
ABSTRACT
Acrylamide (AA) is an important product of the Maillard reaction. Studies have demonstrated that AA caused oxidative stress damage in liver, which in turn triggered an inflammatory response, and the activation of NOD-like receptor protein-3 (NLRP3) inflammasome played a key factor in regulating inflammation. Therefore, we hypothesized NLRP3 inflammasome activation was involved in AA-induced inflammatory response. AA induced the reactive oxygen species (ROS) overproduction, accompanied by the MAPK pathway activation, which resulted in NLRP3 inflammasome formation, and eventually increased IL-1β and IL-18 release. The activation of the MAPK pathway was inhibited when using ROS scavenger (NAC). And when MAPK selective inhibitors were used, KCs viability was increased though AA-treated. Meanwhile the NLRP3 inflammasome activation was inhibited, which decreased the release of the cellular inflammatory secretion factors IL-1β and IL-18. Overall, the activation of ROS-MAPK-NLRP3-IL-1β signalling axis induced by AA plays an important role in the process of inflammation.
Signaling pathways involved in inflammation induced by AA. After AA stimulated KCs, a large amount of ROS were released. ROS activated MAPK signaling pathway to promote the activation of NLRP3 inflammasome. Activation of NLRP3 signaling activated Caspase-1, resulting in the maturation of IL-1β and IL-18. The activation of NLRP3 inflammasome was inhibited and the levels of cytoinflammatory factor IL-1β and IL-18 were decreased when using MAPK selective inhibitors.
GRAPHICAL ABSTRACT
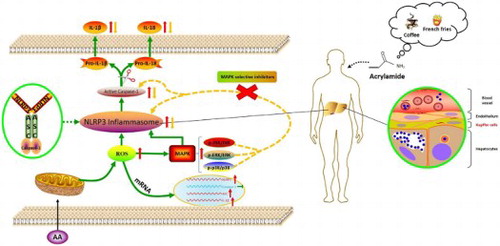
1. Introduction
Acrylamide (AA), a carcinogenic vinyl monomer with toxicity, is widely used in industrial production (Ruenz et al., Citation2016; Xintong et al., Citation2018). AA has neurotoxicity (Erkekoglu & Baydar, Citation1984; Palus et al., Citation2019), genotoxicity (Dearfield et al., Citation1988), reproductive toxicity (Wei et al., Citation2014) and potential carcinogenicity (Rice, Citation2005), and is classified in Group 2A carcinogens by the International Agency for Research on Cancer (IARC) (Zuckerman, Citation1995). AA does not naturally occur, food is prone to the Maillard reaction during high-temperature processing, which tends to form AA (Stadler et al., Citation2002), especially when food contains high levels of glucose and asparagine (Friedman, Citation2003; Mottram, Wedzicha, & Dodson, Citation2002). The exposure of people to AA is multi-channel, including smoking, air or water pollution and food consumption. Some surveys have shown that the industrial environment (occupational exposure) had a certain toxic effect on the liver. In the process of long-term exposured to AA in the occupational population (even low dose), the liver has been damaged by AA, which eventually lead to change biochemical indicators of the liver and blood lipids in occupationally exposed people (Liu, Citation1989; Wang et al., Citation2015). However, ingestion from the mouth is considered to be the fastest and most complete pathway for the body to absorb AA. The contents of AA in some commonly consumed food (including fried potato chips and coffee) are much higher than the daily limited intake (2 µg/day) (Esposito et al., Citation2017; WHO and FAO, Citation2002). Furthermore, Swedish scholars found that high-temperature fried and baked foods contained a large amount of AA, and its content exceeded the maximum allow able amount of drinking water more than 500 times (Tareke et al., Citation2002). Survey data indicated that the average intake of AA in the population ranges from 0.3 to 0.8 μg/kg BW d−1, the intake of children is 2–3 times that of adults. And the average intake of high AA intake is up to 4 μg/kg BW d−1 (Erkekoglu & Baydar, Citation2010). The concerns raised by these exposure doses have led researchers to gradually shift their focus to the safety of AA. Research showed that AA induces oxidative stress damage and produces inflammatory response (Pan et al., Citation2018). It is confirmed that the activation of the NLRP3 inflammasome is one of the most important factors in triggering inflammatory responses. Therefore, the detailed molecular mechanism accounting for the effect of AA-induced activation of the NLRP3 inflammasome deserves to be investigated.
The liver is the starting site for AA metabolism after oral or intraperitoneal injection. Research showed that a high concentration of AA is found in the liver, it indicating that liver can be used as a target tissue for AA (Exon, Citation2006). Hepatotoxicity is reported to be one of the adverse effects mediated by AA. Previous studies have shown that after AA enters the body, the epoxidation of AA to GA passes through the cytochrome P450 2E enzyme in the liver, which in turn damages the body (Watzek et al., Citation2013). In addition, Kupffer cells (KCs) are a key component of the mononuclear phagocytic system and critical for the liver and system’s response to pathogens. KCs are in turn a large number of hepatic macrophages present in the sinusoids, which are the largest tissue-resident macrophage population. KCs are important for liver homeostasis and protective immune responses. KCs’ functions include phagocytosis of exogenous antigens, antigen presentation to T cells, and secretion of several key inflammatory mediators (Laura et al., Citation2013). Another study also showed that KCs were closely related to inflammation (Stănculeţ et al., Citation2013). Therefore, in this study, we used KCs as a trial target to explore the activation of NLRP3 inflammasome and the secretion of inflammatory factors induced by AA.
