
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The development of vaccines and drugs is very important in combating the coronavirus disease 2019 (COVID-19) virus. The effectiveness of these developed vaccines and drugs has decreased as a result of the mutation of the COVID-19 virus. Therefore, it is very important to combat COVID-19 mutations. The majority of studies published in the literature are studies other than COVID-19 mutation prediction. We focused on this gap in this study. This study proposes a robust transformer encoder based model with Adam optimizer algorithm called TfrAdmCov for COVID-19 mutation prediction. Our main motivation is to predict the mutations occurring in the COVID-19 virus using the proposed TfrAdmCov model. The experimental results have shown that the proposed TfrAdmCov model outperforms both baseline models and several state-of-the-art models. The proposed TfrAdmCov model reached accuracy of 99.93%, precision of 100.00%, recall of 97.38%, f1-score of 98.67% and MCC of 98.65% on the COVID-19 testing dataset. Moreover, to evaluate the performance of the proposed TfrAdmCov model, we carried out mutation prediction on the influenza A/H3N2 HA dataset. The results obtained are promising for the development of vaccines and drugs.
1. Introduction
Coronaviruses contain a single-stranded positive polarity Ribonucleic Acid (RNA) genome sequence. Coronaviruses were first discovered in the 1960s (Haimed et al., Citation2021). The first found coronaviruses were HCoV-229E and HCoV-OC43 coronaviruses. Later, Severe Acute Respiratory Syndrome Coronavirus 1 (SARS-CoV-1) coronavirus in 2003, HCoV-NL63 coronavirus in 2004, HCoV-HKU1 coronavirus in 2005, MERS (Middle East Respiratory Syndrome Coronavirus) coronavirus in 2012, and finally COVID-19, caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), coronavirus found at the end of December 2019 (Haimed et al., Citation2021). COVID-19 virus renamed by the World Health Organization (WHO) (Sohrabi et al., Citation2020), first appeared in the city of Wuhan, the capital of China’s Hubei province, at the end of December 2019 (Wu et al., Citation2020). The COVID-19 virus has spread to many countries, especially China. Countries have had to go into partial or full shutdowns to combat the COVID-19 virus. This caused great fear and panic among the people (Hai-Dong et al., Citation2022). WHO declared a World Emergency for the COVID-19 outbreak on 30 January 2020, and then announced to the world that it had turned into a pandemic on 11 March 2020 (Sharma et al., Citation2021; Tang et al., Citation2024). As of April 28, 2024, worldwide the number of confirmed cases is 775,379,864, while the number of confirmed deaths is 7,047,396 (World Health Organization, Citation2023). The COVID-19 virus has caused mild symptoms in about 80% of infected people, but acute respiratory distress syndrome (ARDS) in some people (Sharma et al., Citation2021). ARDS can cause multiple organ failure and other serious diseases (Suri et al., Citation2020). Many test kits are described for the diagnosis of the COVID-19 virus (Rashid et al., Citation2020). The most widely used and proven real-time reverse transcriptase polymerase chain reaction (rRT-PCR) is frequently used in the detection of COVID-19 virus (Serena Low et al., Citation2021). rRT-PCR test results are usually available in a few hours to 2 days (Sharma et al., Citation2021). Measures such as physical or social distance, quarantine, ventilation of closed areas, covering the mouth and nose in case of coughing and sneezing have been taken to reduce the effectiveness/spread of the COVID-19 virus. Additionally, various vaccines have been developed and the effectiveness of the COVID-19 virus has been reduced to a certain extent. However, the frequent mutation of the COVID-19 virus has either greatly reduced or eliminated the effectiveness of these vaccines and drugs. For this reason, it has become very difficult to fight the COVID-19 virus. To overcome these challenges, it is vitally important to predict mutations that may occur on the COVID-19 virus. If mutations can be predicted on the COVID-19 virus structure, especially in the S protein, vaccines can be updated quickly even if the COVID-19 virus mutates. Especially recently, Transformer based models are used very effectively in sequence-based mutation tasks. Transformer-based models are particularly successful in natural language processing tasks. The main reason for this is that it is completely attention-based and many attention heads work in parallel. In this study, we propose a robust transformer encoder based model with Adam optimizer algorithm, TfrAdmCov, for COVID-19 mutation prediction. we aim to predict mutations on the COVID-19 virus using the proposed TfrAdmCov model. The proposed TfrAdmCov model has the ability to easily capture long-term dependencies. Thanks to the transformer layer of the proposed TfrAdmCov model, it provides the opportunity to perform large-scale parallel computing. In addition, the information learned through the MHA architecture can be remembered more easily. This supports that the proposed TfrAdmCov model is more robust. As a result, it has been observed that the proposed TfrAdmCov model is quite successful in mutation prediction of the COVID-19 virus compared to both other models and the literature. The contributions of this article have been given below.
We propose a robust transformer encoder based model with Adam optimizer algorithm, TfrAdmCov, for COVID-19 mutation prediction.
We use the GridSearchCV hyperparameter tuning algorithm to improve the performance of machine learning-based models.
We use agglomerative clustering algorithm to create Training and Testing datasets.
We use the stratified 10-fold cross validation technique in addition to the holdout technique to evaluate the performance of machine learning-based models in a healthy way.
We perform mutation prediction both on COVID-19 and infulenza A/ H3N2 HA datasets.
We conduct a detailed performance comparison of the proposed TfrAdmCov model with traditional machine learning algorithms and deep learning algorithms on both COVID-19 and influenza A/ H3N2 HA datasets.
To facilitate readability of the article, the organisation of the remaining part of the article is as follows: Related works are mentioned in Section 2. Section 3 provides detailed information about the COVID-19 virus Section 4 presents the proposed TfrAdmCov model. Section 5 presents experimentations (COVID-19 dataset, the GridSearchCV hyperparameter tuning technique, baseline models, etc.). In Section 6 results and discussion are presented. The limitations of the study are mentioned in Section 7. Finally, the findings are discussed in Section 8.
2. Background
When the literature is examined in detail, the majority of studies are studies other than COVID-19 mutation prediction. Additionally, mutation prediction has been frequently performed on influenza virus. Some relevant studies in the literature are given below. Tarek et al. (Citation2023) proposed a convolutional neural network (CNN)- gated recurrent unit (GRU) hybrid model (CNN-GRU) for COVID-19 death prediction. They predicted COVID-19 deaths on the Indian dataset using the CNN-GRU model. When they compared the proposed CNN-GRU model with existing models, they observed that the proposed CNN-GRU model was more successful. ElAraby et al. (Citation2022) proposed the Gray-scale Spatial Exploitation Net (GSEN) model with stochastic gradient descent optimisation technique to classify the COVID-19 Chest X-ray (CXR) images. The GSEN model outperformed other models. Elzeki et al. (Citation2021) proposed the Chest X-Ray COVID Network (CXRVN) model with mini-batch gradient descent and Adam optimizer to detect the COVID-19 virus from Chest X-ray (CXR) images. The proposed model was tested on three datasets. As a result of the test, the proposed model is very successful in detecting the COVID-19 virus. Raheja et al. (Citation2023) proposed a diffusion prediction model for prediction of number of COVID-19 cases in India, France, China and Nepal countries. They compared the proposed model with other state-of-the art models. The proposed model outperformed other state-of-the art models. Elzeki et al. (Citation2021) proposed a novel perceptual two-layer image fusion using deep learning based model (NSCT + CNN_VGG19) to obtain CXR images for imbalanced COVID-19 dataset. They compared the proposed model in detail with other models. The proposed model achieved better performance than other models. Chakraborty et al. (Citation2022) proposed a COVID-19 risk prediction approach for diabetic patients by a fuzzy inference system and machine learning models. They used stratified k-fold cross-validation technique to evaluate the performance of the proposed model. Experimental results showed that the proposed model is more successful in the COVID-19 risk prediction than other existing models. Hassan et al. (Citation2024) proposed a Deep Convolutional Neural Network (DCNN) model for the classification of COVID-19 from CT scan images. Experimental results showed that the proposed model with Adam optimizer outperformed several state-of-the-art models in the COVID-19 classification task. Shrestha et al. (Citation2022) proposed a Deep Learning Based Convolution Neural Network called DCNN model with Adam optimizer to detect Brain Tumour. Experimental results showed that the proposed DCNN model achieved remarkable results in detecting the Brain Tumour. Hassan et al. (Citation2023) proposed a deep-learning based automatic COVID-19 detection model for smart cities. The proposed model could help reduce further spread of COVID-19, especially in crowded places. Cai et al. (Citation2024) proposed an encoder-decoder based FluPMT model to predict the haemagglutinin (HA) protein sequence of the next season’s dominant strain of Influenza A viruses. They used attention mechanisms to investigate dependencies among residues of sequences and used time series to model the evolution of influenza A viruses. As a result, they showed that the performance of the FluPMT model on both the H1N1 dataset and the H3N2 dataset was better than other models. Li et al. (Citation2023) proposed a graph deep learning network-based model, GraphLncLoc for predicting long non-coding RNAs (lncRNAs) subcellular localisation. The GraphLncLoc uses graph convolutional networks to learn latent features and then the high-level features obtained are fed into a fully connected layer to carry out the final prediction. They showed that the GraphLncLoc model achieved better performance than other models. Yin et al. (Citation2022) proposed, named as IAV-CNN, a 2D convolutional neural network (CNN) based model to predict influenza antigenic variants. The IAV-CNN model and other models have been trained and tested on three influenza datasets (H1N1, H3N2 and H5N1). Experimental results show that the IAV-CNN outperforms state-of-the-art models on three influenza datasets. Abbas et al. (Citation2022) applied various sequence-to-sequence deep learning models on antigenic influenza HA sequence pairs and then attempted to generate the antigenic pair of an emerging influenza A virus. They observed that the proposed deep learning models achieved remarkable results on the influenza A virus. Salama et al. (Citation2016) estimated RNA virus mutations by using amino acid sequences of proteins that make up RNA, neural networks (NN) and rough set techniques. In their study, they used a dataset consisting of Newcastle RNA virus sequences obtained from China and South Korea. When the results were examined, they observed that the coarse set technique gave better results than neural networks. They showed that the accuracy rate of the coarse set technique is over 75%. Mohamed et al. (Citation2021) predicted the next DNA sequence using seq2seq LSTM deep learning in their study. In their study, they used the New Castle disease virus dataset and the H1N1 Influenza virus dataset. The New Castle disease virus dataset consists