
Abstract
A competitive, direct, chemiluminescent enzyme-linked immunosorbent assay (CL-ELISA) for chloramphenicol (CAP) residues in milk, milk powder, honey, eggs and chicken muscle has been developed. The method gave a detection limit of 0.7 ng L−1 and a linear range of 2.1–92.4 ng L−1, with the IC50 of 13.6 ng L−1 under optimal conditions, dramatically better than any previously reported ELISA method for CAP detection. Spiked at levels of 5–60 ng L−1 in different food samples, recoveries were in the range of 72.1–116.0%, with coefficient of variations of 4.2–20.2%. In a study of incurred residues, the chicken muscle samples diluted 5-, 10- and 20-fold, results obtained by CL-ELISA correlated well with those obtained by gas chromatography with microcell electron capture detector and traditional ELISA. The developed CL-ELISA method is, therefore, suitable for rapid screening trace CAP residues in food samples.
Introduction
Chloramphenicol (CAP) is a very effective broad-spectrum antibiotic which was widely used in both human and veterinary practice for prevention and treatment of many bacterial infections. However, CAP is a hemotoxic substance for humans and can cause bone marrow depression, aplastic anaemia and acute leukaemia (Shi, Wu, Zheng, Li, & Zhang, Citation2007). These potential hazards led to a prohibition of its use in food-producing animals in many countries, including China, the USA and the EU (235th Bulletin, Citation2002; Stolker & Brinkman, Citation2005). Because of its low cost and excellent antibacterial effect, CAP is still illegally used. Therefore, it is urgent to develop a rapid and sensitive method for determining CAP at trace levels in food samples.
A variety of existing analytical methods to detect and qualify CAP in food matrices, such as liquid chromatography (LC) with an Iron Trap Detector (Moragues, Igualada, & León, Citation2012), LC–mass spectrometry (LC–MS) or LC–MS/MS (Barreto, Ribeiro, Hoff, & Costa, Citation2012; Lu et al., Citation2012; Rezende, Fleury Filho, & Rocha, Citation2012; Taka, Baras, & Chaudhry Bet, Citation2012), enzyme-linked immunosorbent assay (ELISA) (Sai et al., Citation2010; Wang, Zhang, Gao, Duan, & Wang, Citation2010), surface plasmon resonance-based biosensor (Yuan, Oliver, Aguilar, & Wu, Citation2008) and piezoelectric immunosensor (Karaseva & Ermolaeva, Citation2012), have been reported for the determination of CAP residue in animal product. However, these instrumental approaches, although accurate, and fit for multiresidue detection, were costly and very time-consuming, thus not suitable for detection of large number of samples. The chemiluminescent ELISA (CL-ELISA) offers the possibility of improving the sensitivity of the immunoassay by at least 2–3 orders of magnitudes compared with conventional colorimetric detection, which would make it a useful system for detecting trace residue of CAP in animal products. Previous reports (Lin, Han, Liu, Xu, & Guan, Citation2005; Xu, Peng, Hao, Jin, & Wang, Citation2006; Zhang, Zhang, Shi, Eremin, & Shen, Citation2006) have established more sensitive CL-ELISA for detection of CAP than traditional ELISA. However, these methods were only fit for one sample type (either milk or chicken muscle) and required dilution of the extract solution to overcome matrix interference, decreasing the sensitivity of the methods.
In this study, we have developed a more sensitive CL-ELISA for the trace determination of CAP applicable in milk, milk powder, honey, eggs and chicken muscle, simultaneously. Under optimal conditions, the sensitivity of the CL-ELISA was 0.7 ng L−1, and the linear range was 2.1–92.4 ng L−1. Such analytical sensitivity can provide additional data on the occurrence of CAP residues in foods that may be missed by current methodologies. The reaction time was just 15 min, the shortest time for ELISA's detection of CAP. This method was confirmed by gas chromatography with microcell electron capture detector (GC-µECD) and, compared with traditional ELISA, in chicken muscle spiked with CAP or containing incurred residues.
Materials and methods
Apparatus
Chemiluminescence reader – Veritas Microplate Luminometer (Turner BioSystems, Sunny Vale, CA).