The inflammasome is a specific inflammatory signalling molecule in the cell, which mediates the maturation and secretion of the pro-inflammatory cytokines IL-1β and IL-18 by regulating caspase-1 dependent proteolysis. Among various inflammasomes, NLRP3 is the most widely studied. It is a polyprotein complex of approximately 700 kDa, composed of nucleotide-binding oligomerization domain-like receptors (NLRs) family member NLRP3, effector proteins caspase-1 and an apoptosis-associated speck-like protein containing CARD (ASC) (Pétrilli et al., Citation2007). NLRP3 activation is affected by many factors, including pathogen-associated molecular patterns (PAMPs), such as viruses, bacteria, fungus; damage-associated molecular patterns (DAMP), such as ATP, crystals, particles and reactive oxygen species (ROS) in both cells (parenchymal or nonparenchymal) (Qu et al., Citation2019). ROS is known as a key factor in mediating inflammatory response and its production is increased when stimulated by external substances. Excessive ROS could activate the mitogen-activated protein kinase (MAPK) signalling pathway, which finally results in NLRP3 activation. The cell growth and differentiation are regulated by the MAPK pathway (Berghe, Citation1998), which controls cellular responses to cytokines (Debnath et al., Citation2017; Ghonime et al., Citation2014). In addition, the MAPK pathway influences NLRP3 inflammasome activation (Okada et al., Citation2014; Yu et al., Citation2013) and is involved in the inflammatory reaction (Ali et al., Citation2011). Liang et al. revealed that the NLRP3 inflammasome was activated through the ROS-mediated activation of p38, ERK1/2 (Liang et al., Citation2013). Our previous findings also indicated that AA could increase ROS production and cause the activation of the MAPK pathway in BRL-3A cells. These novel findings raise the possibility that AA induces the NLRP3 inflammasome activation via the ROS-MAPK axis. Exogenous substance stimulation plays a key factor in NLRP3 inflammasome formation, however, there is little research on foodborne AA-induced NLRP3 inflammasome activation. In this study, we aimed to determine how AA regulated NLRP3 expression and activated NLRP3 inflammasome, and whether AA-induced NLRP3 activation went through the ROS mediated MAPK pathway. In order to do this, we validated the hypothesis using the MAPK pathway selective inhibitor. This will provide an important theoretical basis for elucidating how AA induces NLRP3 inflammasome activation and triggers inflammatory response.
2. Materials and methods
2.1. KCs culture and drug treatment
KCs (BeNa Culture Collection, BNCC, Beijing, China) were cultured in 1640 complete medium, with 89% RPMI 1640 medium (Hyclone Corporation, Logan, UT, USA), 10% (v/v) fetal bovine serum (Gemini Bio-products, Woodland, USA), 0.5% L-glutamine (Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China), as well as 0.5% penicillin and streptomycin (Hyclone Corporation, Logan, UT, USA). KCs were revived in 10 cm sterile cell culture dishes (Corning Incorporated, New York, USA), and then incubated at 37°C for 24 h with 5% CO2.
AA (Sigma Chemical Co, St Louis, MO, USA, CAS: 79 – 06 – 1, purity > 99.8%) was dissolved in purified water and filtered through a 0.22 μm membrane (Millipore, Billerica, MA, USA), and then was diluted to different concentrations using the basic medium. Up to the logarithmic growth phase, subcultured KCs were incubated in 4 mL 1640 medium with different concentrations of AA (0, 0.5, 1 mM) at 37°C for 24 h with 5% CO2. Where MAPK selective inhibitors (SB202190 10 μM, SP6000125 10 μM and SCH772984 4 μM, Selleck Chemicals, Houston, TX, USA) and ROS scavenger (N-acetyl-L-cysteine, NAC 10 mM, Beyotime Biotechnology, Shanghai, China) were used. KCs were pre-treated with MAPK selective inhibitors or ROS scavenger for 2 h prior to culture.
2.2. Cell viability assay
The Cell Counting Kit-8 (CCK-8)(Dojindo Laboratories; Kumamoto, Japan) was used to evaluate the viability of KCs. To ensure the same cell density, KCs were plated in 96-well microtiter plates (7.5 × 104 cells per well) and then cultured for 24 h. AA solutions were diluted with RPMI 1640 basic medium. Then KCs were cultured with 100 μL AA solutions (0, 0.5, 1, 2, 4 mM) for 24 h before 10 μL CCK-8 solution was added into each well, and then the 96-well microtiter plates were incubated for another 4 h. The effects of MAPK selective inhibitors on the KCs activity were investigated following the same steps as mentioned above, except for pre-treatment with MAPK selective inhibitors for 2 h and treatment with AA for 22 h. The KCs were divided into 8 groups: the control group, 1 mM AA group, 1 mM AA + 10 μM SB202190 group, 1 mM AA + 10 μM SP6000125 group, 1 mM AA + 4 μM SCH772984 group, only 10 μM SB202190 group, only 10 μM SP6000125 group and only 4 μM SCH772984 group. The absorbance density was determined at 490 nm by enzyme-linked immunometric meter (Synergy HT, BioTek, USA).
2.3. ROS measurement
The levels of ROS were determined using fluorescent probe, which was 2′, 7′ – dichlorofluorescin diacetate (DCFH-DA, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). KCs were plated in 6-well microtiter plates and the density was 3 × 105 cells per well. After 24 h adherent growth, KCs were treated with different concentrations of AA (0, 0.5, 1 mM) for 6 h. Then KCs were washed three times with pre-cooling PBS and DCFH-DA (10 μM) was added to the cell culture prior to incubation at 37°C for 30 min. The fluorescence intensity of ROS was observed by fluorescence microscope (Nikon TS100, Japan).
2.4. Real-time quantitative PCR
KCs were inoculated at a density of 5 × 105 cells per well in 6-well microtiter plates. After 24 h incubation, KCs were treated with different concentrations of AA (0, 0.5, 1 mM) for 24 h. Total RNA was extracted from KCs using Trizol (Shanghai Yisheng Biological Technology Co., Ltd., Shanghai, China). RNA was determined by UV spectrophotometer. 1 μg RNA was reverse-transcribed to complementary DNA (cDNA) via PrimeScript RT reagent kit (Monad Biotechnology Co., Ltd., Suzhou, China). Real-time quantitative PCR (RT-qPCR) was used with 2 × ChamQ SYBR green master mix (Vazyme biotech Co., Ltd., Nanjing, China) on a CFX96™ Real-Time System (Bio-Rad Laboratories, California, USA). The RT-qPCR conditions were 95°C for 30 s, followed by 40 cycles of denaturing at 95°C for 10 s, annealing at 60°C for 30 s, extending at 95°C for 15 s, 60°C for 60 s and 95°C for 15 s, respectively. The primers are displayed in . The 2−ΔΔCT method was used to analyze the relative changes in gene expression.
