Abstract
A monoclonal antibody (MAb) generated against Ochratoxin A (OTA) has been used in a competitive direct enzyme linked immunosorbent assay (cdELISA) for the detection of OTA in dried figs obtained from the Spanish retail market. Fifty per cent inhibition of the maximum binding was obtained with an OTA concentration of 2 ng/mL, and the detection limit for OTA in solution was 0.18 ng/mL, corresponding to 3.15 ng OTA per gram of sample. OTA was detected in 19 (54.3%) out of 35 samples of dried figs analysed, with concentrations that ranged from 3.15 to 277 ng/g. Five samples contained OTA concentrations above the tolerable level set by EC regulations for dried vine fruits (10 ng/g). The MAb-based cdELISA assay developed in this work could be effectively applied for OTA screening in dried figs.
Introduction
Ochratoxin A (OTA) is a mycotoxin produced primarily by Penicillium verrucosum, Aspergillus ochraceus and Aspergilli of the section Nigri, especially A. carbonarius. (Blesa, Soriano, Molto, & Mañes, Citation2006; Esteban, Abarca, Bragulat, & Cabañes, Citation2004; European Food Safety Authority [EFSA], Citation2006). In Spain, A. carbonarius has been reported as the principal responsible for OTA contamination of grapes, wine and dried vine fruits (Abarca, Accensi, Bragulat, Castellá, & Cabañes, Citation2003; Cabañes et al., Citation2002). The occurrence of this toxin has been reported in several foodstuffs including cereals, coffee, cocoa, spices, beer, wine, grape juice, dried fruits and blood derived meat products (Angelini, Bazzo, Savino, & Borgo, 2008; de Magalhães, Sodré, Viscogliosi, & Grenier-Loustalot, 2011; Karbancioglu-Guler & Heperkan, Citation2008; Visconti & De Girolamo, Citation2005).
Toxicological studies indicate that OTA is generally absorbed from the gastrointestinal tract in animals and show strong toxic effects in their livers and kidneys. Exposure to OTA has been associated with distinct renal diseases endemic in the Balkans, referred to as Balkan endemic nephropathy and urinary tract tumours. In addition, the toxin is also considered to be teratogenic, mutagenic and immunosuppressive in certain animal models (Castegnaro et al., Citation2006; Creppy, Citation2002; Duarte, Pena, & Lino, Citation2010; Liu, Tsao, Wang, & Yu, 2008). Thus, the International Agency for Research on Cancer (IARC) evaluated OTA in 1993, and classified it as possibly carcinogenic to humans (group 2B), based on sufficient evidence for carcinogenicity in animal studies and inadequate evidence in humans (IARC, 1993).
Studies carried out in Europe reported the presence of ochratoxigenic fungi and OTA in dried fruits (Bircan, Citation2009; Karbancioglu-Guler & Heperkan, Citation2008; MacDonald et al., Citation1999; Ministry of Agriculture, Fisheries and Food [MAFF], Citation1999; Möller & Nyrberg, Citation2003). Presence of OTA in these commodities is a recognised problem due to toxin formation during sun drying of the fruits (Karbancioglu-Guler & Heperkan, Citation2008; Stefanaki, Foufa, Tsatsou-Dritsa, & Dais, 2003). The European Commission (Citation2010) has established maximum levels of OTA for raw cereal grains, cereal products, dried vine fruit, roasted coffee, wine, grape juice, spices (pepper, chilli pepper, nutmeg, ginger and turmeric), liquorice and foods for infants and young children. Moreover, it is foreseen setting maximum limits for OTA in dried fruits other than dried vine fruits, as well as a review of the existing maximum levels, in particular for OTA in dried vine fruit (10 µg/kg) and grape juice (2 µg/kg).