Microtitre plate reader – Sunrise Microtiter Plate Reader (TECAN, Groedig, Austria).
Transparent 96-well microtitre ELISA plates and White Opaque high-binding plates for chemiluminescent measurements – Costar (Cambridge, USA).
Milli-Q System – (Millipore, USA).
UV-Vis spectrophotometer – Model 751GW (Shanghai Analytical Instrument, Shanghai, China).
Buffers
Coating buffer (CB, pH 9.6) – 0.05 M carbonate buffer made with 1.59 g Na2CO3 and 2.93 g NaHCO3 in 1 L of purified water.
Blocking buffer – 0.01 M phosphate-buffered saline (PBS) containing 0.5% casein.
Washing solution (PBST) – 0.01 M PBS containing 0.05% Tween 20.
A 0.2 M sodium phosphate solution (pH 7.2) – containing 11.0 g NaH2PO4·2H2O, 51.6 g Na2HPO4·12H2O in 1 L purified water.
(e) PBS (pH 7.4) – 0.01M PBS was prepared by dissolving 8.0 g NaCl, 0.2 g KCl, 0.24 g KH2PO4 and 3.63 g Na2HPO4·12H2O in 1 L purified water.
Solution A: 0.36 M K4Fe(CN)6·3H2O; Solution B: 1.04 M ZnSO4·7H2O.
Substrate buffer (pH 5.0) – 1 mg/mL tetramethylbenzidine (TMB) and 0.01% (v/v) H2O2 in 4 mM citric acid and 10 mM Na2HPO4·12H2O solution.
Stop solution – 2.0 M H2SO4.
Reagents
Standards – CAP and CAP succinate (99% purity, Sigma Aldrich, St. Louis, MO); florfenicol (99% purity, Schering-Plough Corp., Kenilworth, NJ); thiamphenicol (97.6% purity, Schering-Plough Corp.); Levonitro base, clenbuterol, ractopamine, sulfadiazine, ciprofloxacin, penicillin were purchased from Shanghai Caienfu Technology Co., Ltd. (Shanghai, China). All of the stock solution concentration is 2 mg/mL and stored at −0°C, and working standards in the 0.5–180 ng/L range were prepared from the 2 mg/mL stock solution by serial dilution in assay buffer. The CAP, CAP succinate, florfenicol and Levonitro base stock solution were prepared in methanol; clenbuterol, ractopamine and sulfadiazine were prepared in ethanol; ciprofloxacin and penicillin were prepared in purified water; thiamphenicol was prepared in dimethylfomamide.
Analytical grade regents – 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene sulphonate, N-hydroxysuccinimide (NHS), bovine serum albumin (BSA), horseradish peroxidase (HRP), dicyclohexylcarbodiimide (DCC), dimethylformamide (DMF) and TMB were purchased from Sigma (St. Louis, USA). All other chemicals and organic solvents were of reagent grade and were purchased from Beijing Chemical Company (Beijing, China).
The chemiluminescent substrate solution called SuperSignal was purchased from Pierce (Rockford, IL).
Production of polyclonal antibody
Protein–CAP conjugates were synthesised by the activated ester method, using the previously reported procedure (Kolosova, Samsonova, & Egorov, Citation2000). In brief, 700 mg (1.6 mmol) CAP succinate was dissolved in 50 mL distilled water. Then 635 mg (1.5 mmol) 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene sulphonate in 10 mL distilled water and 175 mg (1.5 mmol) NHS in 10 mL distilled water were added (pH 5.3). The reaction mixture was incubated with stirring for 1 h at room temperature.
To 200 mg (3 mmol) BSA in 20 mL of PB, 20 mL of activated CAP succinate (630 mmol) was added. The reaction mixture was incubated with stirring for 2 h at room temperature followed by overnight incubation at 4°C. Subsequently, dialysis against distilled water was carried out for 5 days. The dialysed solution was characterised by UV-vis spectrophotometry. CAP–BSA conjugate was used as immunogen. The procedure of antibody production and characterisation of PAb was the same as that was reported by Zhang et al. (Citation2006). Especially, after five booster injections, the sera were collected and purified by saturated (NH4)2SO4, then supplemented with an equal volume of glycerol and stored at −20°C until testing.