Table 1. Sequence of primers used for quantitative RT-PCR.
2.5. Preparation of cell protein samples
After AA treatment, the culture medium was discarded and KCs were washed three times using pre-cooled PBS (Hyclone Corporation, Logan, UT, USA). KCs were lysed on ice with 400 μL RIPA lysis buffer containing protease inhibitor (4 μL) and phosphatase inhibitor (4 μL) for 30 min (Beyotime Biotechnology, Shanghai, China). The lysate was collected and centrifuged at 13,620 × g for 10 min at 4°C. For uniform concentrations, KCs protein samples were adjusted using BCA assay kit (Beyotime Biotechnology, Shanghai, China) and then were denatured in loading buffer (Beyotime Biotechnology, Shanghai, China) at 100°C for 10 min.
2.6. Western blotting
Based on protein concentration determination, KCs protein samples were loaded onto 10% or 12% SDS-PAGE gel. The electrophoresis conditions were 90 V 10 min and then 120 V 60 min. The target proteins were identified by Rainbow Maker (Thermo Fisher Scientific Inc, New York, USA). Then the proteins on the SDS-PAGE gel were transferred to the polyvinylidene difluoride membranes (PVDF)(Millipore, Billerica, MA, USA) using a semi-dry transmembrane instrument (Bio-Rad Laboratories, California, USA). After the transmembrane, the PVDF membranes were incubated in blocking reagent (Beyotime Biotechnology, Shanghai, China) at room temperature for 1 h and then hatched with primary antibodies against NLRP3 (ab210491, 1:1000), Caspase-1 (ab1872, 1:100), ASC (ab175449, 1:500), IL-1β (ab9787, 1:100), JNK (ab179461, 1:1000), p-JNK (ab124956, 1:1000), ERK (ab17942, 1:1000), p-ERK (ab201015, 1:1000), p38 (ab31828, 1:1000), p-p38 (ab4822, 1:1000) and GAPDH (ab181602, 1:10,000) (Abcam, Cambridgeshire, UK) overnight at 4°C.
After washing twice with TBST buffer solution and once with TBS buffer solution (Beijing Solarbio Science and Technology Co., Ltd., Beijing, China), PVDF membranes were hatched with IgG secondary antibodies (Bioss antibodies, Beijing, China) at room temperature for 2 h. Then the PVDF membranes were washed according to the previous method. Target proteins reacted with enhanced chemiluminescence (ECL) chromogenic substrate (Beyotime Biotechnology, Shanghai, China). The bands were detected using Scan LiDE 100 (Canon, Japan). The grey values were analyzed by the Image-J software.
2.7. Bioactive IL-1β and IL-18 ELISA
To evaluate the effects of AA on pro-inflammatory cytokine release, the KCs-culture medium was collected in 4 mL EP tubes. The supernatants were collected from KCs-culture medium after centrifugation at 1000 × g for 20 min at 4°C. The release of pro-inflammatory cytokine IL-1β and IL-18 in KCs supernatants was detected using rat IL-1β and IL-18 ELISA kits (Wuhan USCN Business Co., Ltd., Wuhan, China) according to the protocol.
2.8. Statistical analysis
The data were expressed as the mean ± SD. Statistical calculations were performed with GraphPad Prism 7 (GraphPad Software, Inc., San Diego, USA). The data were analyzed by ANOVA and the Newman–Keuls multiple-comparison test. Values of P < 0.05 were considered statistically significant.
3. Results
3.1. Effects of AA on Kupffer cells viability
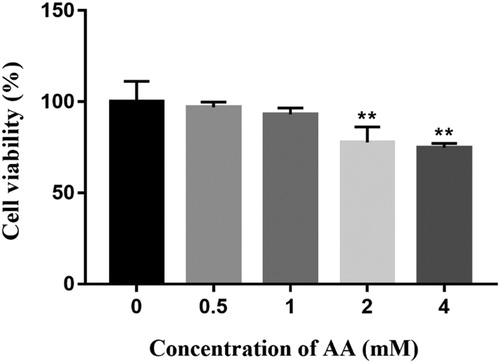
AA has toxic effects on KCs viability, which was detected by CCK-8 viability assay. As shown in , the viability was suppressed with the increase of AA. AA had no distinct influence on the viability of KCs incubated with doses ranging from 0 to 1 mM, but higher concentrations at 2 and 4 mM had obvious cytotoxicity and the viability was reduced to 77.66% and 71.44%, respectively (P < 0.01). Therefore, higher concentrations were eliminated. AA levels at 0, 0.5 and 1mM were adopted for further study.
Figure 1. Effects of AA on Kupffer cells viability. The cells viability of AA-treated Kupffer cells was determined using CCK-8 method. The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as **P < 0.01.
3.2. Effects of AA on ROS release
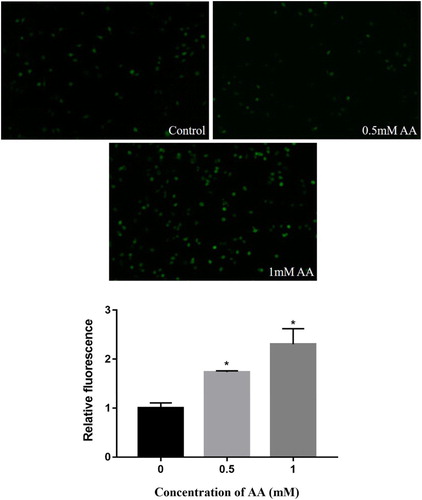
We first investigated ROS production in KCs, and the effects of AA-induced ROS production are shown in . The results of fluorescence microscope illustrated that ROS production was obviously elevated in AA-treated KCs compared with un-treated cells, and the release of ROS was in a dose-dependent manner (P < 0.01). In this study, AA caused oxidative stress and released a large amount of ROS, which might activate the MAPK pathway and NLRP3 inflammasome.
Figure 2. Effects of AA on ROS release in Kupffer cells treated for 6 h with AA (0, 0.5, 1 mM). Fluorescent pictures were acquired by fluorescence microscope. The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as *P < 0.05.
