithm XRLattCPred, nine algorithm combination models are established in the experiment, which shows that there are order restrictions between different algorithm combinations, and different arrangement orders may lead to different model results. In this paper, the combined model with the best prediction performance is named XRLattCPred as the final prediction model.
3.6. Evaluation index
The evaluation index is an intuitive performance that reflects the quality of the model. In the experiment, we used four different evaluation indicators to evaluate this model. They are Mean Absolute Error (MAE), Root Mean Square Error (RMSE), Pearson Correlation Coefficient (PCC) and Explained Variance Score (VarScore), respectively.
MAE represents the sum of the average absolute value of the predicted value and the actual value. The smaller the value, the better the fit between the two sets of data. The calculation formula of MAE is shown in formula (4):
(4)
(4)
RMSE is the square root of the variance of the residual (the predicted value minus the actual value). The fitting effect of the two sets of data increases with decreasing RMSE values. The calculation formula of RMSE is shown in formula (5):
(5)
(5)
PCC indicates the degree of correlation between two variables, and the value is usually between [0, 1]. The stronger the correlation between the two sets of variables, the closer the value is to 1, on the contrary, the weaker the correlation, the closer the value is to 0 (Shah & Zaveri, Citation2021; Wiedermann & Hagmann, Citation2016). Here, we expect the model to obtain a higher PCC score, which means that the model has higher prediction accuracy, better prediction performance, and higher data correlation. The calculation formula of PCC is shown in formula (6):
(6)
(6)
The value of VarScore is located at [0,1]. When this value is closer to 1, the independent variable can clearly explain the variance of the dependent variable. If this value is smaller, its effect will be worse. The calculation formula of VarScore is shown in formula (7):
(7)
(7)
Among them, yi and ui represent the actual recombination frequency and the predicted recombination frequency respectively, and
are their mean values, n is the total number of data points, and VarScore is the variance of each distribution.
4. Results and discussion
4.1. Experimental environment configuration
All the algorithms used in this paper are available in the Python scikit-learn library, and the experimental environment is python 3.6.
4.2. Parameter settings
Parameter setting is one of the necessary conditions for the model to have good performance. In this paper, we utilised parameter optimisation to find the optimal hyperparameters for each model. The specific parameters are shown in Table .
Table 2. Parameter setting of each model.
The parameter n_estimators represents the number of decision trees in the forest. The larger the n_estimators, the stronger the learning ability of the model and the easier the model is to overfit. The parameter max_depth represents the maximum depth of the decision tree, which can effectively limit overfitting. The parameter learning_rate is the learning rate, that is, the step size of the decision tree iteration. The larger the learning_rate, the faster the iteration speed, but it may not converge to the real optimum. The smaller the learning_rate, the more likely it is to find a more precise optimal value, but the iteration speed will be slower. Their search scopes are shown in Table .
Table 3. Search scopes for each parameter.
4.3. Efficient prediction of XRLattCPred
In this paper, XRLattCPred is used to predict the recombination rate of attCr0 mutants, and a prediction algorithm based on structural features is established. In the algorithm XRLattCPred, the input is used to describe the structural characteristics of attC sites, and the output is the recombination rate. By learning the relationship between structural features and recombination, the algorithm XRLattCPred can infer which important structural features the recombination is based on, which is very helpful for improving the method of synthesising recombination sites. The algorithm XRLattCPred is based on the improvement of the combination optimisation strategy. It establishes a prediction model through the first 20 important features in the feature sequence, and cross-uses Random forest algorithm, XGBoost algorithm and LightGBM algorithm. A total of nine algorithm combinations are constructed and selected the best performance algorithm combination of Random forest + XGBoost (Table ).
Table 4. Evaluation index score table of nine algorithm combinations.
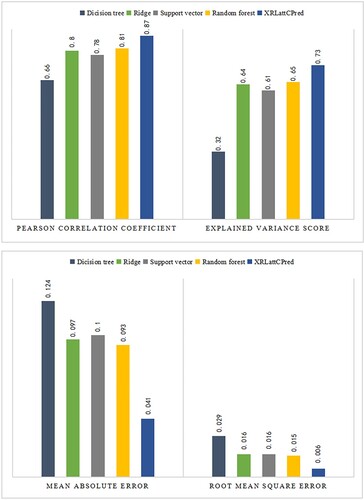
The experimental results show that the scores of the algorithm XRLattCPred on the four evaluation indicators are PCC = 0.87, VarScore = 0.73, MAE = 0.041 and RMSE = 0.006, respectively. Among them, a high PCC score indicates a high correlation between the real results and the predicted results, a high VarScore indicates that the algorithm can better reflect the changes of the data set, a low MAE score indicates that there is a smaller error between the predicted results and the real results, and a low RMSE score indicates that the predicted results are closer to the real results. In conclusion, the algorithm XRLattCPred achieves high precision prediction of attC sites and achieves smaller prediction error. We can conclude that the use of smaller feature sequences can still achieve effective prediction of attC sites from this experiment.