Synthesis of HRP-conjugated CAP
HRP-conjugated CAP was synthesised using the previously reported procedure with minor revision (Lin et al., Citation2005). Briefly, the CAP succinate (3.1 mg), NHS (5.5 mg) and DCC (4.6 mg) were mixed and stirred in 0.5 mL DMF for 6 h at room temperature. The HRP (4.3 mg) in 1.5 mL of 0.2 M NaHCO3 was then added to the reaction mixture with stirring at room temperature for 16 h. The reaction solutions were then dialysed against PBS for three days and mixed with 2 mL of 50% glycerol and stored at −20°C.
Procedure of direct competitive CL-ELISA
Plates were coated overnight at 4°C with 100 µL of the PAb dissolved in buffer A (1.5 µg/mL). The plates were washed with 260 µL/well buffer C manually three times, blocked with 150 µL/well of buffer B and incubated at 37°C for 1 h. After the plates were washed as described above (conditioned ELISA plates can be stored at −4°C for one week), then 80 µL/well of standard in buffer D or sample solution, followed by 20 µL/well of HRP-conjugated CAP at a dilution of 1/160,000 in buffer D were added, respectively. The competitive reaction took place for 15 min at room temperature. After washing five times, finally the HRP tracer activity was revealed by adding 100 µL/well of a freshly prepared substrate mixture of SuperSignal substrate solution. The intensity of light emission was measured at 425 nm, using a chemiluminesence reader, immediately after the addition of the substrate, and the results were expressed in relative light units (RLUs). The procedure of traditional ELISA was the same as the described above. The reaction took place 15 min after addition of 100 µL/well TMB substrate, and absorbance was measured at 450 nm, using a microtitre plate reader, after addition of 50 µL/well 2.0 M H2SO4 within 10 min.
Optimisation of chemiluminescent ELISA
Several physicochemical factors influencing immunoassay performance were studied in CL-ELISA. Modifications of RLUmax and IC50 parameters of the standard curves were evaluated under different conditions. In order to assess the influence of Tween 20, buffer ionic strength, pH and competitive time and temperature, standard curves and PAbs were prepared as follows: (1) PAbs and standard curves were added to serial dilutions of Tween 20 (from 0% to 0.1%, v/v) in 0.1 M PB. (2) Standard analyte curves and a constant concentration of PAb were diluted by PB at different concentrations (0.01, 0.02, 0.05, 0.1, 0.2 mol/L). (3) Competitive analyte curves and a constant concentration of PAb were diluted in 0.1 M PB at different pH values (from 6.6 to 8.0). Meanwhile, the competitive reaction was taking place at room temperature or 37°C for 30, 45, 60, 75 min, respectively.
Data analysis
Standards and samples were run in quintuplicate wells, and mean chemiluminescence intensity values were divided by RLUmax (chemiluminescence intensity in the absence of analyte). The ratio is defined as B/B 0. Standard curves were obtained by plotting B/B0 against the logarithm of analyte concentration and fitted to a four-parameter logistic equation, using either Origin (version 8.0, Microcal, USA) software packages
Cross-reactivity study
The specificity of the antisera was assessed by evaluating the extent of cross-reactivity (CR) studies with three compounds structurally related and five other structurally unrelated to CAP in optimised traditional ELISA, and their IC50 values were compared with IC50 of CAP. CR was calculated as follows:
Sample preparation and GC-µECD analysis of CAP
Spiked milk sample analysis
For extraction of CAP from milk, 500 µL Solution A and 500 µL Solution B was added to 10 mL milk, mixed thoroughly and then centrifuged for 10 min at 4000g in 4°C; 4.4 mL of aqueous supernatant (amount to 4 mL milk) was thoroughly mixed with 8.0 mL ethyl acetate for 10 min in a new tube. Centrifuged at 4000g for 10 min, 4 mL of organic supernatant (amount to 2 mL milk) was transferred to a new tube and dried by nitrogen at 60°C. The residue was dissolved in 2 mL of buffer D. The sample solution was for determination.