The availability of reliable methods for the determination of OTA in wine is therefore required in order to fulfil the need to protect consumer health from the risk of exposure to the toxin (Visconti, Pascale, & Centonze, Citation1999). The analytical methods used to determine OTA in food are based mainly on high-performance liquid chromatography with fluorescence detection using cleanup and preconcentration procedures based on solid-phase extraction (SPE) and immunoaffinity columns (Bircan, Citation2009; Iamanaka, Tanawaki, Menezes, Vicente, & Fungaro, 2005; MacDonald et al., Citation1999; Stefanaki et al., Citation2003; Zinedine et al., Citation2007). The instrumental methods provide sensitive and specific techniques, but they are time-consuming, very laborious, need skilled personnel and require expensive and sophisticated equipment. A rapid, sensitive and specific assay is therefore highly desirable for routine analysis of large number of samples. The competitive enzyme-linked immunosorbent assay provides a simple, fast, inexpensive and reliable alternative to instrument methods. Many samples can be analysed simultaneously, and this assay is commonly used to analyse the presence of many mycotoxins in foodstuffs (Angelini et al., Citation2008; Fujii et al., Citation2006; Park, Kimy, Shon, & Kim, 2002; Thirumala-Devi et al., Citation2000; Wang, Liu, Xu, Zhang, & Wang, Citation2007; Zheng et al., Citation2005).
In the present study a cdELISA based on a monoclonal antibody (MAb) was developed for the detection of OTA in naturally contaminated dried figs obtained from the Spanish retail market.
Materials and methods
Preparation of OTA-protein conjugates
OTA–protein conjugates were prepared by coupling OTA to keyhole limpet hemocyanin (KLH, Pierce Biotechnology, Rockford, USA) or horseradish peroxidase (HRP, Sigma) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Pierce) following manufacturer instructions. OTA was dissolved in 0.1 M MES (2-[N-morpholino]ethane sulfonic acid) at a concentration of 2 mg/mL, and 0.1 mL of freshly prepared EDC solution (10 mg/mL in MilliQ water) were added to 0.5 ml of the mycotoxin solution. This mixture was added to 0.2 mL (2 mg) of either KLH or HRP dissolved in MES. The reaction was allowed to proceed at room temperature for 2 h, and then was dialysed overnight at 4 °C against 0.1 M phosphate-buffered saline (PBS, pH 7.4). The dialysed solution was collected in a dark flask and stored at 4 °C until used.
Production of MAbs
Four 6-week-old female BALB/c mice were injected intraperitoneally with 0.2 mL of an emulsion containing 0.1 mL of 1 mg/mL OTA-BSA conjugate (Sigma, Steinheim, Germany) and 0.1 mL of complete Freund's adjuvant (Sigma). Mice were boosted four times by intraperitoneal injections with the same dose of antigen emulsified in incomplete Freund's adjuvant (Sigma) at 15-day intervals. Sera were collected by tail vein bleeding and tested for specific antibody production by indirect ELISA. The mice were finally boosted intravenously with 0.2 mL of conjugated OTA (without adjuvant) three days before the cellular fusion. The immunised mouse with the highest titre of serum antibodies was sacrificed by ether and cervical dislocation for hybridoma production. The spleen was removed aseptically and mechanically dissociated by passage through a sterile nylon mesh. Spleen cells were mixed with myeloma cells (P3X63-Ag 8653) in a 5:1 ratio and fused in polyethylene glycol (molecular weight, 1500; Roche Diagnostics, Mannheim, Germany) as described by Köhler and Milstein (Citation1975). The cells were handled and maintained according to a previously described procedure (Gutiérrez et al., Citation1997). Cell growth was apparent 10–14 days after fusion. The supernatants of cell cultures were screened by indirect ELISA for the presence of antibodies binding to OTA. The hybridomas of interest were cloned by limiting dilution in 96-well plates with DMEM medium (GIBCO, Paisley, UK) containing 15% bovine calf serum (PAA Laboratories, Liuz, Austria). The clones of interest were then allowed to multiply in triple-flasks (Nunc, Roskilde, DK) to obtain a high enough concentration of the MAb for further experiments, and the hybridomas were frozen at –80 °C in bovine calf serum-DMSO (90:10) (Sigma).