3.3. Effects of AA on the activation of MAPK pathway
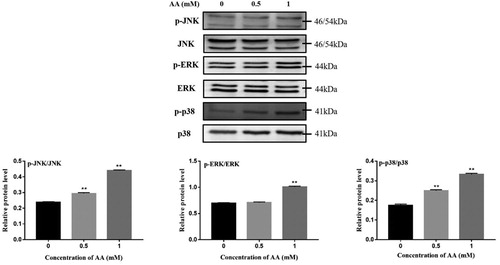
The MAPK pathway, which has three major participants (p38, ERK, and JNK23), may play an important role in the activation of NLRP3 inflammasome17. To investigate whether AA induced the changes of the MAPK pathway molecular mechanisms in KCs, the changes in the activation (phosphorylation) of JNK, ERK1/2 and p38 MAPK in AA-stimulated KCs were determined. As shown in , the phosphorylated protein levels were significantly elevated in KCs by AA stimulation (P < 0.01), which suggested that AA activated the MAPK pathway and up-regulated the phosphorylated protein levels of JNK, ERK1/2 and p38 MAPK.
Figure 3. Effects of AA on activation of MAPK pathway. p-JNK, JNK, p-ERK, ERK, p-p38 and p38 protein expression were tested by Western blot – analysis. The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as **P < 0.01.
3.4. Effects of NAC on AA induced MAPK pathway activation
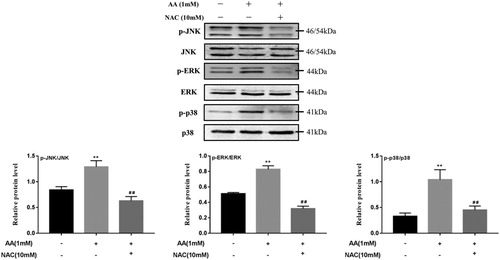
To determine if ROS mediates the activation of the MAPK pathway, ROS scavengers (NAC) were used to test. As shown in , the protein levels of the p-JNK/JNK, p-ERK/ERK and p-p38/p38 were significantly lower than those in AA-stimulated KCs after pre-treatment with NAC (10 mM) for 2 h, which were reduced by 51.2%, 61.5% and 56.6%, respectively. The addition of NAC blocked the activation of MAPK pathway, which demonstrated that ROS mediates the activation of the MAPK pathway.
Figure 4. Effects of NAC on AA induced MAPK pathway activation. p-JNK, JNK, p-ERK, ERK, p-p38 and p38 protein expression were tested by Western blot – analysis. The values are presented as means ± of SD (n = 3). **P < 0.01, versus the un-treated KCs; ##p < 0.01, versus the AA-treated KCs.
3.5. Effects of AA on NLRP3 inflammasome-associated mRNA expression
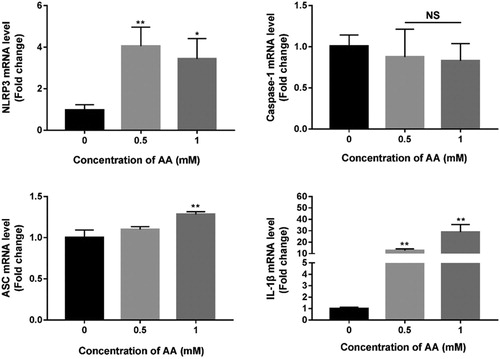
In this study, mRNA expression levels of NLRP3, Caspase-1, ASC and IL-1β were measured by real-time qPCR (As shown in ). After AA treatment for 24 h, NLRP3, ASC and IL-1β mRNA expression levels were significantly elevated compared with un-treated KCs (P < 0.05). The mRNA expression levels were dependent on the concentration of AA, but there were no significant changes in Caspase-1 mRNA expression levels. In the follow-up experiment, we determined the changes in protein levels of NLRP3, Caspase-1, ASC and IL-1β.
Figure 5. Effects of AA on NLRP3 inflammasome mRNA expression. The mRNA expression of NLRP3, Caspase-1, ASC and IL-1β were detected by real-time PCR, normalized by U6 and expressed as 2−ΔΔCT. The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as *P < 0.05 or **P < 0.01.
3.6. Effects of AA on NLRP3 inflammasome activation
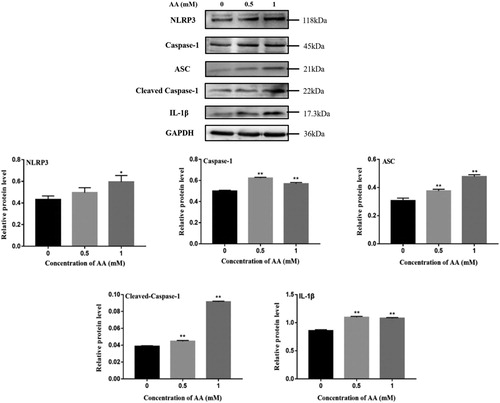
AA caused inflammatory response, which is associated with immune cells. KCs were used to explore the formation of NLRP3 inflammasome. To study the variation of NLRP3 inflammasome-associated molecules, the expression levels of NLRP3, Caspase-1, ASC, cleaved-Caspase-1 and IL-1β in KCs were determined by Western Blotting. Consistent with NLRP3 inflammasome-associated mRNA expression, the expression levels of NLRP3, ASC and pro-Caspase-1 in KCs were significantly increased upon AA stimulation (As shown in ). In particular, there was a significant difference at 1 mM AA (P < 0.05). As expected, cleaved-Caspase-1 and IL-1β expression levels were markedly increased in KCs stimulated with AA, which corroborated that NLRP3 inflammasome was activated.
Figure 6. Effects of AA on NLRP3 inflammasome activation. NLRP3, Caspase-1, ASC, cleaved-Caspase-1 and IL-1β protein expression were tested by Western blot – analysis. The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as *P < 0.05 or **P < 0.01.
