
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Compound K (CK) is the metabolite and final active ingredient of diol-type ginsenosides. In this study, we investigated the effect of CK on mitochondrial apoptosis in SMMC-7721 and BEL-7404 cells and the regulatory mechanism through in vitro and in vivo experiments. The results demonstrated that CK inhibited Hepatocellular carcinoma (HCC) cells proliferation and arrested the cells in G0/G1 phase. CK induces mitochondrial apoptosis in HCC cells and inhibited p-ERK expression. Bcl-2 associated transcription factor 1 (Bclaf1) was distributed in the nucleus and cytoplasm, and CK inhibited its expression. Treatment of a nude mouse xenograft model bearing SMMC-7721 cells with CK decreased the expression of Bclaf1, p-ERK, and Bcl-2 but increased that of Bax. In summary, ginsenoside CK downregulated Bclaf1 expression, inhibited the activation of the ERK pathway, and triggered mitochondrial apoptosis in HCC. These findings uncovered a potential therapeutic strategy leveraging the anti-tumor effects of CK against HCC.
Introduction
Hepatocellular carcinoma (HCC), accounting for approximately 90% of all primary liver cancers, is the fourth leading cause of cancer mortality globally (Chidambaranathan-Reghupaty et al., Citation2021). At present, comprehensive therapy based on chemoradiotherapy, targeted drugs, immunization, and traditional Chinese medicine (TCM) remains an effective treatment strategy to prolong the survival and improve the quality of life of patients with liver cancer (Fan et al., Citation2022). In recent years, breakthroughs have been made in the treatment of liver cancer using targeted drugs. However, with increases in the drug concentration and duration of treatment, the risk of drug resistance increases, leading to poor prognoses and severe side effects. The treatment of advanced liver cancer thus needs to be improved. Because of the advantages of TCM in suppressing various cancer-related signaling pathways and molecular targets with few toxic and side effects, increasing numbers of TCM compounds, extracts, and active ingredients have been widely used in the prevention and treatment of liver cancer. Panax ginseng C. A. Mey, a member of the Araliaceae family, is a rare TCM. Compound K (CK, (A)) is the metabolite and final active ingredient of diol ginsenosides in microorganisms, and it has a wide range of pharmacological activities (Liu et al., Citation2022). Mitochondrial apoptosis is the main regulatory pathway of endogenous apoptosis (Chu et al., Citation2021). When cells encounter internal apoptosis-stimulating factors, such as DNA damage, hypoxia, and a loss of cell growth factors, the inner mitochondrial apoptosis pathway can be activated, and apoptosis can proceed (Tian et al., Citation2022). The anti-apoptotic protein Bcl-2 mainly exists in the mitochondrial membrane, whereas the pro-apoptotic protein Bax generally exists in the cytoplasm (Aouacheria et al., Citation2017). When cells received apoptotic stimuli, Bax is repositioned on the mitochondrial surface, the permeability of the outer mitochondrial membrane is increased, and mitochondrial permeability transition pores are opened. Cytochrome C (Cyt-C) and other pro-apoptotic factors located in the inner mitochondrial membrane are released into the cytoplasm. Cyt-C binds with apoptotic protease activator to form Cyt-C/apoptotic protease activator apoptotic bodies, which activate the cascade reaction. The downstream caspase 3 and other apoptotic executor proteins are activated (Dong et al., Citation2020), causing irreversible cell apoptosis. The mitochondrial apoptosis pathway plays a central role in initiation and amplification and an important role in the occurrence and development of tumors. Bcl-2 associated transcription factor 1 (Bclaf1, also known as BTF) is a multifunctional protein rich in arginine–serine and DNA-binding domains. It plays important roles in ontogeny as well as cancer and other diseases by regulating the transcription and post-transcriptional processing of specific genes, apoptosis, DNA damage response, cell differentiation, and other processes. Studies reported that Bclaf1 regulates the expression of ubiquitin specific peptidase 22 mRNA in cisplatin-resistant lung cancer cells and controls G1 phase arrest by targeting P21, indicating that Bclaf1 can mediate cisplatin resistance in lung cancer by regulating DNA damage repair and P21-mediated G1 phase arrest (Jiang et al., Citation2020). Wen et al. found that Bclaf1 can promote angiogenesis by regulating HIF-1α transcription in HCC, suggesting that Bclaf1 is related to HCC (Wen et al., Citation2019). Our previous study found that CK could reduce the binding of Bclaf1 to HIF-1α and inhibit the glycolytic pathway, thereby inhibiting liver cell proliferation (Zhang et al., Citation2020). Bclaf1 has different physiological functions in different stages of liver cancer, but its mechanism is unclear.