Indirect ELISA
Screening for hybridoma cells producing anti-OTA antibodies was carried out using indirect ELISA. Flat-bottomed micro-ELISA plates (Costar, New York, USA) were coated with 0.1 mL per well of OTA-KLH (1 mg/mL) diluted 1:750 in PBS and incubated for 1 h at 37 °C. The wells were washed five times with PBS containing 1% Tween 20 (PBST). Then, 50 µl of the culture supernatants and 50 µl of PBS were added to the wells. The plates were incubated for 1 h at room temperature (22 °C) with shaking, and washed again with PBST. One hundred microlitres of peroxidase-conjugated rabbit anti-mouse immunoglobulins (DAKO, Glostrup, Denmark) diluted 1:1000 in PBST were added to the wells and the plates were incubated with shaking for 1 h at room temperature before being washed with distilled water. Finally, 0.1 mL of 2,2’-azino-bis-[3-ethylbenzthiazoline-6-sulfonate] substrate (ABTS, Roche Diagnostic, Mannheim, Germany) was added to each well, the plates were incubated for 20 min at room temperature and the colorimetric reaction was measured at 405 nm using a iEMS Reader MF Spectrophotometer (Labsystems, Oy, Helsinki, Finland).
Selection of positive anti-OTA MAb by competitive indirect ELISA
Hybridomas showing binding to OTA in the indirect ELISA were screened for displacement with OTA in solution using a competitive indirect ELISA (ciELISA). ciELISA differs from indirect ELISA in that 50 µl of the hybridoma supernatants (diluted 1:500 in PBS) were added to the wells together with 50 µl of OTA standard (0.15 ng/mL–10 ng/mL), and incubated with shaking for 1 h at room temperature. To increase sensitivity, 50 µl of the substrate solution consisting of ready-to-use 3,3’, 5,5’-tetramethylbenzidine (TMB, Roche Diagnostic, Indianapolis, USA) was added to the wells, instead of ABTS. The plates were incubated at room temperature (22 °C) for 15 min before addition of 25 µl stop solution (1 M H2SO4), and absorbance measurements were read at 450 nm on a plate reader. The sensitivity of MAb for detection of OTA was determined from a standard curve built by plotting the absorbance against the concentration of OTA in solution.
Determination of isotypes of anti-OTA MAb
The isotypes of anti-OTA MAb were determined by the Mouse Monoclonal Antibody Isotyping Reagents Kit (Sigma, Missouri, USA) using class and subclass specific anti-mouse immunoglobulins (IgG, IgA and IgM), following the antigen mediated method recommended by the manufacturer.
Purification of anti-OTA MAb
Immunoglobulins were purified from the hybridoma supernatants using Hi-Trap Protein G HP affinity columns (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Protein concentration of the purified antibodies was measured using a NanoDrop Spectrophotometer ND-1000 (NanoDrop Technologies, INC, Wilmington, USA).
Measurement of cross-reactivity
Cross-reactivity of anti-OTA MAb towards ochratoxin B (OTB), citrinin (CTN), phenylalanine (Phe) and BSA was evaluated through cdELISA as described below for analysis of dried fig samples, and using the following equation (Cho et al., Citation2005):
OTA extraction from dried fig samples
Thirty-five dried fig samples (approximately 500 g per sample) were purchased from local food stores located in Madrid (Spain), and stored at –20 °C in airtight containers until used. OTA extraction was carried out essentially according to Senyuva, Gilbert, Ozcan, & Ulken (Citation2005). One part of dried figs (100 g) was homogenised at high speed with one part of water (100 mL) in a blender (Sunbeam Oster, FL, USA). One hundred milliliters of methanol was added to 40 g of the slurry (equivalent to 20 g of sample), and the sample was blended at high speed for 1 min. Then, 80 mL of 2% sodium bicarbonate was added, and the sample was blended again for 2 min at high speed. The sample was then centrifuged at 10,000x g for 20 min at 4 °C, and the supernatant filtrated through nitrocellulose filter (0.22 µm GSWP) (Millipore, Cork, Ireland). The filtrate was collected in a glass flask wrapped in aluminium foil, and stored at 4 °C until used.