3.7. Effects of AA on IL-1β and IL-18 secretion
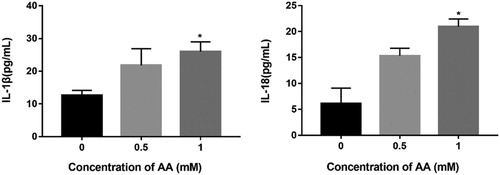
Mature IL-1β and IL-18 are produced through cleavage of inactive pro-IL-1β and pro-IL-18 by Caspase-1, which is activated by NLRP3 inflammasome. In order to validate whether AA activated NLRP3 inflammasome further, the secretion of IL-1β and IL-18 in KCs cultures was determined by Enzyme-linked immunosorbent assay. As shown in , the cytoinflammatory factor levels of IL-1β and IL-18 in KCs cultures were significantly increased in AA-stimulated KCs compared to un-treated KCs (P < 0.05). These results suggested that AA induced the NLRP3 inflammasome formation and secreted cytoinflammatory factors IL-1β and IL-18.
Figure 7. Effects of AA on IL-1β and IL-18 secretion. The release of IL-1β and IL-18 from Kupffer cells treated for 24 h with AA (0, 0.5, 1 mM). The values are presented as means ± of SD (n = 3). Significant differences with control group were designated as *P < 0.05.
3.8. Effects of MAPK selective inhibitors on KCs viability
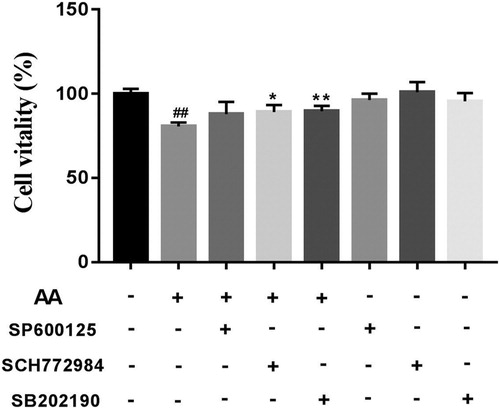
To explore the effects of MAPK selective inhibitors on un-treated and AA-treated KCs viability, we also measured KCs viability using the CCK-8 method. As shown in , treatment with the MAPK selective inhibitors alone had no effects on KCs viability compared with un-treated KCs, which demonstrated that the concentrations of MAPK selective inhibitors (SB-202190 10 μM, SP6000125 10 μM and SCH772984 4 μM) were proper. Compared with un-treated KCs, KCs viability was significantly reduced after 1 mM AA treatment. However, KCs viability was increased by MAPK selective inhibitors pre-treatment for 2 h and then 1 mM AA stimulation for 22 h, showing that blocking the MAPK signalling pathway could improve KCs viability. To continue with, we investigated the effects of the MAPK pathway on the NLRP3 inflammasome activation.
Figure 8. Effects of MAPK selective inhibitors on cells viability. The cells viability of MAPK selective inhibitors-treated Kupffer cells was determined using CCK-8 method. The values are presented as means ± of SD (n = 3). ##P < 0.01, versus the un-treated KCs; **p < 0.01, versus the AA-treated KCs.
3.9. MAPK pathway mediate the activation of NLRP3 inflammasome
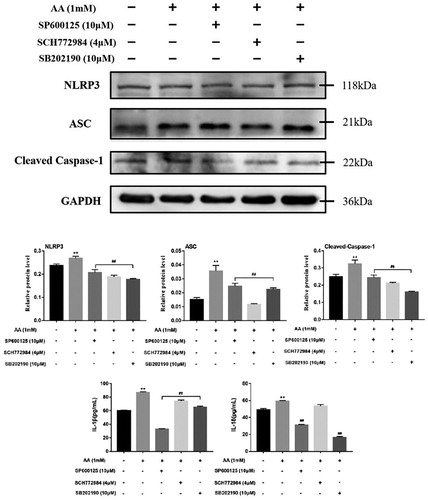
To prove the effects of the MAPK pathway on the activation of NLRP3 inflammasome, the MAPK selective inhibitors were used. As shown in , the phosphorylation protein levels of JNK, ERK and p38 were inhibited in KCs pre-treated with MAPK selective inhibitors for 2 h and then stimulated with AA for 22 h, showing that MAPK selective inhibitors were effective. We further determined the changes in the components of NLRP3 inflammasome proteins after MAPK selective inhibitors treatment. When MAPK selective inhibitors (SB-202190 10 μM, SP6000125 10 μM and SCH772984 4 μM) were added, the expression levels of NLRP3, ASC and cleaved-Caspae-1 were decreased (As shown in ), so were the cytoinflammatory factor levels of IL-1β and IL-18, showing that the MAPK pathway could play an important regulatory role in AA-induced inflammation and AA activate NLRP3 inflammasome via the MAPK-NLRP3 axis.
Figure 9. Effects of MAPK selective inhibitors on MAPK pathway. Kupffer cells were exposed to AA (1 mM) 22 h after pre-treatment of SP6000125 (10 μM), SCH772984 (4 μM) or SB202190 (10 μM) for 2 h, and the expression of p-JNK, JNK, p-ERK, ERK, p-p38 and p38 were estimated using western blot. The values are presented as means ± of SD (n = 3). **P < 0.01, versus the un-treated KCs; ##p < 0.01, versus the AA-treated KCs.
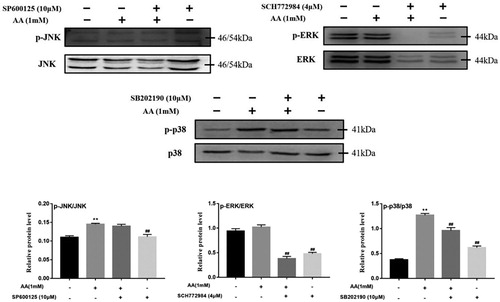
Figure 10. Effects of MAPK selective inhibitors on activation of NLRP3 inflammasome. Kupffer cells were exposed to AA (1 mM) 22 h after pre-treatment of SP6000125 (10 μM), SCH772984 (4 μM) or SB202190 (10 μM) for 2 h, and the expression of NLRP3, ASC and cleaved-Caspase-1 were estimated using western blot. The release of IL-1β and IL-18 from Kupffer cells were determined by ELISA kits. The values are presented as means ± of SD (n = 3). **P < 0.01, versus the un-treated KCs; ##p < 0.01, versus the AA-treated KCs.