of 83 samples with a sequence length of 1778. The H1N1 Influenza virus dataset consists of 4609 samples with a sequence length of 535. While the success rate (accuracy) of the proposed seq2seq LSTM model on the New Castle disease virus dataset was 96.9%, the success rate (accuracy) on the H1N1 Influenza Virus dataset was 98.9%. Yin et al. (Citation2020) predicted whether mutations would occur in the next flu season using the haemagglutinin (HA) protein sequences of influenza A/H1N1, H3N2, H5N1 viruses in their study. They proposed the Tempel model, which is an efficient and robust time series mutation prediction model for mutation prediction of influenza A viruses. In their study, when the experimental results on three influenza datasets (H1N1, H3N2, H5N1) are examined, it is seen that the proposed Tempel model is compatible with other approaches commonly used in the literature (baseline, LR, SVM, Recurrent Neural Network (RNN), Gated Recurrent Unit (GRU), LSTM). They showed that it can significantly improve predictive performance and provide new insights into viral mutation and evolutionary dynamics. The proposed Tempel model achieved a performance value of 0.991. Yin et al. (Citation2023) proposed ViPal model, a general framework, for virulence prediction. They used a posterior regularisation technique to transform prior viral knowledge into constraint features. The ViPal model could improve virulence prediction performance compared to other models. As far as we have researched in the literature, there are very few studies on the mutation prediction of the COVID-19 virus. We carried out this study to contribute to the literature. Some studies in the literature on COVID-19 mutations and other coronavirus mutations that have appeared before can be summarised as follows. Saha et al. (Citation2020) used 566 COVID-19 genome sequences isolated in India for mutation analysis in their study. Alignment of the sequences was performed using the multiple sequence alignment method (MSA) CLUSTALW (Anonymous, Citation2023a) using the reference (NC_45512.2) sequence from the National Center for Biotechnology Information (NCBI). Once the sequences were aligned, they created a consensus sequence to locate the mutation site and analyse each COVID-19 genome. As a result of the study, they identified a total of 3384 mutation points, including 933 substitution/point mutations, 2449 deletion mutations and 2 insertion mutations in 566 genome sequences isolated in India. Wang et al. (Citation2020) used 31,421 COVID-19 genome sequences for mutation analysis in their study. In this study, they calculated the mutation rate and mutation h-index of all COVID-19 genes. They showed that among the genes that make up the structure of the COVID-19 virus, the N gene was mutated the most. They also stated that the N gene is the most vulnerable gene in the COVID-19 genome. Haimed et al. (Citation2021) proposed a reverse engineering approach to reveal the patterns and evolutionary behaviour of the COVID-19 virus using AI and Big Data. They used the Long Short Term Memory (LSTM) method to predict the next evolved instance of the COVID-19 virus. Also, they used the amino acid sequence ORF7a, which is a 29 amino acid long small protein of the COVID-19 virus. At the end of the study, they predicted the possible evolved sample of the ORF7a protein with a success rate of 40–50%. In order to increase this success rate, they increased the success rate to 72% by using consistent patterns. Nawaz et al. (Citation2021) obtained detailed information from COVID-19 genome sequences by using AI techniques in their study. They experimented with sequential pattern mining (SPM) in a computer environment to see if there are hidden patterns that reveal the frequent patterns of nucleotide bases and their relationships with each other. They also proposed an algorithm for mutation analysis in genome sequences to find the places where nucleotide bases were changed in genome sequences and calculate the mutation rate. When the results obtained were examined, it was seen that SPM revealed important information and patterns in the COVID-19 genome sequences to study the evolution or variations in COVID-19 strains. Hossain et al. (Citation2021a) estimated the mutation rate of the 2000th variant, which may occur in the future, by applying the LSTM deep learning model to the COVID-19 genome sequence in their study. they used a total of 259,044 COVID-19 whole genome sequences, including other regions. They identified a total of 3,334,545 mutations from these used samples. In their study, Zhou et al. (Citation2023) proposed the TEMPO model, called a transformer-based mutation prediction framework, for COVID-19 mutation prediction in their study. They designed a phylogenetic tree-based sampling method to generate viral sequences assembled with temporal information. In addition, they stated that the proposed TEMPO model could successfully predict 22 mutations that had not occurred before. The TEMPO model they proposed obtained an accuracy value of 0.655 on the COVID-19 dataset. As result, when the literature has been examined in detail, the majority of the studies are on other aspects of the COVID-19 virus. However, there are very few studies on mutation prediction of the COVID-19 virus using artificial intelligence-based models.
In this study, we focused on this gap in the literature. This presents a robust transformer encoder based model with Adam optimizer algorithm, TfrAdmCov, for COVID-19 mutation prediction. The proposed TfrAdmCov model has the ability to easily capture long-term dependencies. The attention mechanisms in the transformer encoder layer focuses only on the most important features in the feature set to maximise the performance of the model during training. In this way, it reduces unnecessary computational resources and allows the model to achieve better generalisation performance. The transformer encoder model can easily capture long-term dependencies by its attention mechanism in the input sequence and perform large-scale parallel calculation unlike other deep learning models. As a result, the proposed TfrAdmCov model is quite successful in genetic sequence mutation prediction of the COVID-19 virus, compared to both other models and the state-of-the-art models in the literature.
3. COVID-19 (SARS-CoV-2)
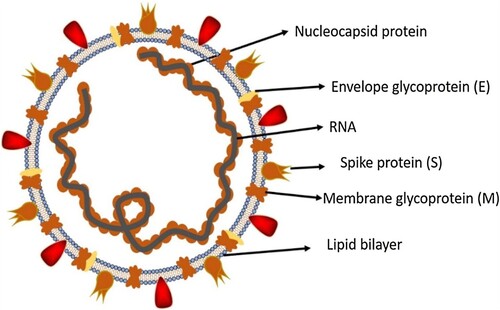
COVID-19 virus is a genus of positive-sense betacoronavirus belonging to the Coronaviridae family with enveloped, single-stranded RNA genomes (de Wit & Cook, Citation2020). Coronaviruses have four types, alpha, beta, gamma and delta (Jaimes et al., Citation2020). Human coronaviruses are in alpha and beta genera (Shereen et al., Citation2020). Some human corona viruses (HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1) infect the simple seasonal upper respiratory tract, while others (SARS-CoV-1, MERS and COVID-19) cause pneumonia and severe ARDS can cause illness (Cui et al., Citation2019). The genome of the COVID-19 virus has the highest genome similarity with the bat-RaTG13 coronavirus of all coronaviruses over 96%. It also shows genomic similarity to SARS-CoV-1 over 79% and MERS coronavirus over 50% (Sharma et al., Citation2021). The genome of the first COVID-19 virus to appear in China has a length range of 29,903 kilobases (Nawaz et al., Citation2021). The structure of the COVID-19 virus consists of structural proteins (S, Envelope (E), Membrane (M) and Nucleocapsid (N)), non-structural proteins (NSP1-NSP16) and auxiliary proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c and ORF10) (C. rong Wu et al., Citation2022). Figure shows the structure of the COVID-19 virus.
Figure 1. Structure of the COVID-19 virus (Shereen et al., Citation2020).
3.1 COVID-19 S (Spike) protein
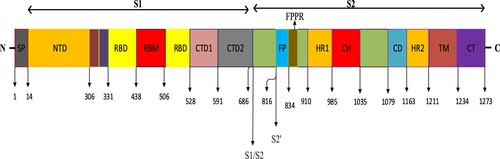
The S protein on the surface of the COVID-19 virus used as the dataset is expressed as a transmembrane glycoprotein (Zhang et al., Citation2021). The S protein consists of 1273 amino acids in total (Anonymous, Citation2023b). As seen in Figure , the COVID-19 S protein consists of Signal Peptide (SP), S1, S1/S2, S2 subunits (Huang et al., Citation2020). The S1 subunit consists of N-Terminal Domain (NTD), Receptor-Binding Domain (RBD), and C-Terminal Domain 1 (CTD1) and C-Terminal Domain 2 (CTD2), while the S2 subunit consists of Fusion Peptide (FP), Fusion-Peptide Proximal Region (FPPR), Heptad Repeat 1 (HR1), Central Helix (CH), Connector Domain (CD), Heptad Repeat 2 (HR2), Transmembrane Domain (TM) and Cytoplasmic Tail (CT) domains (Barnes et al., Citation2020). Via the RBD domain in the COVID-19 S protein structure, the virus binds to the Angiotensin Converting Enzyme 2 (ACE-2) protein on the host cell surface. Then, using the S2 subunit, fusion with the host cell takes place, and then the COVID-19 virus enters the host cell. Upon binding to the ACE2 receptor, the S protein undergoes conformational changes, enabling cleavage of the S protein by furin proteases in the S1/S2 region to produce the S1 and S2 subunits. In order to facilitate the entry of the COVID-19 virus into the host cell, the transmembrane serine protease-2 (TMPRSS2S2) on the cell surface plays a role in the preparation of the S protein by dividing the S2′ domain in the S2 subunit. COVID-19 S RBD contains a receptor binding motif (RBM) and a core structure (Jackson et al., Citation2022). Figure shows the detailed structure of the COVID-19 S protein.
Figure 2. Detailed structure of the COVID-19 S protein (Jackson et al., Citation2022).
3.2. Mutation
Mutation can be briefly expressed as permanent changes that occur in the Deoxyribonucleic Acid (DNA) or RNA sequence in a living thing’s genome. RNA viruses mutate more than DNA viruses (Shaikh et al., Citation2021). In particular, the virus frequently mutates while copying its RNA genome in the host cell (Qin et al., Citation2021). The COVID-19 virus has mutated many times (Hossain et al., Citation2021b). Developed test kits cannot fully capture the dominant COVID-19 variants. The effectiveness of existing vaccines is also significantly reduced against mutations. Understanding the genome sequence of the COVID-19 virus, its behaviour and origin, and how quickly it mutates is very important for the development of vaccines/drugs (Haimed et al., Citation2021). The COVID-19 virus has mutated in different regions over time, revealing new variants. Although the majority of these new variants did not cause any adverse effects, they changed the course of the epidemic due to the contagiousness/fatality of some dominant variants such as delta/omicron (Shiehzadegan et al., Citation2021). Detailed analysis of the genome sequence of the COVID-19 virus and mutation analysis will contribute to the development of vaccines or drugs (Ahmed & Jeon, Citation2022). So far, some dominant variants caused by the COVID-19 virus can be expressed as follows; B.1.1.7 (Alpha) variant detected in the UK, B.1.351 (Beta) variant and B.1.1.529 (Omicron) variant detected in South Africa, P.1 (Gamma) detected in Brazil variant can be referred to as variant B.1.617.2 (Delta) detected in India (Qin et al., Citation2021; Lopez-Rincon et al., Citation2021; Gage et al., Citation2021; Sokhansanj & Rosen, Citation2022). Based on available data, variant B.1.1.529 (Omicron) is the predominant variant worldwide (Madhi et al., Citation2022). Updates are also made in the Covid-19 test kits in accordance with the detected variants. In this way, negative results can be prevented in updated test kits in patients infected with mutated viruses. With the emergence of COVID-19 mutations, the effects of available vaccines have decreased significantly. Analysis of the genome sequencing of the COVID-19 coronavirus and the use of advanced machine learning-based models can help physicians understand the genetic makeup of the COVID-19 virus. In addition, understanding the genome sequencing of the COVID-19 virus will contribute to the development of vaccines or drugs to be developed (Ahmed & Jeon, Citation2022).