OTA purification by immunoaffinity column cleanup
Immunoaffinity columns ‘Neocolum for Ochratoxin’, (Neogen, Auchincruive, UK) were used to purify OTA from the figs extracts, following the manufacturer directions. For conditioning, 10 mL of PBS was passed through the column by gravity. Ten mL of the clear filtrate were mixed with 40 mL of PBS and the diluted filtrate was passed through the column by gravity. Then 20 mL of PBST (0.01% v/v) and 10 mL of PBS were passed and the column was dried by passing air through it with the aid of a syringe. OTA was eluted using a two step-procedure. First 1.5 mL of methanol was passed through the column by gravity followed by 1.5 mL of distilled water and the eluate was collected in a dark flask and stored at 4 °C until used.
OTA analysis: competitive direct ELISA
A cdELISA was used for the OTA analysis in dried fig samples. The wells of a microtitre plate were coated with the purified anti-OTA MAb (1.5 mg/mL) diluted 1:200 in PBS and incubated for 1 h at 37 °C. After washing the plate with PBST, 50 µl of an OTA standard (0.15 ng/mL–10 ng/mL) or purified sample extract diluted 1:5 in PBS were added to the wells together with 50 µl OTA-HRP (1 mg/mL) diluted 1:500 in PBS. Plates were incubated for 1 h with shaking at room temperature, and then washed with distilled water. Fifty microlitres of TMB substrate were added to the wells and the plate was incubated 20 min at room temperature before stopping the reaction with 25 µl of 1 M H2SO4. Absorbance measurements were read at 450 nm on a plate reader. Average absorbance was calculated from individual absorbance obtained from triplicate wells and results were expressed as the percentage of inhibition:
where A 1 was the mean absorbance in the presence of the sample or OTA standard and A 0 was the mean absorbance in their absence. The OTA concentration was determined using a standard curve (7.8–500 pg OTA/well), plotting percent inhibition against the log of OTA amount. Detection limit was determined as the concentration corresponding to 10% of inhibition of OTA–HRP conjugate.
Results and discussion
Production and characterisation of monoclonal anti-OTA antibodies
OTA is a low molecular weight molecule (403.8 Da), that has to be conjugated to a carrier protein to elicit immune response in animals. In this work, a commercial OTA–BSA conjugate was used to immunise mice. Screening for antibody production started 14 days after fusion of sensitised mouse spleen cells with myeloma cells. A clone was selected as positive when the measured absorbance yielded a value above 1.0. Three hybridoma cultures secreting antibodies specific to OTA (named 5D3, 5E3 and 1C6) were identified by indirect ELISA from a total of 288 wells tested. The analysis of the isotype of the anti-OTA antibodies showed that the three hybridomas produced IgG1 antibodies. In addition, ciELISA was implemented in the screening steps. Evidence of free OTA binding was exhibited by the antibodies produced by hybridoma 5E3, whereas all other antibodies showed no binding to free OTA in solution. Cells of hybridoma 5E3 were further cloned by limiting dilution, and finally, a stable hybridoma cell line, named MAP1, producing antibodies against OTA was established and used for subsequent tests.
Production of useful quantities of the MAP1 MAb was achieved by culturing in large flasks. The culture supernatant was harvested after 15 days, and immunoglobulins purified by affinity chromatography in a Protein G column. The eluted fractions containing the purified immunoglobulins were pooled and the protein concentration of the purified MAP1 was 1.5 mg/mL. It should be stated that the purified MAP1 preparation also contains some immunoglobulins from the bovine calf serum used for hybridoma culture.