4. Discussion
AA is a typical harmful substance produced during the thermal processing of food with reproductive, neurological, genotoxic and potential carcinogenicity as well as threat to human health (Mottram et al., Citation2002). Seydi et al. have confirmed that AA has hepatotoxicity, because AA is involved in oxidative stress and induces mitochondrial/lysosomal damage and cell death signalling (Seydi et al., Citation2015). Our previous findings also indicated that AA could cause oxidative stress in hepatocytes and induce hepatotoxicity. However, there is no in-depth study of its toxic effects on immune cells. Whether AA can cause inflammatory response is worth exploring.
The NLRP3 inflammasome was mainly expressed in neutrophils, macrophages, Kupffer cells (KCs), monocytes and dendritic cells (Guarda et al., Citation2011). Macrophages, as major regulatory cells in the inflammatory response, play a vital role in the innate immune and adaptive immune system. KCs account for 20–35% of all non-parenchymal cells in liver, and 80–90% of tissue macrophages in vivo. Thence, KCs are called liver-derived macrophages (Li et al., Citation2014; Wei-Qun et al., Citation2013). Upon stimulation, NLRP3 recruits ASC to compose NLRP3-ASC oligomers and then cleaves pro-Caspase-1 to cleaved-Caspase-1. Cleaved-Caspase-1 triggers the maturation and secretion of interleukin-1β (IL-1β) and interleukin-18 (IL-18) (Qu et al., Citation2019). Studies have found that IL-1β is mainly derived from KCs. Therefore, we explored how AA activated the NLRP3 inflammasome in KCs.
ROS regulates inflammation responses, and serves as second messenger in the process. Some downstream pathways are involved in ROS-induced inflammation. Shi and his colleagues have certificated that NLRP3 inflammasome activation was inhibited via Nrf2-mediated anti-oxidant pathway (Shi et al., Citation2018). In addition, other pathways, including AMPK and NF-κB pathways, are also involved in ROS-mediated activation of the NLRP3 inflammasome. ROS not only causes oxidative stress response, but also triggers an inflammatory cascade involved in the MAPK pathway that produces various pro-inflammatory cytokines. Wang et al. have put forward that proinflammatory activity was correlated with the ROS-MAPK pathway and was involved in the NLRP3 inflammasome activation (Wang et al., Citation2019). In our experiment, AA increased the production of ROS dose-dependently compared with un-treated KCs. With 1 mM AA, ROS levels were increased by 1.3 times compared to the un-treated KCs. This proved that KCs stimulated by AA could induce a large amount of ROS production. We presume the overproduction of ROS caused MAPK pathway and NLRP3 inflammasome activation.
MAPKs (JNK, ERK1/2 and p38) are a family of serine/threonine protein kinases that regulates basic biological processes and cellular responses to external stressors. JNK, ERK1/2 and p38 are activated by phosphorylation in this family of proteins involved in regulating cell proliferation and death (Pearson et al., Citation2001). Our findings indicated that the levels of the MAPK pathway-associated proteins including p-JNK, p-ERK1/2 and p-p38 were increased in AA-induced KCs compared with un-treated KCs. In particular, when AA was 1 mM, the expression of MAPK pathway-associated proteins was the highest. Increased levels of phosphorylated JNK, ERK1/2 and p38 indicated that AA activated the MAPK pathway. However, compared with AA-treated KCs, the key protein levels of the MAPK pathway (p-JNK/JNK, p-ERK/ERK and p-p38/p38) were significantly reduced after pre-treatment with NAC (10 mM) for 2 h. It demonstrated that AA mediated the activation of the MAPK pathway through excessive release of ROS.
Previous studies have proved that the MAPK pathway family, especially JNK, ERK1/2 and p38, regulate the inflammatory response and immune response (Ajizian, English, & Meals, Citation1999; Daniele et al., Citation2015; Smith et al., Citation2018). In recent years, the role of the NLRP3 inflammasome in immune regulation has received increasing attention (Chessa, Ganau, & Mazzarello, Citation2015). Now that we have determined that AA activated the MAPK pathway in KCs, we next investigated whether AA affected NLRP3 inflammasome activation. In this experiment, NLRP3, ASC, Caspase-1 and IL-1β mRNA expression levels were detected by RT-qPCR. The results indicated that mRNA expression levels of NLRP3, ASC and IL-1β were enhanced in AA-treated KCs compared with the un-treated KCs. There was no significant change in the mRNA expression level of Caspase-1. To further investigate the NLRP3 inflammasome activation induced by AA, the changes of protein levels were determined. Western Blotting results demonstrated that protein levels of NLRP3, Caspase-1, ASC, Cleaved-Caspase-1 and IL-1β were elevated in AA-treated KCs compared with normal KCs. The activated Caspase-1 contributed to the maturation of IL-1β and IL-18. The results of ELISA showed that the levels of inflammatory cytokine IL-1β and IL-18 secreted by KCs were significantly increased compared to un-treated KCs. And the levels of inflammatory cytokines IL-1β and IL-18 were enhanced with increasing AA dosage. The result indicated that AA could activate the NLRP3 inflammasome and release a large amount of inflammatory cytokines IL-1β and IL-18, which was consistent with previous results (Chunteng et al., Citation2018). To identify if the MAPK pathway was involved in AA-induced NLRP3 inflammasome activation in KCs, the MAPK selective inhibitors were used. When KCs were pretreated with MAPK inhibitors for 2 h and then were stimulated with 1 mM AA for 22 h, the viability of KCs was significantly improved compared with the cells stimulated with 1 mM AA only, suggesting that blocking the MAPK pathway could improve the viability of KCs. Then, the effects of MAPK pathway on the NLRP3 inflammasome activation were investigated. The protein levels of NLRP3, ASC and Cleaved-Caspase-1 were significantly reduced in MAPK selective inhibitors-pretreated KCs compared to AA-treated KCs. At the same time, the levels of cellular inflammatory secretion factors IL-1β and IL-18 were markedly inhibited as well. The experiment demonstrated that the formation of the NLRP3 inflammasome was inhibited when MAPK selective inhibitors were used. It can therefore be concluded that the MAPK pathway was involved in NLRP3 inflammasome activation.