4. The proposed TfrAdmCov model
The transformer encoder model is a fully attention mechanism-based architecture and is frequently used in natural language processing (NLP) tasks (Kalyan et al., Citation2021). The attention mechanism in the transformer encoder layer focuses only on the most important features in the feature set to maximise the performance of the model during training. In this way, it reduces unnecessary computational resources and allows the model to achieve better generalisation performance. The transformer encoder model can easily capture long-term dependencies by its attention mechanism in the input sequence and perform large-scale parallel calculation (Zhou et al., Citation2023). The standard transformer architecture is composed of transformer encoder-decoder layers. Because we will not make machine translation, we only used the transformer encoder layer. Each transformer encoder layer has two sublayers: multi-head attention (MHA) and feedforward network (FFN). Moreover, the transformer encoder layer has a residual connection around each of the two sub-layers, followed by layer normalization (Vaswani et al., Citation2017; Pacal, Citation2024a). Scaled-dot product attention has been shown in Figure . Scaled-dot product attention mechanism utilises weight matrices for adjusting model parameters during training. The
and
vectors are obtained via matrix multiplication between the weight matrices
and the embedded inputs
: The index
presents the token index position in the input sequence, which has length
.
. An attention function makes a query
and a set of key-value pairs
to an output. The output is computed as a weighted sum of the values (Vaswani et al., Citation2017; Galassi et al., Citation2021). The attention function has been given in Equation (1).
(1)
(1) where
, is the key dimensionality,
is the value dimensionality.
is used to scale the attention weights.
Figure 3. Scaled-dot product attention. Where is the number of tokens in the input sequence and
is the dimensionality of those tokens.
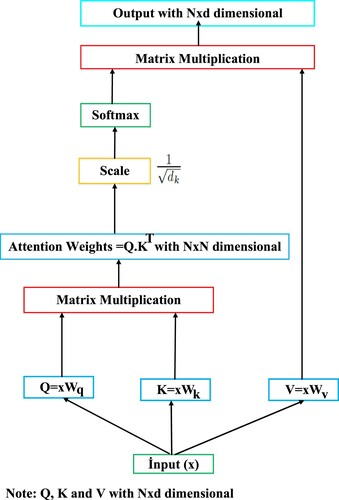
As seen in Figure , matrices are obtained using the input sequence
. Then, attention weights are obtained as a result of multiplying
. The data obtained is scaled by multiplying the attention weights by
. The scaled data is given as input to the softmax function. The data is normalised with the softmax function and the final output is obtained by multiplying the normalised data with the
matrix. Where
is the number of tokens in the input sequence and
is the dimensionality of those tokens. MHA can be expressed as a mechanism that allows the model to jointly participate in information from different representational subspaces at different locations. In other words, MHA allows each token task in the input sequence to be shared with different self-attention heads by using one or more self-attentions running simultaneously or in parallel. This allows multiple operations to be performed at the same time, unlike RNN-based models which in the output depends on the previous input. The MHA mechanism relies on one or more scaled dot-product attention (self-attention) operating on a key
, a value
and a query
(Voita et al., Citation2020). Now, let MHA be indexed by
, then we calculate the multi-head attention function using the function in Equations (2) and (3).
(2)
(2)
(3)
(3)
(Vaswani et al., Citation2017). where
,
,
are projection matrices.
is a final linear projection matrice (Voita et al., Citation2020). MHA has been shown in Figure . In this work, we employ h = 2 parallel running scaled-dot product attention layers or heads in MHA. For each scaled-dot product attention layer, we use
.
Figure 4. MHA. is the number of total scaled-dot product attention running in parallel.
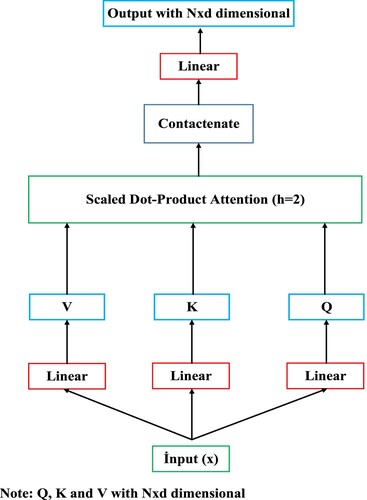
As seen in Figure , To obtain matrices, the input sequence
is passed through the linear layer. Then,
matrices are given as input to scaled-dot product attention layers. The outputs obtained from the scaled-dot product attention layers are combined and the outputs are passed through the linear layer to obtain the final output. The transformer encoder layer consists of one fully connected FFN layer which consists of two linear trasformations and one RELU activation between these two linear trasformations. The formulation of FNN layer has been given in Equation (4).
(4)
(4) where
, (
,
),
represent the input embedding, weights and biases, respectively (Vaswani et al., Citation2017). In addition, in the transformer encoder, residual or skip connection helps preserve the input sequence, allowing the transformer model to learn more complex functions. Additionally, residual connection helps avoid the vanishing gradient problem in the transformer encoder and improves the transformer model’s performance. In this study, because we did not perform a machine translation task, we only used the transformer encoder layer. For this purpose, the proposed TfrAdmCov model only learns the features of the input sequence and performs COVID-19 mutation prediction based on these learned features. The workflow of the proposed TfrAdmCov model for mutation prediction of COVID-19 virus has been shown in Figure .
Figure 5. The workflows of the proposed TfrAdmCov model for mutation prediction of COVID-19 virus. Where is the number of tokens in the input sequence and
is the dimensionality of those tokens. The size of the input sequence is
.
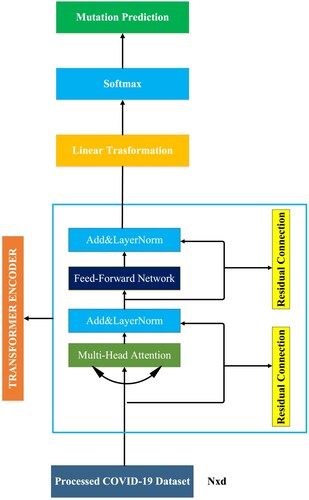
As seen in Figure , the processed COVID-19 dataset is given as input to MHA. The data obtained from the MHA is given as input to the layer normalisation. The data obtained from the layer normalisation is given as input to the FFN layer. The data obtained from FFN layer is given as input to the layer normalisation. The data obtained from layer normalisation is given as input to the linear transformation layer. The data obtained from linear transformation layer is given as input to the softmax layer. Then, the new dataset obtained has been passed through the softmax layer and finally mutation prediction of COVID-19 virus has been carried out.
4.1. Softmax layer
The softmax function is a type of activation function often used in deep learning tasks. It maps actual values to probability values between 0 and 1. This function has been used in attention mechanisms recently. The softmax formula has been given in Equation (5).
(5)
(5) where
presents the
-th value of input sequence
.
denotes other sequences in
data.
is the dimension of sequence
.
4.2. Loss function
Binary classification tasks use a loss function called binary cross-entropy, which compares each predicted probability with the true class output and updates the probabilities depending on the distance from the expected value. The task addressed in this study is a two-class problem (mutation, no mutation). Therefore, we use binary cross-entropy for calculating the loss value between the true and the predicted
. The loss function
is calculated in Equation (6):
(6)
(6) where
is the number of input samples and
is the set of selected residue sites.
is the number of selected positions of
-th training sample for COVID-19 mutation prediction.
5. Experimentations
The COVID-19 S protein dataset, the COVID-19 pre-processing steps, holdout method and stratified 10-fold cross validation method, GridSearchCV hyperparameter tuning technique and baseline models have been explained below.
5.1. COVID-19 S protein dataset
The COVID-19 S protein dataset consists of S protein sequences. The COVID-19 S protein dataset consists of 1273 amino sets in total (Anonymous, Citation2023b; Zhang et al., Citation2021). In this study, a total of 15,000 COVID-19 S protein sequences have been downloaded for each year from the reference address (Anonymous, Citation2023b) between 2020 and 2022. After downloading all S protein sequences, all sequences have been aligned by year using the CLUSTALW (Anonymous, Citation2023a) multiple sequence alignment (MSA) method.
5.2. Preparation and pre-processing steps of the COVID-19 S protein dataset
Apart from the amino acids that are directly encoded by the 20 universal genetic codes in some strains between 2020 and 2022, some strains have a few ambiguous amino acids. In order to eliminate the ambiguity in these strains, one of the letters “D” or “N” has been randomly assigned instead of the indefinite letter “B”. Instead of the indefinite letter “Z” has been randomly assigned one of the letters “E” or “Q”. Finally, a random amino acid assignment has been made among 20 universal amino acids instead of the ambiguous letter “X”. In this way, all ambiguities have been removed (Yin et al., Citation2020). In this study, the dataset generation method presented by Yin et al. (Citation2020) has been used. This method was obtained using the KMeans clustering algorithm. In this study, we used the KMeans clustering algorithm in the first place, but we could not achieve the performance of machine learning based algorithms at the desired level. Therefore, agglomerative clustering (Sasirekha & Baby, Citation2013) algorithm, which is an important factor in increasing the success rate, has been preferred instead of KMeans clustering algorithm while creating the dataset.
5.3. Agglomerative clustering
Agglomerative clustering is a variant of the hierarchical clustering method (Sasirekha & Baby, Citation2013). Agglomerative clustering is referred to as the (part-to-whole or bottom-up) approach. Clusters in the entire dataset are made into clusters. These clusters, which are created later, are combined with the clusters that are close depending on the distance, and a new cluster is obtained. In other words, the agglomerative clustering algorithm starts by treating each object/instance as a single cluster. Then the pairs of clusters are successively merged until all the clusters are combined into one large cluster containing all the objects (Sasirekha & Baby, Citation2013). At the stage of creating the training dataset, COVID-19 strains have been divided according to years and the agglomerative clustering algorithm has been used to divide the strains in each year into clusters. In addition, the parameter of the agglomerative clustering algorithm and the values of these parameters are shown in Table .
In this study, Training, Testing and Kfold datasets have been used. The amounts of Training, Testing and Kfold datasets by years have been shown in Table . As seen in Table , For the Training dataset, 30 strains have been randomly selected for each year among 11,250 COVID-19 S protein strains. For the testing data set, 10 strains have been randomly selected for each year among 3750 COVID-19 S protein strains. For the Kfold dataset, 40 strains have been randomly selected for each year among 15,000 COVID-19 S protein strains. The reason we chose the data quantities for each dataset like this is because we use the GridSearchCV hyperparameter tuning method. Because it takes a lot of time to get the results when choosing the best parameter values through the GridSearchCV method of each machine learning based model. For the datasets used in this study, two (2) clusters have been created for each year using the agglomerative clustering algorithm, which is also expressed in Figure . For example, a strain selected from the B1 cluster in 2020 year, a random strain from the A1 or A2 cluster in 2021, which has the lowest hamming distance (Norouzi et al., Citation2012) to this strain, has been selected. Similarly, a strain selected from the B1 cluster in 2020 year, a random strain from the C1 or C2 cluster in 2022, which has the lowest hamming distance to this strain has been selected. This process continues until all strains have been included in the datasets. Ultimately, datasets have been obtained by combining data from different years of COVID-19 strains one by one (Yin et al., Citation2020).
Figure 6. Example of creation of COVID-19 S protein datasets (Yin et al., Citation2020).
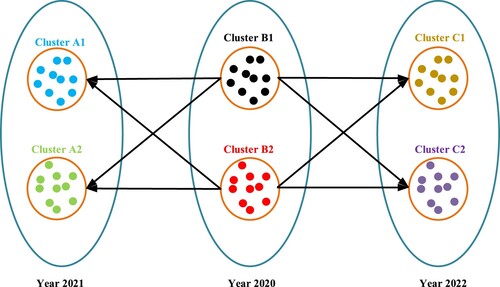
Table 1. Number of strains of COVID-19 S protein datasets by years.