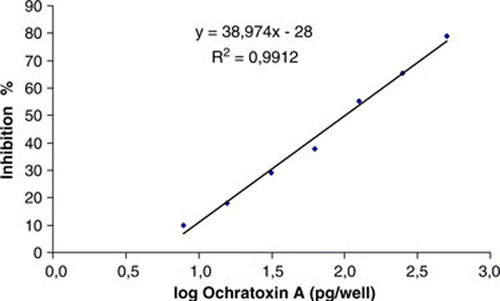
Competitive direct ELISA (cdELISA) was used to determine the sensitivity and specificity of MAP1 MAb. From the standard curve of cdELISA using MAP1 and OTA–HRP conjugate, the detection range was between 7.8 and 500 pg OTA/well. The detection limit of the cdELISA, based on a 90% confidence interval at 10% inhibition of OTA–HRP conjugate, was 9.4 pg OTA/well (0.18 ng/mL OTA in solution), corresponding to 3.15 ng OTA per gram of dried fig, and the 50% inhibition concentration (IC50) was 100.3 pg OTA/well (2 ng/mL), corresponding to 33.4 ng/g dried fig (). The cdELISA developed here showed a detection limit comparable to the detection limits reported for OTA detection by ELISA in other studies. Fujii et al. (Citation2006) reported a detection limit of 3.75 ng OTA per gram of coffee beans. Park et al. (Citation2002), Liu et al. (Citation2008) and Flajs, Domijan, Ivic, Cvjetkovic, and Peraica (Citation2009) recorded a detection limit of 5 ng OTA per gram of cereals, coffee beans and red wines, respectively. Lower detection limit (1.9 ng/g) was reported by Zheng et al. (Citation2005) and Díaz, Torres, Vega, & Latorre (Citation2009) in corn and grapes.
Figure 1. Standard curve for OTA detection by cdELISA. Each point represents the mean inhibition data from duplicate values of three standard curves performed in different days.
OTA is the most prominent member of the ochratoxin family, followed by OTB, and their chemical structures are very similar. OTB lacks only the chlorine atom in the isocoumarin ring that has the OTA molecule (Heussner, Moeller, Day, Dietrich, & O'Brien, Citation2007). Other molecules with chemical structures similar to OTA that could be recognised by antibodies against the mycotoxin are Phe and CTN. Also, because mice were immunised with an OTA–BSA conjugate, immunisation results in antibodies specific for the hapten, the carrier and also for various mixtures of these molecules. Here, cross-reactivities to the carrier were avoided because antibodies against BSA found no compatible antigen in the indirect ELISA screening procedure. Nevertheless, the presence of antibodies that recognise BSA epitopes should be tested. The cross-reactivity experiment against OTB, Phe, CTN and BSA showed that this antibody had a moderate cross-reaction with OTB (30.5%), whereas there was no reactivity with CTN, Phe and BSA, indicating that the antibody recognises neither the Phe nor the BSA molecules but an epitope specific for the entire structure of ochratoxins.
Application of the ELISA assay to the analysis of OTA in dried figs
Thirty-five dried fig samples were obtained from local food stores and analysed by cdELISA for detection of OTA contamination. The ELISA analysis of dried fig samples showed that 19 out of 35 samples analysed in the survey (54.3%) contained OTA at levels above the detection limit of 3.15 ng/g, with OTA concentrations ranged from 3.15 to 245.3 ng/g. Fourteen samples (40%) contained OTA levels from 3 to 10 ng/g, and five samples (14.3%) contained more than the maximum tolerable level of OTA set by EC regulations for dried vine fruits (10 ng/g).