4.4. Comparison with other algorithms
Traditional regression algorithms usually have good predictive performance, so we also compare the algorithm XRLattCPred with traditional Decision tree regression, Ridge regression, Support vector regression and Random forest regression algorithms, and the experimental results show the strong performance of the algorithm XRLattCPred (the comparison result is shown in Figure ). It can be seen from the overall comparison of the experimental results that in the four evaluation index dimensions, the algorithm XRLattCPred has achieved good scores. Among them, the Pearson correlation coefficient and the explained variance score both obtained the highest scores, PCC = 0.87 and VarScore = 0.73, respectively. This shows that the correlation between the value predicted by the algorithm XRLattCPred and the actual value is higher than other models. The scores of mean absolute error and root mean square error scores were the lowest, MAE = 0.041 and RMSE = 0.006, respectively. This shows that the algorithm XRLattCPred has a smaller error. To sum up, the algorithm XRLattCPred with higher prediction accuracy and better performance. Meanwhile, according to the results, we can find that there are features that have opposite effects on recombination in the feature sequences describing the attC site.
Figure 5. Comparison of the results in the histogram.
4.5. Important features filtered out
Regression algorithm is a supervised learning technique commonly used in machine learning. Data prediction can learn the relationship between independent and dependent variables and estimate unknown continuous functions based on limited data points. For the above experiments, it can be seen that the regression algorithm XGBattCPred has achieved a good score on the attCr0 mutant data set, which indicates that the regression algorithm has learned the relationship between the structural characteristics of attC sites and the recombination rate.
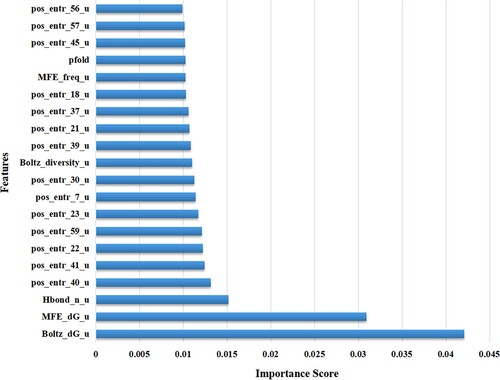
Therefore, this paper judges the importance of the feature by calculating the correlation between the relevant feature and the target value, so as to realise the scoring output of the feature sequence. This can not only screen out important features that contribute to recombination, but also be of great help in improving the synthesis method of attC sites. In this paper, accumulating and summing the 10 feature scoring sequences output by Random forest, XGBoost and LightGBM, not only effectively reduces the randomness and uncertainty caused by a single result, but also makes the output feature scoring sequence obtain higher reliability and stability. The importance score of each feature in the feature sequence is the value between the interval [0,1], and the sum of the scores of all features is equal to 1, and the important features will get higher scores. The top 20 important features obtained through XRLattCPred are the final result of the feature sequence output by the Random forest (Figure ). From the output feature score map, we can see that the recombination rate of attC sites is a multi-factor function, and the final result is based on a series of important features. Therefore, we propose the algorithm XRLattCPred based on important features to establish a predicting model.
Figure 6. Score map of the top 20 important features output by Random forest.
5. Conclusions
Due to the high importance of DNA recombination sites for gene therapy and drug development, we studied the prediction problem of DNA recombination sites and proposed an attC site prediction algorithm XRLattCPred based on combination optimisation strategy. Algorithm XRLattCPred adopts combination optimisation strategy, which combines different algorithms to give full play to the advantages of each algorithm and improve the accuracy of prediction. In addition, the XRLattCPred algorithm is also suitable for other specific sites and other genetic elements based on sequence features, with good portability.
In this work, the data set we used is the structural feature data of attC site mutants, and the recombination of attC site may also be influenced by the environment, integron and other factors. Therefore, in the future, we will consider more features and establish an attC site prediction algorithm based on multiple features to further improve prediction accuracy and credibility.