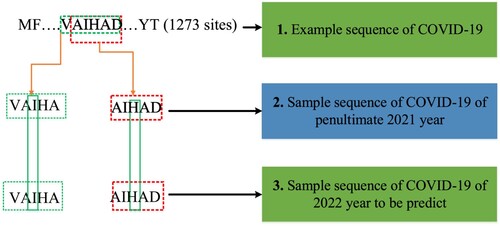
In this study, training examples have been created for Training, Testing and Kfold datasets using strains in 2020 and 2021 years. It has been obtained using the years 2021 (penultimate year) and 2022 (year to predict) to generate Label/target data samples of the Training, Testing and Kfold datasets. Our aim is to predict the mutations of the COVID-19 virus in 2022 using the 2020 and 2021 strains. The stages of creating training examples for Training, Testing and Kfold datasets have been shown in Figure . As seen in Figure , While creating the training datasets, 5 sites/residues have been used to represent each amino acid. For example, the sequence “VAIHA” has been used to represent the “I” amino acid. The “VAIHA” sequence has been then split into 3 overlapping 3-gram [VAI, AIH, IHA] small sequences. The sequence “AIHAD” has been used to represent the “H” amino acid. The “AIHAD” sequence has been then split into 3 overlapping 3-gram [AIH, IHA, HAD] small sequences. This process has been continued in this way for all sites found in the whole COVID-19 S protein structure. After dividing all strains used in the study into 3 overlapping 3-gram sites, each 3-gram is represented by a 100-dimensional embedding vector based on ProtVec presented by Asgari & Mofrad (Citation2015). Then, a single 100-dimensional vector has been obtained by taking the sum of 3 vectors with 100-dimensional. This process has been continued until the last strain. To create label data samples of training and testing datasets for each year, 2021 (penultimate year) and 2022 (last year to be predicted) have been used. Creation phase of label samples for training and testing datasets has been shown in Figure . As seen in Figure , in order to check whether the F amino acid taken from 2021 (penultimate year) has been mutated, the centre position (3rd position, I amino acid) of the “VAIHA” sequence has been checked. If the I amino acid in this central position in 2022 (last year) has changed, the mutation label is “1”, and if it has not changed, then the mutation label is “0”. Similarly, in order to check whether the T amino acid taken from 2021 (penultimate year) has been mutated, the centre position (3rd position, H amino acid) of the “AIHAD” sequence has been checked. If the H amino acid in this central position in 2022 (last year) has changed, the mutation label is “1”, and if it has not changed, then the mutation label is “0”. This process has been continued until the last strain (Yin et al., Citation2020).
Figure 7. Creation phases of training and testing samples for training and testing datasets.
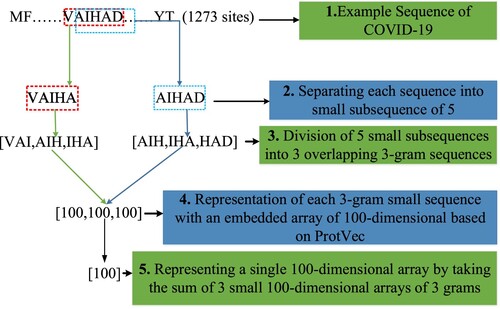
Figure 8. Creation phase of label samples for training and testing datasets.
The processed COVID-19 virus dataset and its details have been shown in Figure . As seen in Figure , the processed COVID-19 dataset consists of label and input data. If the label value is 1, it means there is a “mutation”, and if it is 0, it means there is “no mutation”. Each input data consists of 5 training samples (3 overlapping – 3 grams). Every three grams is represented by a 100-dimensional embedding vector based on of ProtVec presented by Asgari & Mofrad (Citation2015). In the training phase of the model, by summing each three-gram 100-dimensional vector is used by a single 100-dimensional vector.
Figure 9. Processed COVID-19 Dataset.

In this study, the total amount of data in Training, Testing and Kfold data sets, which are given as an input to deep learning and machine learning based models, have been shown in Tables , respectively. In Table , the total amounts of the Training dataset by years have been shown. In Table , the total amounts of the Testing dataset by years have been shown. In Table , the total amounts of the Kfold dataset by years have been shown.
Table 2. Total amount of Training dataset by years.
Table 3. Total amount of Testing dataset by years.
Table 4. Total amounts of Kfold dataset by years.
5.4. Influenza A/ H3N2HA dataset
In this study, the previously emerging influenza A/ H3N2 HA protein dataset has been used to measure the performance of the proposed TfrAdmCov model. The Infulenza A/ H3N2 dataset presented by Yin et al. (Citation2020) consist of HA protein sequences between 1991 and 2016. This dataset consists of a total of 132,000 sequence samples (3 overlapping 3-grams) (Yin et al., Citation2020). Class quantities and approximate percentages for Training and Testing datasets of the Influenza A/H3N2 HA protein dataset presented by Yin et al. (Citation2020) have been shown in Table .
Table 5. Class quantities and approximate percentages for Training and Testing datasets of the Influenza A/H3N2 HA protein dataset.
5.5. Holdout method and stratified 10-fold cross validation method
In this study, holdout and stratified 10-fold cross validation techniques have been used to evaluate the performances of machine learning based models. In the holdout technique (Kohavi, Citation1995), Training and Testing datasets have been used. Deep learning and machine learning based models have been first trained on the Training dataset. Then, deep learning and machine learning based models have been tested on the Testing dataset, which he had never seen before, and performance measurements have been obtained for each algorithm. In the stratified 10-fold cross validation technique (Kohavi, Citation1995), Kfold dataset has been used. The Kfold dataset has been divided into 10 parts. The performances of machine learning based models have been measured, with 90% of each piece being training and 10% test data. As seen in Table in this study, there is a class imbalance in the COVID-19 S protein datasets. Samples from all classes are not guaranteed when the holdout technique is used in model evaluation. This is a big problem. To overcome this problem, the stratified 10-fold cross validation technique, which allows the use of data by preserving the sample percentages of each class of the COVID-19 S protein datasets, has been also preferred (Thölke et al., Citation2022; Mbow et al., Citation2021). As seen in Table , the total amounts of the Training and Testing dataset for the holdout technique by years have been shown. Table shows the amounts of the COVID-19 S protein Kfold dataset by years for the stratified 10-fold cross validation technique. Table shows the class amounts and approximate percentages for the Training, Testing and Kfold datasets.
Table 6. Total amount of Training and Testing datasets by years for holdout technique.
Table 7. Total amounts of Kfold dataset by years for stratified 10-fold cross validation technique.
Table 8. Class quantities and approximate percentages for Training, Testing, and Kfold datasets.
5.6. GridSearchCV hyperparameter tuning
In this study, GridSearchCV (Ahmad et al., Citation2022) hyperparameter tuning method with 5-fold cross-validation has been used to select the best parameter values of each machine learning based model. The default values of the GridSearchCV algorithm have been used in the study. Among the features of each AI model, 3 random features have been chosen. A total of 5 parameter values, including the default values, have been selected for each preferred feature. Then, the best parameter values have been obtained for each feature using the GridSearchCV algorithm. Hyperparameters for the machine learning models have been shown in Table .
Table 9. Hyperparameters for the machine learning models.
As seen in Table , for each machine learning model, three hyperparameters have been randomly selected among all hyperparameters. Hyperparameter values have been set for each machine learning in Table . Then, the best values among these hyperparameter values have been selected using the GridSearchCV algorithm. The hyperparameters selected for each machine learning based model and their values have been shown in Table for SVM, Table for KNN, Table for XGBoost and Table for LR.
5.7. Baselines models
In this study, support vector machine (SVM), k-nearest neighbours (KNN), eXtreme gradient boosting (XGBoost), logistic regression (LR), RNN, gated recurrent unit (GRU) have been explained below.
5.7.1. SVM model
The SVM model is a supervised learning approach that is frequently used for solving classification and regression problems. The SVM model has the ability to generalise quite high. The purpose of the SVM model is to find a linear optimal hyperplane to maximise the margin of separation between the two classes. One of the most important advantages of the SVM model is that it achieves successful results at a high rate, and the disadvantage is that it gives very late results (Cortes & Vapnik, Citation1995).
5.7.2. KNN model
The KNN model is a frequently used learning approach for solving classification and regression problems. The KNN model can also be expressed as an effective machine learning based learning model that performs classification by looking at the K close relationship with the previous data (Guo et al., Citation2003). Here, care is taken to choose the K value from single digits.
5.7.3. XGBoost model
The XGBoost model is a machine learning based model expressed as an optimised and performance-enhanced version of the Gradient Boosting model. Among the important advantages of this model are obtaining results quickly, preventing excessive learning (memorising) and providing high performance (Memon et al., Citation2019).
5.7.4. LR model
LR model is expressed as a standard probability statistical classification model that is widely used in different fields (statistics, data mining, etc.). The output of this model on any sample is in terms of probability. In particular, its use in binary classification is common. LR model is a probability-based statistical approach extensively used in different fields (Finance etc.). It is especially used commonly in binary classification problems (Feng et al., Citation2014). Figure shows the workflows of the machine learning models for mutation prediction of COVID-19 virus. As seen in Figure , firstly, the COVID-19 S protein strains have been downloaded from the web address in (Anonymous, Citation2023b). Then, these downloaded strains have been aligned with the CLUSTALW (Anonymous, Citation2023a) MSA method. For each strain taken from the dataset, 1273 small 5-arrays representing 1273 domains (amino acids) forming the COVID-19 S protein sequence have been divided. These 5 small arrays have been split into 3 overlapping 3-gram arrays. Each of the 3 overlapping 3-gram small sequences has been represented by 100-dimensional embedded sequences based on ProtVec. Then, 3 overlapping 3-gram small arrays have been represented by a single vector of 100-dimensional by taking the sum of the arrays. The obtained 100-dimensional single vector data has been standardised by applying the StandardScaler (Thara et al., Citation2019) method. Then, machine learning based models with or without GridSearchCV have been applied to these standardised data. Then, Accuracy, Precision, Recall, F1-Score and MCC performance measurement values were obtained by using holdout and stratified 10-fold cross validation techniques.
Figure 10. The workflows of the machine learning models for mutation prediction of COVID-19 virus.
5.7.5. RNN model
Unlike feed-forward networks, RNN models can provide information from the next layer to the previous layer. Although RNNs can model short-term dependencies, they cannot model long-term dependencies due to the vanishing/exploding gradient problem (Zaremba et al., Citation2014).
5.7.6. LSTM model
LSTM model can be defined as a variant of RNNs that captures long-term dependencies. However, its difference from RNN is that it has its own memory. So, the LSTM is more powerful than RNNs. Additionally, with the introduction of the LSTM, the vanishing/exploding gradient problem in RNNs has been eliminated (Hochreiter & Schmidhuber, Citation1997).
5.7.7. GRU model
GRU model is a simplified version of the LSTM model. It was especially developed to learn long-term dependencies efficiently. In addition, It has fewer gates than the LSTM (Chung et al., Citation2014).