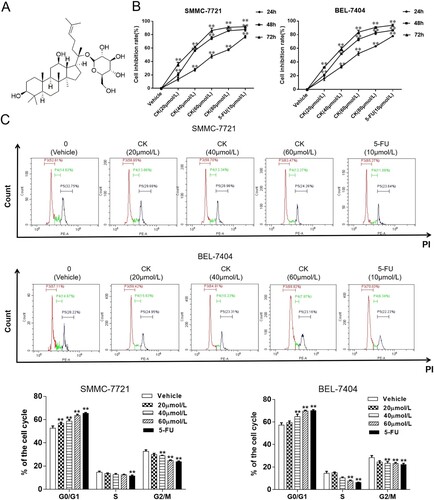
Figure 1. (A) Chemical structure formula of CK. B. In the CCK-8 assay, SMMC-7721 and BEL-7404 cells were treated with CK (20, 40, 60, or 80 µmol/L) or 5-FU (10 µmol/L) for 24 h. Compared with the findings in the blank control group, cell proliferation was significantly inhibited after 48 and 72 h (**P < 0.01). (C) Flow cytometry was used to detect the effects of CK (0, 20, 40, or 60 µmol/L) and 5-FU (10 µmol/L) on the cell cycle progression in SMMC-7721 and BEL-7404 cells after 48 h of treatment. Compared with the blank control group, **P < 0.01.
In this study, HCC cells were selected as the object, and a nude mouse xenograft model of HCC cells was constructed to explore whether CK could regulate the ERK signaling pathway through Bclaf1 and then induce mitochondrial apoptosis in human HCC cells, thereby providing an experimental and theoretical basis for the development and utilization of CK in the treatment of liver cancer.
Materials and methods
Reagent
Human hepatoma cell lines SMMC-7721 and BEL-7404 were cryopreserved by our laboratory. Ginsenoside CK, 5-fluorouracil (5-FU)and cell cycle and apoptosis detection kit were purchased from China Yuanye Biotechnology Co., LTD. (Shanghai, China). Annexin V/PI apoptosis kits was purchased from Becton, Dickinson and Company (NSW, USA). Cyt-c, caspase 3, Cleaved caspase 3, Bclaf1, Bcl-2, Bax, ERK, p-ERK, GAPDH antibodies were purchased from Abcam (Cambridge, MA, USA). β-actin was purchased from Bioss (Beijing, China). Dulbeccos modified Eagle’s medium (DMEM), calf serum, 0.25% Trypsin-EDTA and penicpstreptomycin were purchased from Gibco (Grand Island, NY, USA). CCK-8 and JC-1 mitochondrial membrane potential detection kits were purchased from Biyuntian Biotechnology Co., LTD. (Shanghai, China). U0126 was purchased from Med Chem Express (MCE; Shanghai, China). Horseradase labeled goat anti-rabbit IgG and horseradase labeled goat anti-Mouse IgG were purchased from Zhongshan Jinqiao Co., LTD. (Beijing, China).
Cell culture
Human liver cancer cells SMMC-7721 and BEL-7404 were cultured with DMEM medium containing 10% fetal bovine serum and 100 U/mL dual antibody, and incubated at 37 °C in an incubator (Shanghai Zhicheng Company; China; ZXDR) with 5% CO2 and 95% humidity. The proliferation ability of the cells was detected at logarithmic growth stage.
CCK-8 proliferation assay
The concentration of liver cancer cells was adjusted to 5×103 cells/mL, 100 μL was added to each well in a 96-well plate, and 100 μL PBS was added around the loading well, and the cells were incubated in an incubator. The next day, the old culture medium was discarded, and ginsenoside CK (0, 20, 40, 60, 80 μmol/L) was added successively, and they were divided into blank control group, ginsenoside CK administration group and negative control group, with five rewells in each group. After incubation for 24, 48, 72 h, the old culture medium was discarded, 100 μL culture medium and 10 μL CCK-8 (5.0 mg/mL) were added to each well, and the cells were incubated for 1 h. Optical density (OD) was measured at 490 nm.
Cell cycle analysis
SMMC-7721 and BEL-7404 cells in the logarithmic growth phase were centrifuged at 1 500 g for 5 min, the supernatant was removed, 1 mL of pre-cooled 70% ethanol was added overnight at 4 °C The next day, the cells were centrifuged at 1 500 g for 5 min in a centrifuge, the supernatant was removed, and the cells were resuspended with ice-cold PBS. 0.5 mL Propidium iodide staining solution was added to each tube and incubated for 30 min in a constant temperature incubator under darkness and analyzed by flow cytometry (Beckman Coulter, Inc., CA, USA; A00-1-1102).