Spiked milk powder sample analysis
Two grams of milk powder was dissolved in 10 mL distilled water in a 50 mL tube, and 1 mL Solution A and 1 mL extract Solution B were added, mixed thoroughly and then centrifuged for 10 min at 4000g in 4°C. Aqueous supernatant, 3.6 mL (amount to 0.6 g milk powder), was thoroughly mixed with 6.0 mL ethyl acetate for 10 min in a new tube. Centrifuged at 4000g for 10 min, 4 mL of organic supernatant (amount to 0.4 g milk powder) was filtered through an organic phase polytetrafluoroethylene filter membrane (0.22 µm) to a new tube and dried by nitrogen at 60°C. The residue was dissolved in 0.4 mL of buffer D. The sample solution was for determination.
Spiked honey sample analysis
Two grams of honey was dissolved in 4 mL distilled water in a 50 mL tube, 4.0 mL ethyl acetate was added and thoroughly mixed for 10 min and then the mixture was centrifuged at 4000g for 10 min. One millilitre of organic supernatant (amount to 0.5 g honey) was filtered through an organic phase polytetrafluoroethylene filter membrane (0.22 µm) to a new tube and dried by nitrogen at 60°C. The residue was dissolved in 0.5 mL of buffer D. The sample solution was for determination.
Spiked eggs sample analysis
Twelve millilitres of ethyl acetate was added to 2 g homogenised eggs and thoroughly mixed for 10 min. Centrifuged at 4000g for 10 min, 6 mL of organic supernatant (amount to 1 g eggs) was transferred to a new tube and dried by nitrogen at 60°C. The residue was dissolved in 1 mL of buffer D and 1 mL of hexane. The mixture was vortexed gently for 1 min. After centrifugation for 5 min at 4000g, the lower fraction was transferred to a new tube. The sample solution was for determination.
Spiked chicken muscle and incurred chicken muscle analysis
Three millilitres of double distilled water was added to 3 g homogenised chicken muscle sample in a 50 mL tube. After vortexing for 1 min, 6 mL of ethyl acetate was added to the mixture and shaken for 10 min at room temperature. After being centrifuged at 4000g for 10 min, 4 mL of organic supernatant (amount to 2 g chicken muscle) was filtered through an organic phase polytetrafluoroethylene filter membrane (0.22 µm) and dried by nitrogen at 60°C. The residue was dissolved in 2 mL of buffer D and 1 mL of hexane. The mixture was vortexed gently for 1 min. After centrifugation for 5 min at 4000g, the lower fraction was transferred to a new tube. Then 80 µL of the solution was added to the white polystyrene microtitre wells and measured.
Analysis of chicken muscle samples by gas chromatography with microcell electron capture detector (GC-µECD)
Sample extraction and clean-up procedures were an adaptation of previously reported procedures (Zhang et al., Citation2006). An Agilentmodel 6890 gas chromatograph equipped with a capillary column (HP-5, 5% phenyl methyl silicone, 30.0 m×320 µm×0.50 µm nominal, model 19091J-113) and a 63Ni electron capture detector (Agilent Technologies, Palo Alto, CA) were used. Samples (3 µL) were injected splitless. Nitrogen was used as a carrier gas. The following temperature programme was applied: start from 150°C (hold time of 0.5 min), then ramp at 30°C/min to 270°C and hold for 5 min. The detector was operated at 300°C.
Limit of detection and limit of quantification
Limit of detection (LOD) and limit of quantification (LOQ) of the CL-ELISA were estimated as the analyte concentrations that exceeded the values obtained for the mean of 20 blank samples plus 3 or 10 times standard deviation, respectively. The extracts of blank food matrices were filled in five wells of a microtitre plate and subjected to CL-ELISA analysis.