In comparison, previous surveys of OTA in retail samples of dried figs, all of them using HPLC methods, reported incidence rates of OTA between 10 and 95%. In the European Commission's (2002) Scientific Co-operation (SCOOP) project report, 20 samples of dried figs imported to the Netherlands were analysed, and two of them (10%) contained OTA, at a maximum concentration of 0.8 ng/g EU (Miraglia & Brera, Citation2002). In Turkey, Bircan (Citation2009) recorded contaminations of OTA in 18% of the dried fig samples analysed, with a maximum concentration of 24.4 ng/g, and Senyuva et al. (2005) reported 14% and 15% OTA occurrence in a two-year study, with a maximum concentration of 26.3 ng/g. Another two-year study carried out in Turkey revealed that 48% of the dried fig samples analysed contained OTA with a maximum concentration of 15 ng/g (Karbancioglu-Guler & Heperkan, Citation2008). OTA was present in 95% of dried fig samples from Brazil with maximum level detected at 23.4 ng/g (Iamanaka et al. Citation2005). In a survey carried out in Morocco, Zinedine et al. (Citation2007) reported the presence of OTA in 65% of the 20 retail dried fig samples analysed, with a maximum concentration of 1.42 ng/g. High concentrations of OTA in dried figs have been reported by Bayman, Baker, Doster, Michailides, and Mahoney (2002) who examined figs from California for ochratoxigenic fungi infection, and measured OTA content in figs with visible fungal colonies. OTA concentrations as high as 1850 ng/g were reported for individual infected figs. It should be pointed out that the two most contaminated samples in the present survey (237 and 245.3 ng/g) had been elaborated with traditional techniques and were purchased in bulk at a local open market. Traditional methods for drying figs may offer optimal environmental conditions for mould growth and mycotoxin production (Zinedine et al., Citation2007) that could explain why these two home-made fig samples contained high concentrations of OTA. Application of the Hazard Analysis and Critical Control Point (HACCP) system in the food industries makes it very difficult to reach such high levels of moulds contamination and OTA concentration in commercial products. However, many small traditional factories have difficulties to apply high hygienic standards to control the growth of moulds and mycotoxin production during the harvesting, fruit drying, distribution and storage periods.
The sensitivity obtained in the works mentioned above (0.1–0.47 ng/g) is better than that developed in the present work, because of the fact that the detection of OTA in dried frigs in these studies was performed using HPLC. However, this cdELISA can be effectively applied in OTA screening of dried figs, with the advantages over HPLC in terms of simplicity and rapidity; it does not require complicated equipment, and high throughput of large numbers of samples if possible. Quality control using a reliable immunoassay is essential to assure the safety of the dried fig samples associated with good harvesting, drying and storage practices (Fujii et al., Citation2006).
Acknowledgements
This study was supported by grant no. PR 27/05-13995-BSCH from the Banco Santander Central Hispano/Universidad Complutense de Madrid and the Programa de Vigilancia Sanitaria S2009/AGR-1489 from the Comunidad de Madrid (Spain). Miguel Ángel Pavón is recipient of a fellowship from the Ministerio de Educación y Ciencia (Spain).
References
- Abarca , M.L. , Accensi , F. , Bragulat , M.R. , Castellá , G. and Cabañes , F.J. 2003 . Aspergillus carbonarius as the main source of ochratoxin A contamination in dried vine fruits from the Spanish market . Journal of Food Protection , 66 : 504 – 506 .
- Angelini , E. , Bazzo , I. , Savino , M. and Borgo , M. 2008 . Ochratoxin A: Comparison of extraction methods from grapes and quantitative determination by different competitive enzyme-linked immunosorbent assay kits . Journal of Food Protection , 71 : 2488 – 2496 .
- Bayman , P. , Baker , J.L. , Doster , M.A. , Michailides , T.J. and Mahoney , N.E. 2002 . Ochratoxin production by the Aspergillus ochraceus group and Aspergillus alliaceus . Applied Environmental Microbiology , 68 : 2326 – 2329 .
- Bircan , C. 2009 . Incidence of ochratoxin A in dried fruits and co-ocurrence with aflatoxins in dried figs . Food and Chemical Toxicology , 47 : 1996 – 2001 .
- Blesa , J. , Soriano , J.M. , Molto , J.C. and Mañes , J. 2006 . Factors affecting the presence of ochratoxin A in wines . Critical Reviews in Food Science and Nutrition , 46 : 473 – 478 .
- Cabañes , F.J. , Accensi , F. , Bragulat , M.R. , Abarca , M.L. , Castellá , G. Minguez , S. 2002 . What is the source of ochratoxin A in wine? . International Journal of Food Microbiology , 79 : 213 – 215 .
- Castegnaro , M. , Canadas , D. , Vrabcheva , T. , Petkova-Bocharova , T. , Chernozemsky , I.N. and Pfohl-Leszkowicz , A. 2006 . Balkan endemic nephropathy: Role of ochratoxins A through biomarkers . Molecular Nutrition and Food Research , 50 : 519 – 529 .