Apoptosis analysis
SMMC-7721 and BEL-7404 were inoculated in Petri dishes and divided into non-staining group, Annexin V single staining group, PI single staining group, ginsenoside CK (0, 20, 40, 60 µmol/L) administration group, 5-FU (10 µmol/L) administration group, sgRNA group. In sgRNA + CK group, after 48 h in the incubator, the old culture medium was discarded, washed with PBS, and digested with trypsin (without EDTA). After cleaning with PBS, 100 μL of 1× binding buffer was added to each tube and mixed well. Then, 5 µL Annexin V and 5 µL PI were added according to the group, and the mixture was mixed. After incubation at room temperature for 15 min, 400 μL 1× binding buffer was added to each group. The apoptosis rate was detected by flow cytometry within 1 h.
Analysis of mitochondrial membrane potential
SMMC-7721 and BEL-7404 in logarithmic growth phase were inoculated into six-well plates and divided into blank control group, ginsenoside CK (40 µmol/L) administration group, 5-FU (10 µmol/L) administration group, sgRNA group, sgRNA + CK group, and sgRNA + CK group. Carbonyl Cyanide 3-chlorophenylhydrazone (CCCP) group. After 48 h incubation in the incubator, JC-1 staining working solution was prepared, and cells in CCCP group were treated for 40 min. The culture medium and JC-1 staining working solution were added to each well, and incubated in an incubator for 60 min. At the end of incubation, the cells were washed with JC-1 buffer (1×), and the cells were photographed under a 400× fluorescence microscope (Olympus Microscopy Inc., Japan; CX41-32RFL).
Immunofluorescence assay
The cells were seeded into six-well plates with cell crawl plates, and then treated with DMEM culture medium, ginsenoside CK (40 µmol/L) and 5-FU (10 µmol/L) after attachment. After 48 h incubation in an incubator, fixed with 4% paraformaldehyde. Then, 1 mL 0.2% TritonX-100 was added to each well, permeated for 25 min, and 1 mL 5% BSA was blocked at room temperature for 1 h. 150 µL of primary antibody Bclaf1(1:200) was added to the slide and incubated at 4 °C overnight in a wet box shielded from light. The next day, the samples were washed with PBS, and 200 µL fluorescent secondary antibody (1:100) was incubated in a wet box for 1 h at 37 °C in the dark environment. The samples were washed with PBS, stained with DAPI, and placed in the dark for 5 min at room temperature. After cleaning with PBS, the samples were observed under a 400× fluorescence microscope and photographed.
CRISPR/Cas9 mediated Bclaf1 gene knockout experiment
The CRISPR/Cas9-Bclaf1-sgRNA lentiviral vector was provided by Shanghai Jikaigenin Technology Co., LTD. SMMC-7721 and BEL-7404 cells were divided into control group and sgRNA group. The next day, the cell medium was changed, and the sgRNA group was added with 20 μL lentiviral vector. After 12 h, the conventional medium was changed and the culture was continued. 48 h later, 6 µg/mL dual antibody was added to screen Bclaf1 knockout cells.
Tumor implantation
SMMC-7721 cells with 0.1 mL cell concentration of 1 × 107 cells/mL were injected into the left axilla of nude mice and the graft tumor diameter was measured daily, after 7 days of injection, 32 tumorigenic nude mice were randomly divided into 4 groups: CK (5, 10, 20 mg/kg) administration group and vehicle (normal saline), the mode of administration was the tail intravenous injection, following continuous administration for 15 days. After all nude mice were sacrificed under anesthesia, the graft bodies were removed, calculated the tumor volume, growth inhibition rate and recorded weight of mice.
Immunohistochemistry
The xenografts of nude mice were dissected and tissue wax blocks were made. The wax block was sliced, soaked in xylene for 10 min, then treated with ethanol gradient for 5 min, and then treated with antigen repair solution, heated to a slightly boiling state for 30 min. Two drops of 3% hydrogen peroxis-methanol solution were added to the sections and left for 10 min. 200 μL of 5% bovine serum albumin was added and left for 20 min at room temperature. Appropriate amounts of primary antibodies Bclaf1(1:200), Bcl-2(1:150) and Bax(1:200) were added and incubated in a wet box at 37 °C for 2 h. After washing with PBS, 80 μL of booster was dropped and incubated at room temperature for 30 min. Then, 50 μL DAB solution was added to observe the staining under the microscope. Tissue sections were immersed in hematoxylin dyeing solution for 10 min, dehydrated in ethanol gradient, and soaked in xylene for 20 min. Add drops of neutral gum and seal the piece.