6. Results and discussions
6.1. Implementation details
In this study, in order to maximise the performance of deep learning models (RNN, LSTM, GRU, Transformer), hyper-parameter values (hidden size, dropout, batch size, etc.) have been tested many times (trial and error) and the best hyper-parameter values have been selected. For all deep learning models (except traditional machine learning models), Adam with batch size of 32 (256 for H3N2 HA dataset) has been used for model optimisation. The learning rate has been set to 0.001 and hidden size of 128 in the encoder of the proposed TfrAdmCov model and all deep learning models. Cross entropy has been used as the objective function (to minimise the losses). Dropout value of 0.5 and epoch value of 500 (350 for H3N2 HA dataset) have been used for training the proposed TfrAdmCov model and all deep learning models. In addition, the number of multi head attention used in the transformer encoder layer (Including multi head attention default value = 8 in the open source library (Vaswani et al., Citation2017)) has been tested many times and the best hyper-parameter value has been chosen as multi head attention = 2. All the experimental results have been averaged 10 random trails with different random seeds for deep learning models. The hyperparameters of the proposed TfrAdmCov model and other models have been shown in Table .
Table 10. The hyperparameters of the proposed TfrAdmCov model and other models.
6.2. Performance evaluation
In this study, Training, Testing and Kfold datasets have been trained and tested on a PC with a 2-core Intel(R) Core(TM) i5 7200U CPU@ 2.5 GHz processor, 12GB Ram and Intel(R) HD Graphics 620 GPU. All training, testing and simulations have been carried out with Scikit-learn (Pedregosa et al., Citation2011) and PyTorch (Paszke et al., Citation2017) In the study, the performance measurements of deep learning and machine learning-based models (Accuracy, Precision, Recall, F1-Score, MCC) have been obtained by using Confusion Matrix in Table .
Table 11. Confusion matrix (Luque et al., Citation2019).
True Positive (TP) refers to samples that are actually positive (mutation) and are classified as positive (mutation) when predicted. False Negative (FN) refers to samples that are actually positive (mutation) and classified as negative (no mutation) when predicted. False Positive (FP) refers to samples that are actually negative (no mutation) and are classified as positive (mutation) when predicted. True Negative (TN) refers to samples that are actually negative (no mutations) and are classified as negative (no mutations) when predicted (Chicco & Jurman, Citation2020). Accuracy, precision, recall or sensitivity, f1-score and Matthews correlation coefficient (MCC) performance evaluation metrics used in this study have been given in Equations (7)–(11), respectively (Pacal, Citation2024b).
(7)
(7)
(8)
(8)
(9)
(9)
(10)
(10)
(11)
(11)
6.3. Experimental results
In Table , the performance values of the SVM model with or without GridSearchCV have been shown.
Table 12. Performance values of the SVM model with or without GridSearchCV.
As seen in Table , the SVM model with or without the GridSearchCV has been trained on the Training dataset and tested on the Testing dataset. According to the SVM model without GridSearchCV method of the SVM model with GridSearchCV has increased accuracy (from 99.90% to 99.91%), Recall (from 96.22% to 96.51%), F1-Score (from 0.98.07%- to 98.22%), MCC (from 98.04%- to 98.19%) on the Testing dataset. As a result, it has been observed that the SVM model with the GridSearchCV method significantly improves performance on Training dataset and Testing dataset. The performance values of the KNN model with or without GridSearchCV have been shown in Table .
Table 13. Performance values of the KNN model with or without GridSearchCV.
As seen in Table , the KNN model with or without the GridSearchCV has been trained on the Training dataset and tested on the Testing dataset. According to the KNN model without GridSearchCV method of the KNN model with GridSearchCV has increased accuracy (from 99.90%- to 99.91%), Recall (from 96.80%- to 97.09%), F1-Score (from 98.09%- to 98.24%), MCC (from 98.04%- to 98.19%) on the Testing dataset. As a result, it has been observed that the KNN model with the GridSearchCV method significantly improves performance on Training dataset and Testing dataset. The performance values of the XGBoost model with or without GridSearchCV have been shown in Table .
Table 14. Performance values of the XGBoost model with or without GridSearchCV.
As seen in Table , the XGBoost model with or without the GridSearchCV has been trained on the Training dataset and tested on the Testing dataset. According to the XGBoost model without GridSearchCV method of the XGBoost model with GridSearchCV has increased accuracy (from 99.90%- to 99.91%), Precision (from 99.70%- to 100.00%), F1-Score (from 98.08%- to 98.22%), MCC (from 98.08%- to 98.19%) on the Testing dataset. As a result, it has been observed that the XGBoost model with the GridSearchCV method significantly improves performance on the Testing dataset. The performance values of the LR model with or without GridSearchCV have been shown in Table .
Table 15. Performance values of the LR model with or without GridSearchCV.
As seen in Table , the LR model with or without the GridSearchCV has been trained on the Training dataset and tested on the Testing dataset. According to the LR model without GridSearchCV method of the LR model with GridSearchCV has increased accuracy (from 99.57% to -99.79%), Recall (from 85.17%- to 93.90%), F1-Score (from 91.42%- to 95.99%), MCC (from 91.64%- to 95.90%) on the Testing dataset. As a result, it has been observed that the LR model with the GridSearchCV method significantly improves performance on the Training dataset and the Testing dataset. The comparison of the performance values of the Machine learning-based model with or without GridSearchCV has been shown in Table .
Table 16. Comparison of performance values of Machine learning-based models with or without GridSearchCV.
As seen in Table , comparison of Accuracy, Precision, Recall, F1- Score and MCC values on the Kfold dataset using machine learning-based models (SVM, KNN, XGBoost, LR) with or without GridSearchCV has been carried out. The SVM with GridSearchCV achieved the highest performance of 99.893% accuracy value on the Kfold dataset. The SVM model with or without GridSearchCV achieved the highest performance of 99.92% Precision value on the Kfold dataset. The SVM model with GridSearchCV achieved the highest performance of 97.97% f1-score value on the Kfold dataset. The SVM model with GridSearchCV achieved the highest performance of 97.97% f1-score value on the Kfold dataset. On the other hand, The KNN model with GridSearchCV achieved the highest performance of 96.22% f1-score value on the Kfold dataset. the SVM model with GridSearchCV achieved the highest performance of 97.95% MCC value on the Kfold dataset. The performance comparative of the deep learning (RNN, LSTM, GRU, the proposed TfrAdmCov) models on the Testing dataset has been shown in Table .
Table 17. Performance comparative of the deep learning models on the Testing dataset.
As seen in Table , the proposed TfrAdmCov model has achieved better results than other models in terms of accuracy of 99.93%, recall of 97.38%, f1-score of 98.67% and MCC of98.65% on the COVID-19 testing dataset. In addition, the proposed TfrAdmCov model, RNN, LSTM and GRU have achieved the same results in terms of precision of 100.00% on the COVID-19 testing dataset. Performance comparative of the proposed TfrAdmCov model with Adam, RMSprop, AdamW optimizer on the Testing dataset has been shown in Table .
Table 18. Performance comparative of the proposed TfrAdmCov model with Adam, RMSprop, AdamW optimizer models on the Testing dataset.
Table aims to select the optimizer algorithm that achieves the best performance for the proposed TfrAdmCov model amongst three optimizer algorithms (Adam, RMSprop, AdamW). As seen in Table , the proposed TfrAdmCov model with Adam optimizer algorithm performed better than the proposed TfrAdmCov model with RMSprop, AdamW optimizer algorithms. For this reason, the Adam optimizer algorithm was preferred for the proposed TfrAdmCov model. Confusion Matrix obtained using TfrAdmCov model with Adam optimizer algorithm on COVID-19 Testing dataset has been shown in Figure . While Confusion Matrix obtained using TfrAdmCov model with RMSprop optimizer algorithm on COVID-19 Testing dataset has been shown in Figure , Confusion Matrix obtained using TfrAdmCov model with AdamW optimizer algorithm on COVID-19 Testing dataset has been shown in Figure .
Figure 11. Confusion Matrix obtained using TfrAdmCov model with Adam optimizer algorithm on COVID-19 Testing dataset.

Figure 12. Confusion Matrix obtained using TfrAdmCov model with RMSprop optimizer algorithm on COVID-19 Testing dataset.

As seen in Figure , on the COVID-19 testing dataset, the proposed TfrAdmCov model with Adam optimizer algorithm correctly predicted 335 samples out of 344 samples in the mutation class, while it incorrectly predicted 9 samples out of 344 in the mutation class. In addition, the proposed TfrAdmCov model with Adam optimizer algorithm correctly predicted all samples out of 12,386 samples in the no-mutation class.
As seen in Figure , on the COVID-19 testing dataset, the proposed TfrAdmCov model with RMSprop optimizer algorithm correctly predicted 334 samples out of 344 samples in the mutation class, while it incorrectly predicted 10 samples out of 344 in the mutation class. In addition, the proposed TfrAdmCov model with RMSprop optimizer algorithm correctly predicted all samples out of 12,386 samples in the no-mutation class.
As seen in Figure , on the COVID-19 testing dataset, the proposed TfrAdmCov model with AdamW optimizer algorithm correctly predicted 334 samples out of 344 samples in the mutation class, while it incorrectly predicted 10 samples out of 344 in the mutation class. In addition, the proposed TfrAdmCov model with AdamW optimizer algorithm correctly predicted all samples out of 12,386 samples in the no-mutation class. The performance comparative of the deep learning models with 10 random trail with different random seeds on the Testing dataset has been shown in Table .
Figure 13. Confusion Matrix obtained using TfrAdmCov model with AdamW optimizer algorithm on COVID-19 Testing dataset.

Table 19. Performance comparative of the RNN, LSTM, GRU, the proposed TfrAdmCov models with 10 random trail with different random seeds on the Testing dataset.
As seen in Table , the proposed TfrAdmCov model has achieved better results than other models in terms of accuracy of 99.924%, recall of 97.18%, f1-score of 98.57% and MCC of 98.54% on the COVID-19 testing dataset. In addition, the proposed TfrAdmCov model, RNN, LSTM and GRU have achieved the same results in terms of precision of 100.00% on the COVID-19 testing dataset. Performance comparisons of the proposed TfrAdmCov and machine learning-based models with GridSearchCV on the Testing dataset have been shown in Table .
Table 20. Performance comparisons of the proposed TfrAdmCov model and machine learning-based models with GridSearchCV on the Testing dataset.
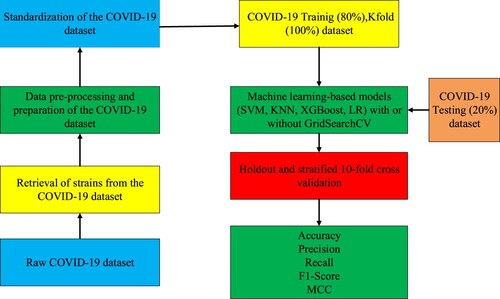
As seen in Table , the proposed TfrAdmCov model has achieved better results than other models in terms of accuracy of 99.93%, recall of 97.38%, f1-score of 98.67% and MCC of 98.65% on the COVID-19 testing dataset. In addition, the proposed TfrAdmCov model, SVM, XGBoost, RNN, LSTM, GRU has achieved the same results in terms of precision of 100.00% on the COVID-19 testing dataset. On the other hand, the LR model has obtained worse results than other models in terms of accuracy of 99.79%, precision of 98.18%, recall of 93.90, f1-score of 95.99% and MCC of 95.90% on the COVID-19 testing dataset. As a result, the proposed TfrAdmCov model is very successful on sequence-based COVID-19 dataset. Accuracy values for the proposed TfrAdmCov model and other models on COVID-19 Testing dataset have been shown in Figure .