- Cho , Y.J. , Lee , D.H. , Kim , D.O. , Min , W.K. , Bong , K.T. Lee , G.G. 2005 . Production of monoclonal antibody against ochratoxin A and its application to immunochromatographic assay . Journal of Agricultural and Food Chemistry , 53 : 8447 – 8451 .
- Creppy , E.E. 2002 . Update of survey, regulation and toxic effects of mycotoxins in Europe . Toxicology Letters , 127 : 19 – 28 .
- de Magalhães , J.T. , Sodré , G.A. , Viscogliosi , H. and Grenier-Loustalot , M. 2011 . Occurrence of Ochratoxin A in Brazilian cocoa beans . Food Control , 22 : 744 – 748 .
- Díaz , G. , Torres , R. , Vega , M. and Latorre , B.A. 2009 . Ochratoxigenic Aspergillus species on grapes from Chilean vineyards and Aspergillus threshold levels on grapes . International Journal of Food Microbiology , 133 : 195 – 199 .
- Duarte , S.C. , Pena , A. and Lino , C.M. 2010 . A review on ochratoxin A occurrence and effects of processing of cereal and cereal derived food products . Food Microbiology , 27 : 187 – 198 .
- Esteban , A. , Abarca , M.L. , Bragulat , M.R. and Cabañes , F.J. 2004 . Effects of temperature and incubation time on production of ochratoxin A by black aspergilli . Research in Microbiology , 155 : 861 – 866 .
- European Commission . 2010 . European Commission regulation No. 105/2010 of 5 February setting maximum levels for certain contaminants in foodstuffs as regards ochratoxin A . Official Journal of the European Union , L 35 , 7 – 8 .
- European Food Safety Authority (EFSA) . 2006 . Opinion of the Scientific Panel on Contaminants in the Food Chain on a Request from the Commission related to ochratoxin A in food. Adopted on 4 April 2006 . The EFSA Journal , 365 , 1 – 56 .
- Flajs , D. , Domijan , A.M. , Ivic , D. , Cvjetkovic , B. and Peraica , M. 2009 . ELISA and HPLC analysis of ochratoxin A in red wines of Croatia . Food Control , 20 : 590 – 592 .
- Fujii , S. , Ribeiro , R.M. , Scholz , M.B. , Ono , E.Y. , Prete , C.E. Itano , E.N. 2006 . Reliable indirect competitive ELISA used for a survey of ochratoxin A in green coffee from the North of Paraná State, Brazil . Food Additives and Contaminants , 23 : 902 – 909 .
- Gutiérrez , R. , González , I. , García , T. , Carrera , E. , Sanz , B. Hernández , P.E. 1997 . Monoclonal antibodies and an indirect ELISA for detection of psychotrophic bacteria in refrigerated milk . Journal of Food Protection , 60 : 23 – 27 .
- Heussner , A.H. , Moeller , I. , Day , B.W. , Dietrich , D.R. and O'Brien , E. 2007 . Production and characterization of monoclonal antibodies against ochatoxin B . Food and Chemical Toxicology , 45 : 827 – 833 .
- Iamanaka , B.T. , Tanawaki , M.H. , Menezes , H.C. , Vicente , E. and Fungaro , M.H.P. 2005 . Incidence of toxigenic fungi and ochratoxin A in dried fruits sold in Brazil . Food Additives and Contaminants , 22 : 1258 – 1263 .
- International Agency for Research on Cancer (IARC) . 1993 . Evaluation of carcinogenic risks of chemical to humans. Some naturally-occurring substances. Food items and constituents. Heterocyclic aromatic amines and mycotoxins . Monographs , 56 , 359 – 362 .
- Karbancioglu-Guler , F.K. and Heperkan , D. 2008 . Natural occurrence of ochratoxin A in dried figs . Analytica Chimica Acta , 617 : 32 – 36 .
- Köhler , G. and Milstein , C. 1975 . Continuous cultures of fused cells secreting antibody of predefined specificity . Nature , 256 : 495 – 497 .