Acknowledgments
The paper was greatly supported by NNSF (National Natural Science Foundation of China), with grant numbers 61672328, 61672323, and the research is also supported by the Science and Research Plan of Luoyang Branch of Henan Tobacco Company No. 2020410300270078.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Abdulkareem, N. M., & Abdulazeez, A. M. (2021). Machine learning classification based on Random forest algorithm: A review. International Journal of Science and Business, 5(2), 128–142. https://doi.org/10.5281/zenodo.4471118
- Bessen, J. L., Afeyan, L. K., Dančík, V., Koblan, L. W., Thompson, D. B., Leichner, C., Clemon, P. A., & Liu, D. R. (2019). Double-slit photoelectron interference in strong-field ionization of the neon dimer. Nature Communications, 10(1), 1–13. https://doi.org/10.1038/s41467-018-07882-8
- Bikard, D., Julié-Galau, S., Cambray, G., & Mazel, D. (2010). The synthetic integron: An in vivo genetic shuffling device. Nucleic Acids Research, 38(15), e153. https://doi.org/10.1093/nar/gkq511
- Bouvier, M., Ducos-Galand, M., Loot, C., Bikard, D., & Mazel, D. (2009). Structural features of single-stranded integron cassette attC sites and their role in strand selection. PLoS Genetics, 5(9), e1000632. https://doi.org/10.1371/journal.pgen.1000632
- Cao, Y., Geddes, T. A., Yang, J. Y. H., & Yang, P. (2020). Ensemble deep learning in bioinformatics. Nature Machine Intelligence, 2(9), 500–508. https://doi.org/10.1038/s42256-020-0217-y
- Cao, Z., Zhou, Y., Yang, A., & Peng, S. (2021). Deep transfer learning mechanism for fine-grained cross-domain sentiment classification. Connection Science, 33(4), 911–928. https://doi.org/10.1080/09540091.2021.1912711
- Chen, T., & Guestrin, C. (2016). Xgboost: A scalable tree Boosting system. Kdd ‘16: Proceedings of the 22nd ACM SIGKDD International conference on knowledge discovery and data mining, 785-794. https://doi.org/10.1145/2939672.2939785
- Chen, Z., Wang, C., Jin, H., Li, J., Zhang, S., & Ouyang, Q. (2022). Hierarchical-fuzzy allocation and multi-parameter adjustment prediction for industrial loading optimisation. Connection Science, 34(1), 687–708. https://doi.org/10.1080/09540091.2022.2031887
- Cury, J., Jové, T., Touchon, M., Néron, B., & Rocha, E. P. C. (2016). Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Research, 44(10), 4539–4550. https://doi.org/10.1093/nar/gkw319
- Domingues, S., Silva1, G. J. d., & Nielsen, K. M. (2012). Integrons. Mobile Genetic Elements, 2(5), 211–223. https://doi.org/10.4161/mge.22967
- Doudna, J. A. (2020). The promise and challenge of therapeutic genome editing. Nature, 578(7794), 229–236. https://doi.org/10.1038/s41586-020-1978-5
- Epum, E. A., & Haber, J. E. (2022). Dna replication: The recombination connection. Trends in Cell Biology, 32(1), 45–57. https://doi.org/10.1016/j.tcb.2021.07.005
- Frumerie, C., Ducos-Galand, M., Gopaul, D. N., & Mazel, D. (2010). The relaxed requirements of the integron cleavage site allow predictable changes in integron target specificity. Nucleic Acids Research, 38(2), 559–569. https://doi.org/10.1093/nar/gkp990
- Ghaly, T. M., Tetu, S. G., & Gillings, M. R. (2021). Predicting the taxonomic and environmental sources of integron gene cassettes using structural and sequence homology of attC sites. Communications Biology, 4(1), 946–946. https://doi.org/10.1038/s42003-021-02489-0
- Grieb, M. S., Nivina, A., Cheeseman, B. L., Hartmann, A., Mazel, D., & Schlierf, M. (2017). Dynamic stepwise opening of integron attC DNA hairpins by SSB prevents toxicity and ensures functionality. Nucleic Acids Research, 45(18), 10555–10563. https://doi.org/10.1093/nar/gkx670
- Häcker, I., Harrell IIR. A., Eichner, G., Pilitt, K. L., O’Brochta, D. A., Handler, A. M., & Schetelig, M. F. (2017). Cre/lox-recombinase-mediated cassette exchange for reversible site-specific genomic targeting of the disease vector, aedes aegypti. Scientific Reports, 7(1), 43883. https://doi.org/10.1038/srep43883