Figure 14. Accuracy values for the proposed TfrAdmCov model and other models on COVID-19 Testing dataset.
As seen in Figure , the proposed TfrAdmCov model reached the best accuracy value of 99.93%, while the LR model reached the worst accuracy value of 99.79%. Comparison of average performance values of the machine learning with stratified 10-fold cross-validation technique, the proposed TfrAdmCov model and deep learning models with 10 random trail with different random seeds on the Testing dataset have been shown in Table .
Table 21. Comparison of average performance values of the machine learning with stratified 10-fold cross-validation technique, the proposed TfrAdmCov model and deep learning models with 10 random trail with different random seeds on the Testing dataset.
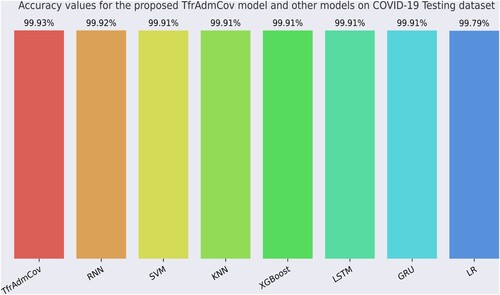
As seen in Table , the proposed TfrAdmCov model has achieved better results than other models in terms of accuracy of 99.924%, recall of 97.18%, f1-score of 98.57% and MCC of 98.54% on the COVID-19 testing dataset by averaging 10 random trail with different random seeds. In addition, the proposed TfrAdmCov model, SVM, XGBoost, RNN, LSTM and GRU have achieved the same results in terms of precision of 100.00% on the COVID-19 testing dataset. On the other hand, the LR model has obtained worse results than other models in terms of accuracy of 99.79%, precision of 98.14%, recall of 94.90%, f1-score of 95.99% and MCC of 95.89% on the COVID-19 testing dataset. As a result, the proposed TfrAdmCov model is quite successful on sequence-based COVID-19 dataset. Average accuracy values for the proposed TfrAdmCov model and other models on the COVID-19 Testing Dataset have been shown in Figure .
Figure 15. Average accuracy values for the proposed TfrAdmCov model and other models on the COVID-19 Testing Dataset.
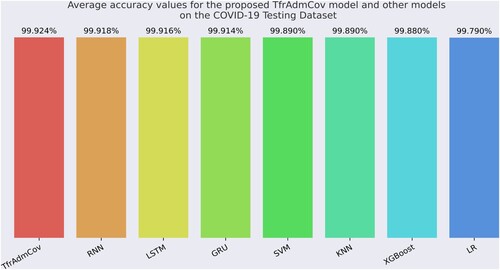
As seen in Figure , the proposed TfrAdmCov model reached the best average accuracy value of 99.924%, while the LR model reached the worst average accuracy value of 99.79%.
6.4. Statistical analyses for the proposed TfrAdmCov model and deep learning models
The results obtained in this study have been obtained by averaging 10 random trials with different random seeds for both the proposed TfrAdmCov model and deep learning models. Detailed analyses have been performed for each model in terms of accuracy, precision, recall, f1-score and MCC performance measurement metric using statistical measures such as average, standard deviation, median, min and max. Statistical analysis of the proposed TfrAdmCov model with 10 random trail with different random seeds on the Testing dataset has been shown in Table .
Table 22. Statistical analysis of the proposed TfrAdmCov model with 10 random trail with different random seeds on the Testing dataset.
As seen in Table , the proposed TfrAdmCov model has been performed as the average of 0.999238, standard deviation of 0.000036, median of 0.999214, min of 0.999214 and max of 0.999293 amongst 10 accuracy values obtained. Statistical analysis of the proposed RNN model with 10 random trail with different random seeds on the Testing dataset has been shown in Table .
Table 23. Statistical analysis of the RNN model with 10 random trail with different random seeds on the Testing dataset.
As seen in Table , the proposed RNN model has been performed as the average of 0.999175, standard deviation of 0.000039, median of 0.999175, min of 0.999136 and max of 0.999214 amongst 10 accuracy values obtained. Statistical analysis of the proposed LSTM model with 10 random trail with different random seeds on the Testing dataset has been shown in Table .
Table 24. Statistical analysis of the LSTM model with 10 random trail with different random seeds on the Testing dataset.
As seen in Table , the proposed LSTM model has been performed as the average of 0.999159, standard deviation of 0.000036, median of 0.999136, min of 0.999136 and max of 0.999214 amongst 10 accuracy values obtained. Statistical analysis of the proposed GRU model with 10 random trail with different random seeds on the Testing dataset has been shown in Table .
Table 25. Statistical analysis of the GRU model with 10 random trail with different random seeds on the Testing dataset.
As seen in Table , the proposed GRU model has been performed as the average of 0.999144, standard deviation of 0.000023, median of 0.999136, min of 0.999136 and max of 0.999214 amongst 10 accuracy values obtained.
6.5. The reason why agglomerative clustering algorithm is preferred instead of K-means in creating training, testing and Kfold datasets
In this study, we used the K-means algorithm to create training, testing and kfold datasets. However, as seen in Table , the performance of the proposed TfrAdmCov model has been higher than that of the Agglomerative clustering algorithm compared to the K-means clustering algorithm, so the Agglomerative clustering algorithm has been preferred to create the training, testing and kfold datasets. Performance comparisons of the proposed TfrAdmCov model on the Testing dataset created using Kmeans and Agglomerative clustering algorithms have been shown in Table .
Table 26. Performance comparison of the proposed TfrAdmCov model on the Testing dataset created using Kmeans and Agglomerative clustering algorithms.
6.6. Performance evaluation of proposed TfrAdmCov on influenza A/ H3N2HA dataset
Performance values of the proposed TfrAdmCov model and other models on the H3N2 HA testing dataset have been shown in Table .
Table 27. Performance values of the proposed TfrAdmCov model and other models on the H3N2 HA testing dataset.
As seen in Table , the proposed TfrAdmCov model has achieved better results than other models on the accuracy of 96.33%, precision of 81.55%, recall of 52.33%, f1-score of 63.75% and MCC of 63.61% on influenza A subtypes H3N2 HA testing dataset. On the other hand, the LR model has obtained the worst result in terms of accuracy of 93.79%, recall of 4.29%, f1-score of 7.87% and MCC of 12.77%. The results on the influenza A subtypes H3N2 HA testing dataset showed that the proposed TfrAdmCov model is quite robust. Confusion Matrix obtained using TfrAdmCov model on H3N2 HA testing dataset has been shown in Figure .
Figure 16. Confusion Matrix obtained using TfrAdmCov model on H3N2 HA testing dataset.

As seen in Figure , on the H3N2 HA testing dataset, the proposed TfrAdmCov model correctly predicted 853 samples out of 1630 samples in the mutation class, while it incorrectly predicted 777 samples out of 1630 in the mutation class. Moreover, the proposed TfrAdmCov model correctly predicted 24,577 samples out of 24,770 samples in the no-mutation class, while it incorrectly predicted 193 samples out of 24,770 in the no-mutation class. Accuracy values for the proposed TfrAdmCov model and other models on the H3N2 HA testing dataset have been shown in Figure .
Figure 17. Accuracy values for the proposed TfrAdmCov model and other models on the H3N2 HA Testing dataset.
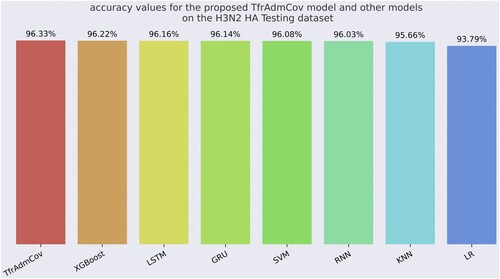
As seen in Figure , the proposed TfrAdmCov model reached the best accuracy value of 96.33%, while the LR model reached the worst accuracy value of 93.79%. Comparison of the proposed TfrAdmCov model with the state-of-the-art works has been shown in Table .
Table 28. Comparison of the proposed TfrAdmCov model with the state-of-the-art works.
As seen in Table , the proposed TfrAdmCov model achieved accuracy of 99.93% on the Testing dataset. In addition, the proposed TfrAdmCov model achieved accuracy of 99.924% by averaging 10 random trail with different random seeds. As seen in Table , the proposed TfrAdmCov model outperformed the state-of-the-art works. The majority of studies published in the literature are either related to other aspects of the COVID-19 virus or mutations of other viruses. We conducted this study by addressing this gap in the literature. In conclusion, the proposed TfrAdmCov model can successfully perform mutation prediction on the COVID-19 dataset.
7. Limitations
When we increased the amount of data used in the study, we experienced problems due to insufficient computer hardware. For this reason, it took a lot of time to obtain the analysis results of each deep learning and machine learning-based models.
As we reviewed the literature while writing this article, the very limited resources available for the prediction of the COVID-19 S protein mutation constituted a major disadvantage for us.
Some difficulties have been encountered while preparing the datasets (data preprocessing steps take a lot of time, etc. so that raw COVID-19 data can be given as input to deep learning and machine learning-based models).
8. Conclusion
This study addressed the gap in the literature regarding COVID-19 mutation. Because predicting mutations in advance will facilitate the development of vaccines and drugs. Therefore, this study proposes a robust transformer encoder based model with Adam optimizer algorithm, TfrAdmCov, for COVID-19 mutation prediction. We aimed to predict mutations that may occur in the COVID-19 S (Spike) protein in the 2022 year using the proposed TfrAdmCov model. We used agglomerative clustering algorithm to create datasets. In addition, we used the GridSearchCV hyperparameter tuning method to improve the performance of machine learning-based models. Holdout technique and stratified 10-fold cross-validation technique have been used to evaluate the performances of each machine learning based models. We also performed statistical analyses to verify the performance of the proposed TfrAdmCov model and deep learning-based models. When the experimental results have been examined, the proposed TfrAdmCov model outperformed both baseline and several state-of-the-art methods. The proposed TfrAdmCov model reached accuracy of 99.93%, precision of 100.00%, recall of 97.38%, f1-score of 98.67% and MCC of 98.65% on the COVID-19 testing dataset. Besides, the proposed TfrAdmCov model has achieved better results than other models in terms of accuracy of 99.924%, recall of 97.18%, f1-score of 98.57% and MCC of 98.54% on the COVID-19 testing dataset by averaging 10 random trail with different random seeds. Similarly, the proposed TfrAdmCov model has achieved better results than other models on the accuracy of 96.33%, precision of 81.55%, recall of 52.33%, f1-score of 63.75% and MCC of 63.61% on influenza A/ H3N2 HA protein dataset. As a result, the proposed TfrAdmCov model can successfully predict mutations occurring on both the COVID-19 dataset and influenza A/ H3N2 HA dataset. We believe that this will help develop vaccines and drugs.