Western blot
The whole protein of cells and tissues was extracted, the protein concentration was determined by BCA kit, calculated the sample size. According to the experimental group, 2.5 µL Marker and sample were added to the loading well successively, Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (100 V, 120 min), transferred to polyvinylidene fluoride (PVDF) membranes (100 V, 30–90 min). Block in 5% skim milk for 1 h at room temperature. TBST diluted primary antibody: Cyt-c (1:1 000), caspase 3 (1:5 000), Cleaved caspase 3 (1:1 000), Bcl-2 (1:1 000), Bax (1:1 000), β-actin (1: 50 000), ERK(1:3 000), p-ERK (1:1 000), Bclaf1 (1:5 000), GAPDH (1:10 000) and then incubated overnight at 4°C. The next day, incubated with anti-rabbit secondary antibody (1:5 000) at room temperature for 1 h, ECL luminescent solution was dropped. Target proteins were detected by Bio-RAD imaging system (Bio-RAD, Hercules, CA, USA; ChemiDoc MP System)
Statistical analysis
All experiments were repeated three times. Data are expressed as mean ± standard deviation (SD), and differences between groups were analyzed by one-way analysis of variance and Student’s t-test. p < 0.05 was considered to be statistically significant. SPSS version 19.0 and GraphPad Prism 5.0 software were used to analyze the results.
Results
CK inhibited the proliferation of HCC cells
CCK-8 was used to detect the effect of CK on the proliferation of SMMC-7721 and BEL-7404 human liver cancer cells. The results illustrated that the inhibition of SMMC-7721 and BEL-7404 cell proliferation gradually increased in a concentration and time-dependent manner. Compared with the findings in the blank control group, treatment with 40, 60, or 80 µmol/L CK and 5-FU as a positive control for 48 h significantly inhibited HCC cell proliferation (P < 0.01, (B)). The IC50s of CK in SMMC-7721 and Bel-7404 cells after 48 h were 42.35 and 42.86 µmol/L, respectively, and thus, the drug concentration of 40 µmol/L and treatment time of 48 h were used in the subsequent analyses. Flow cytometry illustrated that the number of cells in G0/G1 phase gradually increased following treatment, whereas the numbers of cells in S and G2/M phases gradually decreased, indicating that CK arrested liver cancer cells in G0/G1 phase ((C)).
CK promoted apoptosis in SMMC-7721 and BEL-7404 cells
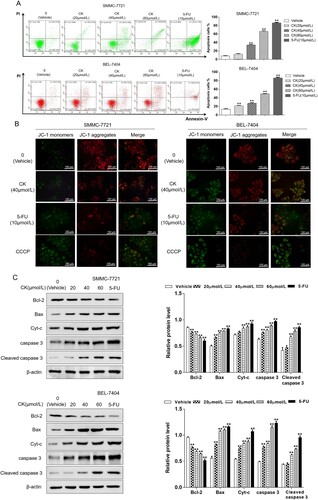
Phosphatidylserine effusion analysis illustrated that after 0, 20, 40, or 60 µmol/L CK or 10 µmol/L 5-FU treatment in SMMC-7721 cells, the apoptosis rates were 8.13 ± 0.85%, 12.43 ± 1.35%, 34.13 ± 1.68%, 66.14 ± 2.16%, and 85.89 ± 1.77%, respectively. Compared with the findings in the blank control group, the apoptosis rate in the 40 µmol/L CK group was significantly increased (P < 0.01). In BEL-7404 cells, the apoptosis rates in the aforementioned groups were 13.47 ± 1.30%, 21.46 ± 1.01%, 27.25 ± 1.64%, 49.05 ± 1.19%, and 85.63 ± 2.25%, respectively ((A)). JC-1 was used as a fluorescent probe to detect the changes of mitochondrial membrane potential. When mitochondrial membrane potential decreases, JC-1 polymers are dispersed into monomers, and the fluorescence changes from red to green. After drug treatment, the red fluorescent spots decreased, and the green fluorescent spots increased, indicating that mitochondrial membrane potential in liver cancer cells was decreased ((B)). Western blotting demonstrated that compared with the findings in the control group, the expression of the mitochondrial apoptosis-related proteins Cyt-C, caspase 3 and cleaved caspase 3 were upregulated in HCC cells (P < 0.01), the expression of the anti-apoptotic protein Bcl-2 decreased as the CK concentration increased, whereas that of the pro-apoptotic protein Bax increased, resulting in a decreasing Bcl-2/Bax ratio ((C)), suggesting that CK can induce apoptosis in human liver cancer cells through the mitochondrial pathway.