Results and discussion
Specificity of the antibody
Molar ratio of hapten to carrier proteins was approximately 16 for CAP–BSA conjugates. The sera from the rabbits showed high titre (1:100,000) in the direct competitive ELISA method. The CR of some related compounds such as Levonitro base, thiamphenicol and florfenicol was tested. There was no significant CR. The CR of other structurally unrelated drugs including clenbuterol, ractopamine, sulfadiazine, ciprofloxacin and penicillin was also tested. No CR was observed ().
Table 1. Percentage of CR of some structurally related and unrelated compounds.
Physicochemical parameter optimisation
Several experimental factors influencing the CL-ELISA performance were studied. The RLUmax/IC50 ratio has been shown to be a useful parameter to estimate the effect of a certain factor on the ELISA performance, the highest ratio indicating highest sensitivity (Mercader & Montoya, Citation1999).
Final assay conditions are summarised in . Selected conditions were antibody concentration (1.5µg/mL), competition time (15 min) and diluents for standard and conjugate (0.2 M PB). They produced a higher RLUmax/IC50 ratio than any other conditions we evaluated.
Table 2. Analytical characteristics of CL-ELISA procedure for CAP detection: ELISA conditions, analytical parameters.
Assay sensitivity
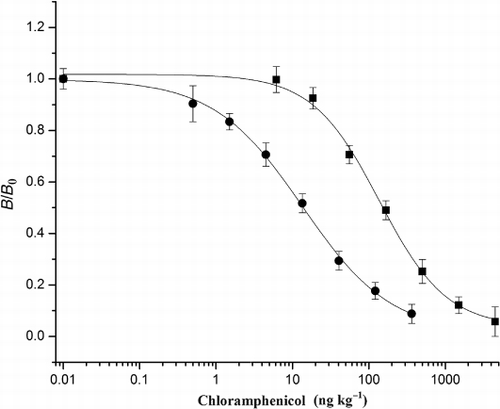
The sensitivity and LOD of the assay for CAP, which are represented by IC50 and IC10 values, were 13.6 ng L−1 and 0.7 ng L−1, respectively. The linear working range determined as the concentrations causing 20–80% inhibition of chemiluminescence intensity was 2.1—92.4 ng L−1. The sensitivity of this CL-ELISA was about 10 times greater than that obtained by the traditional ELISA method with colorimetric detector developed by our own (IC50 =137.0 ng L−1) (), about 70 times more sensitive than obtained by colorimetric ELISA previously reported (Scortichini et al., Citation2005) and even 50 times greater compared with the biotin–streptavidin-amplified BA-ELISA, which represents higher sensitivity in most cases (Wang et al., Citation2010).
Limit of detection and limit of quantification
In the case of the sample extracts in various food matrices, the LOD and LOQ were found to be 0.73–0.86 ng L−1 and 2.2–3.2 ng L−1, respectively (). The LOD for different matrices was consistent with the LOD (IC10) calculated from the standard curve ().
Table 3. The limit of detection and limit of quantification in milk, honey, eggs, milk powder, and chicken muscle matrices.
Table 4. Analytical parameters for CL-ELSIA of chloramphenicol.
Matrix effect examination and analysis of CAP-spiked samples
One of the common challenges of immunoassay for food analysis is matrix interference. The simplest way to overcome this interference is to have the analytical samples and standards in the same medium. Hence, we dried extracts under nitrogen and dissolved the dried residue in 0.2 M PB, the diluent used for standards. To gain information on the matrix effect, a calibration curve generated in 0.2 M PB was compared with that obtained using milk, honey, eggs, milk powder and chicken muscle matrices. The almost identical analytical parameters of the inhibition curves suggested that there were no significant matrix effects from milk, honey, eggs, milk powder and chicken muscle extract (). Compared with the previous reports (Lin et al., Citation2005; Xu et al., Citation2006; Zhang et al., Citation2006), we did not dilute the extract solution to overcome the matrix effect, reducing the sensitivity. In this study, the greatest advantage of the extract method of milk powder, honey and chicken muscle was using the polytetrafluoroethylene filter membrane (0.22 µm). The organic solvent (ethyl acetate and CAP dissolved in ethyl acetate) was filtered through the membrane, and the small-sized suspended substances in milk powder, pollen in honey and macromolecules and dissolved solids such as protein and enzyme in chicken muscle were prevented from going through the membrane. This filtering processing also reduced the background interference to some extent.