In the next study, we plan to apply the proposed TfrAdmCov model in this study to different virus mutation predictions. Additionally, we plan to perform mutation prediction on other proteins of the COVID-19 virus in the next study.
Authors contribution statement
Mehmet Burukanli, Software, Methodology, Conceptualisation, Validation, Data curation, Writing original draft. Nejat Yumusak: Methodology, Conceptualisation, Validation, Investigation, Supervision.
Data availability and access
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Ethical and informed consent for data used
This article does not contain any studies with human participants or animals performed by any of the authors. Informed consent was obtained from all individual participants included in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Abbas, M. E., Chengzhang, Z., Fathalla, A., & Xiao, Y. (2022). End-to-end antigenic variant generation for H1N1 influenza HA protein using sequence to sequence models. PLoS ONE, 17(3 March), 1–14. https://doi.org/10.1371/journal.pone.0266198
- Ahmad, G. N., Fatima, H., Ullah, S., Saidi, A. S., & Imdadullah. (2022). Efficient medical diagnosis of human heart diseases using machine learning techniques with and without GridSearchCV. IEEE Access, 10(March), 80151–80173. https://doi.org/10.1109/ACCESS.2022.3165792
- Ahmed, I., & Jeon, G. (2022). Enabling artificial intelligence for genome sequence analysis of COVID-19 and alike viruses. Interdisciplinary Sciences: Computational Life Sciences, 14(2), 504–519. https://doi.org/10.1007/s12539-021-00465-0
- Anonymous. (2023a). clustalw. https://www.genome.jp/tools-bin/clustalw.
- Anonymous. (2023b). COVID-19 S protein dataset. https://www.ncbi.nlm.nih.gov/datasets/taxonomy/2697049/.
- Asgari, E., & Mofrad, M. R. K. (2015). Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS ONE, 10(11), e0141287. https://doi.org/10.1371/journal.pone.0141287
- Barnes, C. O., West, A. P., Huey-Tubman, K. E., Hoffmann, M. A. G., Sharaf, N. G., Hoffman, P. R., Koranda, N., Gristick, H. B., Gaebler, C., Muecksch, F., Lorenzi, J. C. C., Finkin, S., Hägglöf, T., Hurley, A., Millard, K. G., Weisblum, Y., Schmidt, F., Hatziioannou, T., Bieniasz, P. D., … Bjorkman, P. J. (2020). Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell, 182(4), 828–842.e16. https://doi.org/10.1016/j.cell.2020.06.025
- Cai, C., Li, J., Xia, Y., & Li, W. (2024). FluPMT: Prediction of predominant strains of influenza a viruses via multi-task learning. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 1–11. https://doi.org/10.1109/TCBB.2024.3378468
- Chakraborty, G. S., Singh, D., Rakhra, M., Batra, S., & Singh, A. (2022). Covid-19 and diabetes risk prediction for diabetic patient using advance machine learning techniques and fuzzy inference system. Proceedings of 5th International Conference on Contemporary Computing and Informatics, 14-16 December, IC3I 2022, 2022, 1212–1219. https://doi.org/10.1109/IC3I56241.2022.10073256
- Chicco, D., & Jurman, G. (2020). The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics, 21(1), 1–13. https://doi.org/10.1186/s12864-019-6413-7
- Chung, J., Gulcehre, C., Cho, K., & Bengio, Y. (2014). Empirical evaluation of gated recurrent neural networks on sequence modeling. ArXiv:1412.3555, 1–9. http://arxiv.org/abs/1412.3555.
- Cortes, C., & Vapnik, V. (1995). Support-vector networks. Machine Learning, 20(3), 273–297.
- Cui, J., Li, F., & Shi, Z.-L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. https://doi.org/10.1038/s41579-018-0118-9
- de Wit, J. J., & Cook, J. K. A. (2020). Spotlight on avian coronaviruses. Avian Pathology, 49(4), 313–316. https://doi.org/10.1080/03079457.2020.1761010
- ElAraby, M. E., Elzeki, O. M., Shams, M. Y., Mahmoud, A., & Salem, H. (2022). A novel gray-scale spatial exploitation learning Net for COVID-19 by crawling internet resources. Biomedical Signal Processing and Control, 73(2022), 103441. https://doi.org/10.1016/j.bspc.2021.103441
- Elzeki, O. M., Elfattah, M. A., Salem, H., Hassanien, A. E., & Shams, M. (2021). A novel perceptual two layer image fusion using deep learning for imbalanced COVID-19 dataset. PeerJ Computer Science, 7:e364, 1–35. https://doi.org/10.7717/PEERJ-CS.364
- Elzeki, O. M., Shams, M., Sarhan, S., Elfattah, M. A., & Hassanien, A. E. (2021). COVID-19: A new deep learning computer-aided model for classification. PeerJ Computer Science, 7:e358, 1–33. https://doi.org/10.7717/peerj-cs.358
- Feng, J., Xu, H., Mannor, S., & Yan, S. (2014). Robust logistic regression and classification. Advances in Neural Information Processing Systems, 27, 253–261.
- Gage, A., Brunson, K., Morris, K., Wallen, S. L., Dhau, J., Gohel, H., & Kaushik, A. (2021). Perspectives of manipulative and high-performance nanosystems to manage consequences of emerging new severe acute respiratory syndrome coronavirus 2 variants. Frontiers in Nanotechnology, 3(June), 1–7. https://doi.org/10.3389/fnano.2021.700888
- Galassi, A., Lippi, M., & Torroni, P. (2021). Attention in natural language processing. IEEE Transactions on Neural Networks and Learning Systems, 32(10), 4291–4308. https://doi.org/10.1109/TNNLS.2020.3019893
- Guo, G., Wang, H., Bell, D., Bi, Y., & Greer, K. (2003). KNN Model-based approach in classification. Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2888, 986–996. https://doi.org/10.1007/978-3-540-39964-3_62
- Hai-Dong, L., Ya-Juan, Y., & Lu, L. (2022). In the context of COVID-19: the impact of employees' risk perception on work engagement. Connection Science, 34(1), 1367–1383. https://doi.org/10.1080/09540091.2022.2071839
- Haimed, A. M. A., Saba, T., Albasha, A., Rehman, A., & Kolivand, M. (2021). Viral reverse engineering using artificial intelligence and big data COVID-19 infection with long short-term memory (LSTM). Environmental Technology & Innovation, 22, 101531. https://doi.org/10.1016/j.eti.2021.101531
- Hassan, E., Shams, M. Y., Hikal, N. A., & Elmougy, S. (2023). COVID-19 diagnosis-based deep learning approaches for COVIDx dataset: A preliminary survey. In G. K. Mostefaoui, S. M. R. Islam, & F. Tariq (Eds.), Artificial intelligence for disease diagnosis and prognosis in smart healthcare (pp.107–122)
- Hassan, E., Shams, M. Y., Hikal, N. A., & Elmougy, S. (2024). Detecting COVID-19 in chest CT images based on several pre-trained models. Multimedia Tools and Applications, 1–21. https://doi.org/10.1007/s11042-023-17990-3
- Hochreiter, S., & Schmidhuber, J. (1997). Long short-term memory. Neural Computation, 9(8), 1735–1780. https://doi.org/10.1162/neco.1997.9.8.1735
- Hossain, M. K., Hassanzadeganroudsari, M., & Apostolopoulos, V. (2021b). The emergence of new strains of SARS-CoV-2. What does it mean for COVID-19 vaccines? Expert Review of Vaccines, 20(6), 635–638. https://doi.org/10.1080/14760584.2021.1915140
- Hossain, M. S., Pathan, A. Q. M. S. U., Islam, M. N., Tonmoy, M. I. Q., Rakib, M. I., Munim, M. A., Saha, O., Fariha, A., Al Reza, H., Roy, M., Bahadur, N. M., & Rahaman, M. M. (2021a). Genome-wide identification and prediction of SARS-CoV-2 mutations show an abundance of variants: Integrated study of bioinformatics and deep neural learning. Informatics in Medicine Unlocked, 27, 100798. https://doi.org/10.1016/j.imu.2021.100798
- Huang, Y., Yang, C., Xu, X., Xu, W., & Liu, S. (2020). Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica, 41(9), 1141–1149. https://doi.org/10.1038/s41401-020-0485-4
- Jackson, C. B., Farzan, M., Chen, B., & Choe, H. (2022). Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology, 23(1), 3–20. https://doi.org/10.1038/s41580-021-00418-x
- Jaimes, J. A., André, N. M., Chappie, J. S., Millet, J. K., & Whittaker, G. R. (2020). Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. Journal of Molecular Biology, 432(10), 3309–3325. https://doi.org/10.1016/j.jmb.2020.04.009
- Kalyan, K. S., Rajasekharan, A., & Sangeetha, S. (2021). AMMUS: A Survey of Transformer-based Pretrained Models in Natural Language Processing. ArXiv Preprint ArXiv:2108.05542., 1–42. http://arxiv.org/abs/2108.05542.
- Kohavi, R. (1995). A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence, IJCAI’95, Morgan Kaufmann, United States, Montreal, QC, Canada, 20–25 August, pp. 1137–1143.
- Li, M., Zhao, B., Yin, R., Lu, C., Guo, F., & Zeng, M. (2023). Graphlncloc: Long non-coding RNA subcellular localization prediction using graph convolutional networks based on sequence to graph transformation. Briefings in Bioinformatics, 24(1), 1–12. https://doi.org/10.1093/bib/bbac565
- Lopez-Rincon, A., Perez-Romero, C. A., Tonda, A., Mendoza-Maldonado, L., Claassen, E., Garssen, J., & Kraneveld, A. D. (2021). Design of specific primer sets for the detection of B.1.1.7, B.1.351 and P.1 SARS-CoV-2 variants using deep learning. BioRxiv, 70, 2021.01.20.427043. https://doi.org/10.1101/2021.01.20.427043
- Luque, A., Carrasco, A., Martín, A., & de las Heras, A. (2019). The impact of class imbalance in classification performance metrics based on the binary confusion matrix. Pattern Recognition, 91, 216–231. https://doi.org/10.1016/j.patcog.2019.02.023
- Madhi, S. A., Kwatra, G., Myers, J. E., Jassat, W., Dhar, N., Mukendi, C. K., Nana, A. J., Blumberg, L., Welch, R., Ngorima-Mabhena, N., & Mutevedzi, P. C. (2022). Population immunity and Covid-19 severity with Omicron Variant in South Africa. New England Journal of Medicine, 386(14), 1314–1326. https://doi.org/10.1056/NEJMoa2119658
- Mbow, M., Koide, H., & Sakurai, K. (2021). An intrusion detection system for imbalanced dataset based on deep learning. 2021 Ninth International Symposium on Computing and Networking (CANDAR), 23-26 November 2021, 38–47. https://doi.org/10.1109/CANDAR53791.2021.00013
- Memon, N., Patel, S. B., & Patel, D. P. (2019). Comparative analysis of artificial neural network and XGBoost algorithm for PolSAR image classification. Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 11941(LNCS(Lc)), 452–460. https://doi.org/10.1007/978-3-030-34869-4_49
- Mohamed, T., Sayed, S., Salah, A., & Houssein, E. H. (2021). Long short-term memory neural networks for RNA viruses mutations prediction. Mathematical Problems in Engineering, 2021. https://doi.org/10.1155/2021/9980347
- Nawaz, M. S., Fournier-Viger, P., Shojaee, A., & Fujita, H. (2021). Using artificial intelligence techniques for COVID-19 genome analysis. Applied Intelligence, 51(5), 3086–3103. https://doi.org/10.1007/s10489-021-02193-w
- Norouzi, M., Fleet, D. J., & Salakhutdinov, R. (2012). Hamming distance metric learning. Advances in Neural Information Processing Systems, 2, 1061–1069.