5. Conclusions
In conclusion, we hypothesize that AA induces the activation of the NLRP3 inflammasome via the ROS-mediated MAPK pathway. AA significantly increased the release of ROS, thus activated the MAPK pathway and the NLRP3 inflammasome. NLRP3 inflammasome activation was inhibited in the presence of MAPK selective inhibitors. Thus, the activation of the NLRP3 inflammasome induced by AA is related to ROS-MAPK-NLRP3-IL-1β signalling axis. Further work is required to seek a plant-derived food to inhibit AA-induced activation of the NLRP3 inflammasome, which is of more practical significance.
Acknowledgments
The authors gratefully acknowledge the funds supports.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ajizian, S., English, B. K., & Meals, E. (1999). Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-γ. The Journal of Infectious Diseases, 179(4), 939–944. doi: 10.1086/314659
- Ali, S., Timmer, A., Bilgrami, S., Park, E., Eckmann, L., Nizet, V., & Karin, M. (2011). Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP Leakage. Immunity, 35(1), 34–44. doi:10.1016/j.immuni.2011.04.015.
- Berghe, V. W. (1998). P38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappa B p65 transactivation mediated by tumor necrosis factor. Journal of Biological Chemistry, 273(6), 3285–3290. doi: 10.1074/jbc.273.6.3285
- Chessa, D., Ganau, G., & Mazzarello, V. (2015). An overview of staphylococcus epidermidis and staphylococcus aureus with a focus on developing countries. The Journal of Infection in Developing Countries, 9(6), 547–550. doi: 10.3855/jidc.6923
- Chunteng, J., Liping, J., Qiannan, L., Xiaofang, L., Tianjiao, Z., & Linlin, D. (2018). Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology, 410, 26–40. doi:10.1016/j.tox.2018.09.002.
- Daniele, S., Da Pozzo, E., Zappelli, E., & Martini, C. (2015). Trazodone treatment protects neuronal-like cells from inflammatory insult by inhibiting NF-κB, p38 and JNK. Cellular Signalling, 27(8), 1609–1629. doi:10.1016/j.cellsig.2015.04.006.
- Dearfield, K. L., Abernathy, C. O., Ottley, M. S., Brantner, J. H., & Hayes, P. F. (1988). Acrylamide: Its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutation Research/Reviews in Genetic Toxicology, 195(1), 45–77. doi:10.1016/0165-1110(88)90015-2.
- Debnath, T., Kim, E. K., Nath, N., & Lee, K. G. (2017). Therapeutic effects of Ligularia stenocephala against inflammatory bowel disease by regulating Antioxidant and inflammatory mediators. Food & Agricultural Immunology, 28(6), 1–13. doi:10.1080/09540105.2017.1332008.
- Dixon, L. J., Barnes, M., Tang, H., Pritchard, M. T., & Nagy, Laura. E.. (2013). Kupffer cells in the liver. Comprehensive Physiology, 3(2), 785–797. doi:10.1002/cphy.c120026.
- Erkekoglu, P., & Baydar, T. (1984). Acrylamide neurotoxicity. Food & Chemical Toxicology An International Journal Published for the British Industrial Biological Research Association, 17(2), 49–57. doi: 10.1179/1476830513Y.0000000065
- Erkekoglu, P., & Baydar, T. (2010). Toxicity of acrylamide and evaluation of its exposure in baby foods. Nutrition Research Reviews, 23(2), 323–333. doi: 10.1017/S0954422410000211
- Esposito, F., Nardone, A., Fasano, E., Triassi, M., & Cirillo, T. (2017). Determination of acrylamide levels in potato crisps and other snacks and exposure risk assessment through a margin of exposure approach. Food and Chemical Toxicology, 108(A), 249–256. doi:10.1016/j.fct.2017.08.006.
- Exon, J. H. (2006). A review of the toxicology of acrylamide. Journal of Toxicology & Environmental Health Part B, 9(5), 397–412. doi: 10.1080/10937400600681430
- Friedman, M. (2003). Chemistry, biochemistry, and safety of acrylamide. A review. Journal of Agricultural and Food Chemistry, 51(16), 4504–4526. doi:10.1021/jf030204+ doi: 10.1021/jf030204+
- Ghonime, M. G., Shamaa, O. R., Das, S., Eldomany, R. A., Fernandes, A. T., & Alnemri, E. S. (2014). Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. The Journal of Immunology, 192(8), 3881–3888. doi:10.4049/jimmunol.1301974.
- Guarda, G., Zenger, M., Yazdi, A. S., Schroder, K., Ferrero, I., & Menu, P. (2011). Differential expression of NLRP3 among hematopoietic cells. The Journal of Immunology, 186(4), 2529–2534. doi:10.4049/jimmunol.1002720.
- Li, P. Z., Li, J. Z., Li, M., Gong, J. P., & He, K. (2014). An efficient method to isolate and culture mouse Kupffer cells. Immunology Letters, 158(1-2), 52–56. doi:10.1016/j.imlet.2013.12.002.
- Liang, L., Tan, X., Zhou, Q., Zhu, Y., Tian, Y., & Yu, H. (2013). IL-1β triggered by peptidoglycan and lipopolysaccharide through TLR2/4 and ROS-NLRP3 inflammasome–dependent pathways is involved in ocular Behcet’s disease. Investigative Opthalmology & Visual Science, 54(1), 402–414. doi:10.1167/iovs.12-11047.
- Liu, G. Z. (1989). Effect of acrylamide on liver function of workers. Journal of Hygienic Toxicology, 3, 197.
- Mottram, D. S., Wedzicha, B. L., & Dodson, A. T. (2002). Food chemistry: Acrylamide is formed in the Maillard reaction. Nature, 419(6906), 448–449. doi: 10.1038/419448a
- Okada, M., Matsuzawa, A., Yoshimura, A., & Ichijo, H. (2014). The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. Journal of Biological Chemistry, 289(47), 32926–32936. doi:10.1074/jbc.M114.579961.
- Palus, K., Obremski, K., Bulc, M., & Całka, J. (2019). The impact of low and high doses of acrylamide on the intramural neurons of the porcine ileum. Food and Chemical Toxicology, 132, 110673. doi:10.1016/j.fct.2019.110673.