- Hoerl, A. E., & Kennard, R. W. (2000). Ridge regression: Biased estimation for nonorthogonal problems. Technometrics, 42(1), 80–86. https://doi.org/10.1080/00401706.2000.10485983
- Joss, M. J., Koenig, J. E., Labbate, M., Polz, M. F., Gillings, M. R., Stokes, H. W., Doolittle, W. F., & Boucher, Y. (2009). Acid: Annotation of cassette and integron data. BMC Bioinformatics, 10(1), 118. https://doi.org/10.1186/1471-2105-10-118
- Klopfenstein, Q., & Vaiter, S. (2021). Linear support vector regression with linear constraints. Machine Learning, 110(7), 1939–1974. https://doi.org/10.1007/s10994-021-06018-2
- Lacotte, Y., Ploy, M. C., & Raherison, S. (2017). Class 1 integrons are low-cost structures in escherichia coli. The ISME Journal, 11(7), 1535–1544. https://doi.org/10.1038/ismej.2017.38
- Larouche, A., & Roy, P. H. (2011). Effect of attC structure on cassette excision by integron integrases. Mobile DNA, 2(1), 3. https://doi.org/10.1186/1759-8753-2-3
- Lewis, R. J. (2000). An introduction to classification and regression tree (CART) analysis. San Francisco, CA: Annual meeting of the society for academic emergency medicine, 1-14. https://www.researchgate.net/publication/240719582
- Libbrecht, M. W., & Noble, W. S. (2015). Machine learning applications in genetics and genomics. Nature Reviews Genetics, 16(6), 321–332. https://doi.org/10.1038/nrg3920
- Liu, G., Ma, J., Hu, T., & Gao, X. (2022). A feature selection method with feature ranking using genetic programming. Connection Science, 34(1), 1146–1168. https://doi.org/10.1080/09540091.2022.2049702
- Liu, T., Yin, J., Gao, L., Chen, W., & Qiu, M. (2019). Consensus RNA secondary structure prediction using information of neighbouring columns and principal component analysis. International Journal of Computational Science and Engineering, 19(3), 430–439. https://doi.org/10.1504/IJCSE.2019.10022734
- Liu, Z., Yang, Y., Li, D., Lv, X., Chen, X., & Dai, Q. (2022). Prediction of the RNA tertiary structure based on a Random sampling strategy and parallel mechanism. Frontiers in Genetics, 12, 813604. https://doi.org/10.3389/fgene.2021.813604
- Liu, Z., Zhu, D., & Dai, Q. (2019). Improved predicting algorithm of RNA pseudoknotted structure. International Journal of Computational Science and Engineering, 19(1), 64–70. https://doi.org/10.1504/IJCSE.2019.099641
- Mazel, D., Escudero, J. A., Nivina, A., & Loot, C. (2015). The integron: Adaptation On demand. Microbiology Spectrum, 3(2), MDNA3-0019-2014. https://doi.org/10.1128/microbiolspec.MDNA3-0019-2014
- Mukhortava, A., Pöge, M., Grieb, M. S., Nivina, A., Loot, C., Mazel, D., & Schlierf, M. (2019). Structural heterogeneity of attC integron recombination sites revealed by optical tweezers. Nucleic Acids Research, 47(4), 1861–1870. https://doi.org/10.1093/nar/gky1258
- Nagra, A. A., Han, F., Ling, Q., Abubaker, M., Ahmad, F., Mehta, S., & Apasiba, A. T. (2019). Hybrid self-inertia weight adaptive particle swarm optimisation with local search using C4.5 decision tree classifier for feature selection problems. Connection Science, 32(1), 16–36. https://doi.org/10.1080/09540091.2019.1609419
- Nivina, A., Escudero, J. A., Vit, C., Mazel, D., & Loot, C. (2016). Efficiency of integron cassette insertion in correct orientation is ensured by the interplay of the three unpaired features of attC recombination sites. Nucleic Acids Research, 44(16), 7792–7803. https://doi.org/10.1093/nar/gkw646
- Nivina, A., Grieb, M. S., Loot, C., Bikard, D., Cury, J., Shehata, L., Bernardes, J., & Mazel, D. (2020). Structure-specific DNA recombination sites: Design, validation, and machine learning–based refinement. Science Advances, 6(30), eaay2922. https://doi.org/10.1126/sciadv.aay2922
- Olorunniji, F. J., Rosser, S. J., & Stark, W. M. (2016). Site-specific recombinases: Molecular machines for the genetic revolution. Biochemical Journal, 473(6), 673–684. https://doi.org/10.1042/BJ20151112