Milk, milk powder, honey, eggs and chicken muscle samples were spiked with CAP at 5, 20, and 60 ng/L and then analysed with the CL-ELISA. Each sample was evaluated five times in duplicate and on three different days to verify the repeatability.
In sample analysis, the intra-assay recoveries were in the range of 92.5–111.7% (milk), 76.0–116.0% (milk powder), 98.0–107.0% (honey), 85.2–116.0% (chicken muscle) and 84.0–104.0% (eggs) (). The coefficients of variation (CVs) were all <15%. The inter-assay recoveries were in the range of 86.2–114.1% (milk), 72.1–112.6% (milk powder), 102.8–111.0% (honey), 72.1–112.3% (chicken muscle) and 78.8–92.3% (eggs). The CVs were all <15% in milk and egg analysis, while in milk powder, honey and chicken muscle matrices analysis, the CVs were in the range of 15–20%.
Table 5. Intra- and inter-assay variations of food samples spiked with CAP.
In general, the CL-ELISA showed good results in milk, milk powder, honey, chicken muscle and eggs, and laid the foundation for the commercialisation of CL-ELSA kits for CAP.
Although the detection range of the CL-ELISA is narrow, and the upper detection limit is far lower than minimum required performance level (MRPL) of 0.3 µg kg−1, the extreme low detection limit fills the need for detecting ultra-trace CAP residues in foods that may not be detected by current methodologies. Meanwhile, simply diluting the extract 10-fold will enable the analysis of samples containing higher levels of CAP.
Analysis of CAP in incurred chicken muscle samples
The incurred chicken muscle sample was bought from meat market, confirmed by GC-µECD after preliminary screening with traditional ELISA. The positive sample (319.5 ng/kg) was diluted 5-fold (material A), 10-fold (material B), 20-fold (material C) and analysed with CL-ELISA and traditional ELISA, respectively (). Materials A, B and C were produced with a two-/three-step dilution of the positive chicken muscle homogenate with blank homogenate and mixed by a three-dimensional mixer. The CL-ELISA gave a good correlation with GC-µECD and ELISA results, although there was a slight tendency for CL-ELISA values to be slightly higher than GC-µECD values, which may ascribe to some loss of CAP resulted from the clean-up procedure used in GC-µECD analysis. Therefore, the CL-ELISA method showed that it was capable of detecting the ultra-trace CAP in chicken muscle precisely.
Table 6. Linearity of dilutions.
Conclusion
In conclusion, a very sensitive direct competitive CL-ELISA method was established in this study. The sensitivity of this CL-ELISA was dramatically better than any previously reported ELISA method for CAP detection. The reaction time was only 15 min, the shortest reported to date. Moreover, this was the first report of a method that fit for five food samples simultaneously. The satisfactory recoveries in the range of 72.1–116.0% proved that measurements of CAP in the five food matrices were valid. In the analysis of actual chicken muscle sample, the results obtained by CL-ELISA correlated well with those calculated from GC-µECD and traditional ELISA. Therefore, the CL-ELISA in this study is suitable for detecting trace of CAP residues in food samples and lays the foundation for the commercialisation of CL-ELISA kits for CAP residue screening.
Safety consideration
Minimise inhalation of organic reagents by working in fume hood. Dispose of organic solvents properly.
Acknowledgements
This study was supported by Beijing Science and Technology Research Project (D101105046110004) and Technology Pillar Program in the Twentieth Five-Year Plan Period (2011BAZ0319816).
References
- Barreto, F., Ribeiro, C., Hoff, R. B., & Costa, T. D. (2012). Determination and confirmation of chloramphenicol in honey, fish and prawns by liquid chromatography – tandem mass spectrometry with minimum sample preparation: Validation according to 2002/657/EC Directive. Food Additive & Contaminants, 29, 550–558.
- Karaseva, N. A., & Ermolaeva, T. N. (2012). A piezoelectric immunosensor for chloramphenicol detection in food. Talanta, 93, 44–48.