- Liu , B.H. , Tsao , Z.J. , Wang , J.J. and Yu , F.Y. 2008 . Development of a monoclonal antibody against ochratoxin A and its application in enzyme-linked immunosorbent assay and gold nanoparticle immunochromatographic strip . Analytical chemistry , 80 : 7029 – 7035 .
- MacDonald , S.J. , Wilson , P. , Barnes , K. , Damant , A. , Massey , R. Mortby , E. 1999 . Ochratoxin A in dried vine fruit: Method development and survey . Food Additives and Contaminants , 16 : 253 – 260 .
- Ministry of Agriculture, Fisheries and Food (MAFF) . 1999 . Survey of aflatoxins and ochratoxins A in cereals and retail products (November 1997) . Food Surveillance Information . Sheet N0. 185. London: Ministry of Agriculture, Fish and Foods .
- Miraglia , M. & Brera , C. 2002 . Assessment of dietary intake of ohratoxin A by the population of EU member states . Reports on tasks for scientific cooperation. Reports of experts participating in SCOOP Task 3.2.7. Rome: Directorate-General Health and Consumer Protection .
- Möller , T.E. and Nyrberg , M. 2003 . Ochratoxin A in raisins and currants: Basic extraction procedure used in two small marketing surveys of the occurrence and control of the heterogeneity of the toxins in samples . Food Additives and Contaminants , 20 : 1072 – 1076 .
- Park , J.W. , Kimy , E.K. , Shon , D.H. and Kim , Y.B. 2002 . Natural co-occurrence of aflatoxin B1, fumonisin B1 and ochratoxin A in barley and corn foods from Korea . Food Additives and Contaminants , 19 : 1073 – 1080 .
- Senyuva , H.Z. , Gilbert , J. , Ozcan , S. and Ulken , U. 2005 . Survey for co-occurrence of ochratoxin A and Aflatoxin B1 in dried figs in Turkey by using a single laboratory-validated alkaline extraction method for ochratoxin A . Journal of Food Protection , 68 : 1512
- Stefanaki , I. , Foufa , E. , Tsatsou-Dritsa , A. and Dais , P. 2003 . Ochratoxin A concentrations in Greek domestic wines and dried wine fruits . Food Additives and Contaminants , 20 : 74 – 83 .
- Thirumala-Devi , K. , Mayo , M.A. , Reddy , G. , Reddy , S.V. , Delfosse , P. and Reddy , D.V.R. 2000 . Production of polyclonal antibodies against ochratoxin A and its detection in chillies by ELISA . Journal of Agricultural and Food Chemistry , 48 : 5079 – 5082 .
- Visconti , A. and De Girolamo , A. 2005 . Fitness for purpose-Ochratoxin A analytical developments . Food Additives and Contaminants , Suppl.1 : 37 – 44 .
- Visconti , A. , Pascale , M. and Centonze , G. 1999 . Determination of ochratoxin A in wine by means of immunoaffinity column clean-up and high-performance liquid chromatography . Journal of Chromatography A , 864 : 89 – 101 .
- Wang , X.H. , Liu , T. , Xu , N. , Zhang , Y. and Wang , S. 2007 . Enzyme-linked immunosorbent assay and colloidal gold immunoassay for ochratoxin A: Investigation of analytical conditions and sample matrix on assay performance . Analytical and Bioanalytical Chemistry , 389 : 903 – 911 .
- Zheng , Z. , Hanneken , J. , Houchins , D. , King , R.S. , Lee , P. and Richard , J.L. 2005 . Validation of an ELISA test kit for the detection of ochratoxin A in several food commodities by comparison with HPLC . Mycopathologia , 159 : 265 – 272 .
- Zinedine , A. , Soriano , J.M. , Juan , C. , Mojemmi , B. , Moltó , J.C. Bouklouze , A. 2007 . Incidence of ochratoxin A in rice and dried fruits from Rabat and Salé area, Morocco . Food Additives and Contaminants , 24 : 285 – 291 .