- Pellegrino, E., Jacques, C., Beaufils, N., Nanni, I., Carlioz, A., Metellus, P., & Ouafk, L. (2021). Machine learning random forest for predicting oncosomatic variant NGS analysis. Scientific Reports, 11(1), 21820. https://doi.org/10.1038/s41598-021-01253-y
- Pereira, M. B., Wallroth, M., Kristiansson, E., & Axelson-Fisk, M. (2016). HattCI: Fast and accurate attC site identification using hidden Markov models. Journal of Computational Biology, 23(11), 891–902. https://doi.org/10.1089/cmb.2016.0024
- Rowe-Magnus, D. A., Guerout, A. M., Biskri, L., Bouige, P., & Mazel, D. (2003). Comparative analysis of superintegrons: Engineering extensive genetic diversity in the vibrionaceae. Genome Research, 13(3), 428–442. https://doi.org/10.1101/gr.617103
- Shah, D., & Zaveri, T. (2021). Hyperspectral endmember extraction using Pearson's correlation coefficient. International Journal of Computational Science and Engineering, 24(1), 89–97. https://doi.org/10.1504/IJCSE.2021.113656
- Song, J., Liu, G., Jiang, J., Zhang, P., & Liang, Y. (2021). Prediction of protein–ATP binding residues based on ensemble of deep convolutional neural networks and LightGBM algorithm. International Journal of Molecular Sciences, 22(2), 939. https://doi.org/10.3390/ijms22020939
- Stalder, T., Barraud, O., Casellas, M., Dagot, C., & Ploy, M. C. (2012). Integron involvement in environmental spread of antibiotic resistance. Frontiers in Microbiology, 3, 119. https://doi.org/10.3389/fmicb.2012.00119
- Tian, X., & Zhou, B. (2021). Strategies for site-specific recombination with high efficiency and precise spatiotemporal resolution. Journal of Biological Chemistry, 296, 100509. https://doi.org/10.1016/j.jbc.2021.100509
- Too, J., Ma, J., Sadiq, A. S., & Mirjalili, S. M. (2021). A conditional opposition-based particle swarm optimisation for feature selection. Connection Science, 34(1), 339–361. https://doi.org/10.1080/09540091.2021.2002266
- Tsafnat, G., Coiera, E., Partridge, S. R., Schaeffer, J., & Iredell, J. R. (2009). Context-driven discovery of gene cassettes in mobile integrons using a computational grammar. BMC Bioinformatics, 10(1), 281. https://doi.org/10.1186/1471-2105-10-281
- Vit, C., Loot, C., Escudero, J. A., Nivina, A., & Mazel, D. (2020). Methods in molecular biology. Methods in Molecular Biology, 2075, 189–208. https://doi.org/10.1007/978-1-4939-9877-7_14
- Wang, C., Wang, Y., & Cheung, Y. (2021). A branch and bound irredundant graph algorithm for large-scale MLCS problems. Pattern Recognition, 119, 108059. https://doi.org/10.1016/j.patcog.2021.108059
- Wei, S., Wang, Y., & Cheung, Y. (2021). A branch elimination-based efficient algorithm for large-scale multiple longest common subsequence problem. IEEE Transactions on Knowledge and Data Engineering, (1), 1. https://doi.org/10.1109/TKDE.2021.3115057
- Weiss, E., Spicher, C., Haas, R., & Fischer, W. (2019). Excision and transfer of an integrating and conjugative element in a bacterial species with high recombination efficiency. Scientific Reports, 9(1), 1–14. https://doi.org/10.1038/s41598-019-45429-z
- Wiedermann, W., & Hagmann, M. (2016). Asymmetric properties of the Pearson correlation coefficient: Correlation as the negative association between linear regression residuals. Communications in Statistics - Theory and Methods, 45(21), 6263–6283. https://doi.org/10.1080/03610926.2014.960582
- Xu, F., Zhao, H., Zhou, W., & Zhou, Y. (2021). Cost-sensitive regression learning on small dataset through intra-cluster product favoured feature selection. Connection Science, 34(1), 104–123. https://doi.org/10.1080/09540091.2021.1970719
- Zhang, F., Gao, D., & Xin, J. (2020). A decision tree algorithm for forest fire prediction based on wireless sensor networks. International Journal of Embedded Systems, 13(4), 422. https://doi.org/10.1504/IJES.2020.10031194
- Zhu, H., Jiang, T., Wang, Y., Cheng, L., Ma, B., & Zhao, F. (2019). A data cleaning method for heterogeneous attribute fusion and record linkage. International Journal of Computational Science and Engineering, 19(3), 311–324. https://doi.org/10.1504/IJCSE.2019.10022717