- Kolosova, A. Y., Samsonova, J. V., & Egorov, A. M. (2000). Competitive ELISA of chloramphenicol: Influence of immunoreagent structure and application of the method for the inspection of food of animal origin. Food and Agricultural Immunology, 12, 115–125.
- Lin, S., Han, S. Q., Liu, Y. B., Xu, W. B., & Guan, G. Y. (2005). Chemiluminescence immunoassay for chloramphenicol. Journal of Agricultural and Food Chemistry, 382, 1250–1255.
- Lu, Y. B., Zheng, T. L., He, X., Lin, X. J., Chen, L. Y., & Dai, Z. Y. (2012). Rapid determination of chloramphenicol in soft-shelled turtle tissues using on-line MSPD-HPLC–MS/MS. Food Chemistry, 134, 533–539.
- Mercader, J. V., & Montoya, A. (1999). Development of monoclonal ELISAs for azinphos-methyl. 2. Assay optimization and water sample analysis. Journal of Agricultural and Food Chemistry, 47, 1285–1293.
- Ministry of Agriculture (2002). No. 235 Bulletin of the Ministry of Agriculture of the People's Republic of China.
- Moragues, F., Igualada, C., & León, N. (2012). Validation of the determination of chloramphenicol residues in animal feed by liquid chromatography with an Ion Trap Detector based on European decision 2002/657/EC. Food Analytical Methods, 5, 416–421.
- Rezende, D. R., Fleury Filho, N., & Rocha, G. L. (2012). Simultaneous determination of chloramphenicol and florfenicol in liquid milk, milk powder and bovine muscle by LC–MS/MS. Food Additive & Contaminants, 29, 559–570.
- Sai, N., Chen, Y., Liu, N., Yu, G., Su, P., Feng, Y., Ning, B. A., … (2010). A sensitive immunoassay based on direct hapten coated format and biotin-streptavidin system for the detection of chloramphenicol. Talanta, 82, 1113–21.
- Scortichini, G., Annunziata, L., Haouet, M. N., Benedetti, F., Krusteva, I., & Galarini, R. (2005). ELISA qualitative screening of chloramphenicol in muscle, eggs, honey and milk: Method validation according to the Commission Decision 2002/657/EC criteria. Analytica Chimica Acta, 535, 43–48.
- Shi, X. Z., Wu, A. B., Zheng, S. L., Li, R. X., & Zhang, D. B. (2007). Molecularly imprinted polymer microspheres for solid-phase extraction of chloramphenicol residues in foods. Journal of Chromatography B, 850, 24–30.
- Stolker, A. A. M., & Brinkman, U. A. T. (2005). Analytical strategies for residue analysis of veterinary drugs and growth-promoting agents in food-producing animals – a review. Journal of Chromatography A, 1067, 15–53.
- Taka, T., Baras, M. C., & Chaudhry Bet, Z. F. (2012). Validation of a rapid and sensitive routine method for determination of chloramphenicol in honey by LC–MS/MS. Food Additive & Contaminants, 29, 596–601.
- Wang, L., Zhang, Y., Gao, X., Duan, Z. J., & Wang, S. (2010). Determination of chloramphenicol residues in milk by enzyme-linked immunosorbent assay: Improvement by biotin-streptavidin-amplified system. Journal of Agricultural and Food Chemistry, 58, 3265–3270.
- Xu, C. L., Peng, C. F., Hao, K., Jin, Z. Y., & Wang, W. K. (2006). Chemiluminescence enzyme immunoassay (CLEIA) for the determination of chloramphenicol residues in aquatic tissues. Luminescence, 21, 126–128.
- Yuan, J., Oliver, R., Aguilar, M. I., & Wu, Y. (2008). Surface plasmon resonance assay for chloramphenicol. Analytical Chemistry, 80, 8329–8333.
- Zhang, S. X., Zhang, Z., Shi, W. M., Eremin, S. A., & Shen, J. Z. (2006). Development of a chemiluminescent ELISA for determining chloramphenicol in chicken muscle. Journal of Agricultural and Food Chemistry, 54, 5718–5722.