- Pan, X., Wu, X., Yan, D. D., Peng, C., Rao, C. L., & Yan, H. (2018). Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: Involvement of the crosstalk between Nrf2 and NF-κB pathways regulated by MAPKs. Toxicology Letters, 288, 55–64. doi:10.1016/j.toxlet.2018.02.002.
- Pearson, G., Robinson, F., Gibson, T. B., Xu, B. E., Karandikar, M., & Berman, K. (2001). Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions 1. Endocrine Reviews, 22(2), 153–183. doi:10.1210/edrv.22.2.0428.
- Pétrilli, V., Dostert, C., Muruve, D. A., & Tschopp , J. (2007). The inflammasome: A danger sensing complex triggering innate immunity. Current Opinion in Immunology, 19(6), 615–622. doi:10.1016/j.coi.2007.09.002.
- Qu, J., Yuan, Z., Wang, G., Wang, X., & Li, K. (2019). The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. International Immunopharmacology, 70, 147–155. doi:10.1016/j.intimp.2019.02.016.
- Rice, J. M. (2005). The carcinogenicity of acrylamide. Mutation Research, 580(1), 3–20. doi: 10.1016/j.mrgentox.2004.09.008
- Ruenz, M., Bakuradze, T., Eisenbrand, G., & Richling, E. (2016). Monitoring urinary mercapturic acids as biomarkers of human dietary exposure to acrylamide in combination with acrylamide uptake assessment based on duplicate diets. Archives of Toxicology, 90(4), 873–881. doi:10.1007/s00204-015-1494-9.
- Seydi, E., Rajabi, M., Salimi, A., & Pourahmad, J. (2015). Involvement of mitochondrial-mediated caspase-3 activation and lysosomal labilization in acrylamide-induced liver toxicity. Toxicological & Environmental Chemistry, 97(5), 563–575. doi:10.1080/02772248.2015.1047671.
- Shi, A., Shi, H., Wang, Y., Liu, X., Cheng, Y., & Li, H. (2018). Activation of Nrf2 pathway and inhibition of NLRP3 inflammasome activation contribute to the protective effect of chlorogenic acid on acute liver injury. International Immunopharmacology, 54, 125–130. doi:10.1016/j.intimp.2017.11.007.
- Smith, T. L., Slyke, P. V., Jones, N., Dumont, D. J., & McGlade, C. J. (2018). Tie2 signalling through Erk1/2 regulates TLR4 driven inflammation. Cellular Signalling, 51, 211–221. doi:10.1016/j.cellsig.2018.08.001.
- Stadler, R. H., Blank, I., Varga, N., Robert, F., Hau, J., & Guy, P. A. (2002). Acrylamide from Maillard reaction products. Nature, 419, 449–450. doi:10.1038/419449a.
- Stănculeţ, N., Grigoraş, A., Avădanei, R., Floarea, S. A., Amălinei, C., & Căruntu, I. D. (2013). Relationship between Kuppfer cells, inflammation, and fibrosis in chronic hepatitis B and C. Revista Medico-chirurgicala a Societatii de Medici si Naturalisti din Iasi, 117(4), 880–889.
- Tan, X. T., Zhao, T., Wang, Z. H., Wang, J., Wang, Y. J., & Liu, Z. G. (2018). Acrylamide defects the expression pattern of circadian clock and mitochondrial dynamics in C57BL/6J mice liver and HepG2 cells. Journal of Agricultural and Food Chemistry, 66, 10252–10266. doi:10.1021/acs.jafc.8b02473.
- Tareke, E., Rydberg, P., Karlsson, P., Eriksson, S., & Törnqvist, M. (2002). Analysis of acrylamide, a carcinogen formed in heated foodstuffs. Journal of Agricultural and Food Chemistry, 50(17), 4998–5006. doi:10.1021/jf020302f.
- Wang, W., Mao, S., Yu, H., Wu, H., Shan, X., & Zhang, X. (2019). Pinellia pedatisecta lectin exerts a proinflammatory activity correlated with ROS-MAPKs/NF-κB pathways and the NLRP3 inflammasome in RAW264.7 cells accompanied by cell pyroptosis. International Immunopharmacology, 66, 1–12. doi:10.1016/j.intimp.2018.11.002.
- Wang, S. Y., Wang, W., Ma, W. W., Chen, J. S., Gao, R., & Pang, Q. S. (2015). Effect of Low concentration acrylamide on serum biochemical parameters of non-neurotoxicity in occupational population. Occup Health & Emerg Rescue, 33(2), 75–77.
- Watzek, N., Scherbl, D., Schug, M., Hengstler, J., Baum, M., & Habermeyer, M. (2013). Toxicokinetics of acrylamide in primary rat hepatocytes: Coupling to glutathione is faster than conversion to glycidamide. Archives of Toxicology, 87(8), 1545–1556. doi: 10.1007/s00204-013-1054-0
- Wei, Q., Li, J., Li, X., Zhang, L., & Shi, F. (2014). Reproductive toxicity in acrylamide-treated female mice. Reproductive Toxicology, 46, 121–128. doi:10.1016/j.reprotox.2014.03.007.
- WHO G, FAO R. (2002). Health implications of acrylamide in food. Report of a Joint FAO/WHO Consultation. Geneva World Health Organization.
- Yu, J. W., Farias, A., Hwang, I., Fernandes-Alnemri, T., & Alnemri, E. S. (2013). Ribotoxic stress through p38 mitogen-activated protein kinase activates in vitro the human pyrin inflammasome. Journal of Biological Chemistry, 288(16), 11378–11383. doi:10.1074/jbc.M112.448795.
- Zeng, W. Q., Zhang, J. Q., Li, Y., Yang, K., Chen, Y. P., & Liu, Z. J. (2013). A new method to isolate and culture rat Kupffer cells. PLoS ONE, 8(8), e70832. doi:10.1371/journal.pone.0070832.
- Zuckerman, J. A. (1995). IARC monographs on the evaluation of carcinogenic risks to humans. Journal of Clinical Pathology, 48(7), 691–691. doi: 10.1136/jcp.48.7.691-a