- Pacal, I. (2024a). A novel Swin transformer approach utilizing residual multi-layer perceptron for diagnosing brain tumors in MRI images. International Journal of Machine Learning and Cybernetics, 1–21. https://doi.org/10.1007/s13042-024-02110-w
- Pacal, I. (2024b). Maxcervixt: A novel lightweight vision transformer-based approach for precise cervical cancer detection. Knowledge-Based Systems, 289(October 2023). https://doi.org/10.1016/j.knosys.2024.111482
- Paszke, A., Gross, S., Chintala, S., Chanan, G., & Yang, E. (2017). Automatic differentiation in pytorch. 31st conference on Neural Information Processing Systems (NIPS 2017), Nips, 1–4. https://openreview.net/forum?id=BJJsrmfCZ .
- Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., Blondel, M., Prettenhofer, P., Weiss, R., Dubourg, V., Vanderplas, J., Passos, A., Cournapeau, D., Brucher, M., Perrot, M., & Duchesnay, D. (2011). Scikit-learn: Machine learning in Python Fabian. Journal of Machine Learning Research, 12, 2825–2830.
- Qin, L., Ding, X., Li, Y., Chen, Q., Meng, J., & Jiang, T. (2021). Co-mutation modules capture the evolution and transmission patterns of SARS-CoV-2. Briefings in Bioinformatics, 00(May), 1–10. https://doi.org/10.1093/bib/bbab222
- Raheja, S., Kasturia, S., Cheng, X., & Kumar, M. (2023). Machine learning-based diffusion model for prediction of coronavirus-19 outbreak. Neural Computing and Applications, 35(19), 13755–13774. https://doi.org/10.1007/s00521-021-06376-x
- Rashid, Z.Z., Othman, S. N., Abdul Samat, M. N., Ali, U. K., & Ken, W. K. (2020). Diagnostic performance of COVID-19 serology assays. Malaysian Journal of Pathology, 42(1), 13–21.
- Saha, I., Ghosh, N., Maity, D., Sharma, N., Sarkar, J. P., & Mitra, K. (2020). Genome-wide analysis of Indian SARS-CoV-2 genomes for the identification of genetic mutation and SNP. Infection, Genetics and Evolution, 85, 104457. https://doi.org/10.1016/j.meegid.2020.104457
- Salama, M. A., Hassanien, A. E., & Mostafa, A. (2016). The prediction of virus mutation using neural networks and rough set techniques. Eurasip Journal on Bioinformatics and Systems Biology, 2016(1), 1–11. https://doi.org/10.1186/s13637-016-0042-0
- Sasirekha, K., & Baby, P. (2013). Agglomerative hierarchical clustering algorithm – A review. International Journal of Scientific and Research Publications, 3(3), 83–85.
- Serena Low, W. C., Chuah, J. H., Tee, C. A. T. H., Anis, S., Shoaib, M. A., Faisal, A., Khalil, A., & Lai, K. W. (2021). An overview of deep learning techniques on chest X-Ray and CT scan identification of COVID-19. Computational and Mathematical Methods in Medicine, 2021. https://doi.org/10.1155/2021/5528144
- Shaikh, F., Andersen, M. B., Sohail, M. R., Mulero, F., Awan, O., Dupont-Roettger, D., Kubassova, O., Dehmeshki, J., & Bisdas, S. (2021). Current landscape of imaging and the potential role for artificial intelligence in the management of COVID-19. Current Problems in Diagnostic Radiology, 50(3), 430–435. https://doi.org/10.1067/j.cpradiol.2020.06.009
- Sharma, A., Ahmad Farouk, I., & Lal, S. K. (2021). Covid-19: A review on the novel coronavirus disease evolution, transmission, detection, control and prevention. Viruses, 13(2), 1–25. https://doi.org/10.3390/v13020202
- Shereen, M. A., Khan, S., Kazmi, A., Bashir, N., & Siddique, R. (2020). COVID-19 infection: Emergence, transmission, and characteristics of human coronaviruses. Journal of Advanced Research, 24, 91–98. https://doi.org/10.1016/j.jare.2020.03.005
- Shiehzadegan, S., Alaghemand, N., Fox, M., & Venketaraman, V. (2021). Analysis of the Delta Variant B.1.617.2 COVID-19. Clinics and Practice, 11(4), 778–784. https://doi.org/10.3390/clinpract11040093
- Shrestha, H., Dhasarathan, C., Kumar, M., Nidhya, R., Shankar, A., & Kumar, M. (2022). A deep learning based convolution neural network-DCNN approach to detect brain tumor (pp. 115–127). https://doi.org/10.1007/978-981-16-6887-6_11
- Sohrabi, C., Alsafi, Z., O’Neill, N., Khan, M., Kerwan, A., Al-Jabir, A., Iosifidis, C., & Agha, R. (2020). World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). International Journal of Surgery, 76, 71–76. https://doi.org/10.1016/j.ijsu.2020.02.034
- Sokhansanj, B. A., & Rosen, G. L. (2022). Predicting COVID-19 disease severity from SARS-CoV-2 spike protein sequence by mixed effects machine learning. Computers in Biology and Medicine, 149. https://doi.org/10.1016/j.compbiomed.2022.105969
- Suri, J. S., Puvvula, A., Biswas, M., Majhail, M., Saba, L., Faa, G., Singh, I. M., Oberleitner, R., Turk, M., Chadha, P. S., Johri, A. M., Sanches, J. M., Khanna, N. N., Viskovic, K., Mavrogeni, S., Laird, J. R., Pareek, G., Miner, M., Sobel, D. W., … Naidu, S. (2020). COVID-19 pathways for brain and heart injury in comorbidity patients: A role of medical imaging and artificial intelligence-based COVID severity classification: A review. Computers in Biology and Medicine, 124(January), 103960. https://doi.org/10.1016/j.compbiomed.2020.103960
- Tang, L., Wu, T., Chen, X., Wen, S., Zhou, W., Zhu, X., & Xiang, Y. (2024). How COVID-19 impacts telehealth: An empirical study of telehealth services, users and the use of metaverse. Connection Science, 36(1). https://doi.org/10.1080/09540091.2023.2282942
- Tarek, Z., Shams, M. Y., Towfek, S. K., Alkahtani, H. K., Ibrahim, A., Abdelhamid, A. A., Eid, M. M., Khodadadi, N., Abualigah, L., Khafaga, D. S., & Elshewey, A. M. (2023). An optimized model based on deep learning and gated recurrent unit for COVID-19 death prediction. Biomimetics, 8(7), 552. https://doi.org/10.3390/biomimetics8070552
- Thara, D. K., Prema Sudha, B. G., & Xiong, F. (2019). Auto-detection of epileptic seizure events using deep neural network with different feature scaling techniques. Pattern Recognition Letters, 128, 544–550. https://doi.org/10.1016/j.patrec.2019.10.029
- Thölke, P., Jose, Y., Ramos, M., & Abdelhedi, H. (2022). Class imbalance should not throw you off balance: choosing the right classifiers and performance metrics for brain decoding with imbalanced data. BioRxiv, 1–35. https://doi.org/10.1101/2022.07.18.500262
- Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser, Ł, & Polosukhin, I. (2017). Attention is all you need. Advances in Neural Information Processing Systems, 2017-Decem(Nips), 5999–6009.
- Voita, E., Talbot, D., Moiseev, F., Sennrich, R., & Titov, I. (2020). Analyzing multi-head self-attention: Specialized heads do the heavy lifting, the rest can be pruned. ACL 2019 – 57th Annual Meeting of the Association for Computational Linguistics, Proceedings of the Conference, 28 July – 2 August, 5797–5808. https://doi.org/10.18653/v1/p19-1580
- Wang, R., Hozumi, Y., Yin, C., & Wei, G.-W. (2020). Mutations on COVID-19 diagnostic targets. Genomics, 112(6), 5204–5213. https://doi.org/10.1016/j.ygeno.2020.09.028
- World Health Organization. (2023). Novel-Coronavirus-2019 Www.Who.Int. https://www.who.int/es/emergencies/diseases/novel-coronavirus-2019.
- Wu, C., Yin, W., Jiang, Y., & Xu, H. E. (2022). Structure genomics of SARS-CoV-2 and its omicron variant: Drug design templates for COVID-19. Acta Pharmacologica Sinica, December 2021, 1–13. https://doi.org/10.1038/s41401-021-00851-w
- Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. https://doi.org/10.1038/s41586-020-2008-3
- Yin, R., Luo, Z., Zhuang, P., Zeng, M., Li, M., Lin, Z., & Kwoh, C. K. (2023). Vipal: A framework for virulence prediction of influenza viruses with prior viral knowledge using genomic sequences. Journal of Biomedical Informatics, 142(April). https://doi.org/10.1016/j.jbi.2023.104388
- Yin, R., Luusua, E., Dabrowski, J., Zhang, Y., & Kwoh, C. K. (2020). Tempel: Time-series mutation prediction of influenza A viruses via attention-based recurrent neural networks. Bioinformatics (oxford, England), 36(9), 2697–2704. https://doi.org/10.1093/bioinformatics/btaa050
- Yin, R., Thwin, N. N., Zhuang, P., Lin, Z., & Kwoh, C. K. (2022). IAV-CNN: A 2D convolutional neural network model to predict antigenic variants of influenza A virus. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 19(6), 3497–3506. https://doi.org/10.1109/TCBB.2021.3108971
- Zaremba, W., Sutskever, I., & Vinyals, O. (2014). Recurrent Neural Network Regularization. ArXiv:1409.2329, 2013, 1–8. http://arxiv.org/abs/1409.2329.
- Zhang, J., Xiao, T., Cai, Y., & Chen, B. (2021). Structure of SARS-CoV-2 spike protein. Current Opinion in Virology, 50, 173–182. https://doi.org/10.1016/j.coviro.2021.08.010
- Zhou, B., Zhou, H., Zhang, X., Xu, X., Chai, Y., Zheng, Z., Kot, A. C., & Zhou, Z. (2023). TEMPO: A transformer-based mutation prediction framework for SARS-CoV-2 evolution. Computers in Biology and Medicine, 152, 106264. https://doi.org/10.1016/j.compbiomed.2022.106264
Appendix
Table A1. The parameters of the agglomerative clustering algorithm and the values of these parameters.
Table A2. The best values obtained by using the GridSearchCV algorithm for 3 randomly selected features of the SVM model.
Table A3. The best values obtained by using the GridSearchCV algorithm for 3 randomly selected features of the KNN model.
Table A4. The best values obtained by using the GridSearchCV algorithm for 3 randomly selected features of the XGBoost model.
Table A5. The best values obtained by using the GridSearchCV algorithm for 3 randomly selected features of the LR model.