Figure 2. (A) The apoptosis rate of SMMC-7721 and BEL-7404 cells treated with CK (0, 20, 40, or 60 µmol/L) or 5-FU (10 µmol/L) for 48 h was detected by flow cytometry and compared with that of the blank control group (**P < 0.01). (B) A fluorescence probe method was used to detect the changes of membrane potential in SMMC-7721 and BEL-7404 cells treated with CK or 5-FU for 48 h (×400). (C) Western blotting was used to detect the expression of mitochondrial apoptosis-related proteins in SMMC-7721 and BEL-7404 cells treated with CK (20, 40, or 60 µmol/L) or 5-FU in comparison with the blank control group (**P < 0.01).
CK inhibited the ERK signaling pathway
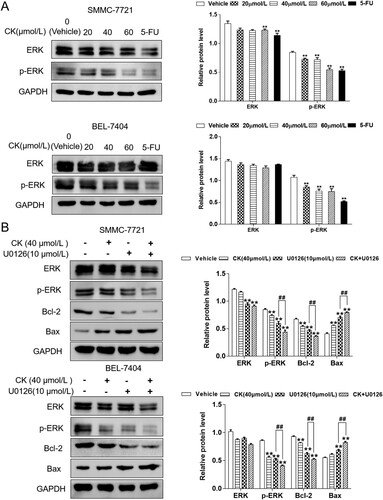
Western blotting revealed no significant difference in ERK expression in SMMC-7721 cells after CK treatment, whereas p-ERK expression gradually decreased as the CK concentration increased. Compared with the findings in the control group, treatment with 20 or 40 µmol/L CK significantly decreased ERK expression (P < 0.01, (A)). To further explore the effect of CK on the ERK pathway in SMMC-7721 and BEL-7404 cells, the ERK inhibitor U0126 was used. The results demonstrated that the inhibitory effect of CK (40 µmol/L) on the ERK pathway was further enhanced compared with that in the control group. The Bcl-2/Bax ratio was significantly decreased by treatment (P < 0.01, (B)).
Figure 3. (A) ERK and p-ERK expression in SMMC-7721 and BEL-7404 cells treated with CK (20, 40, or 60 µmol/L) or 5-FU was detected by western blotting and compared with the findings in the blank control group (**P < 0.01). B ERK, p-ERK, Bcl-2, and Bax expression in SMMC-7721 and BEL-7404 cells treated with CK after U0126 treatment was detected by western blotting and compared with findings in the blank control group (**P < 0.01; comparison of U0126 and U0126 + ginsenoside CK, ##P < 0.01).
CK inhibited Bclaf1 protein expression in SMMC-7721 and BEL-7404 cells
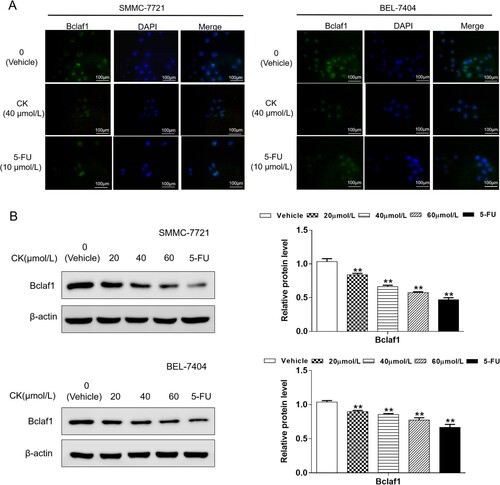
Immunofluorescence analysis revealed that Bclaf1 was expressed in both the nucleus and cytoplasm. After 40 µmol/L CK treatment in SMMC-7721 and BEL-7404 cells, the number of cells and green fluorescence intensity decreased, indicating that CK inhibited Bclaf1 expression ((A)). To investigate the effect of CK on Bclaf1 expression in cells, western blotting revealed that compared with the findings in the control group, Bclaf1 expression decreased as the CK concentration increased (P < 0.01, (B)).
Figure 4. (A) Immunofluorescence analysis was used to detect the expression and localization of Bclaf1 in SMMC-7721 and BEL-7404 cells (×400). (B) Bclaf1 expression in SMMC-7721 and BEL-7404 treated with CK (20, 40, or 60 µmol/L) was detected by western blotting and compared with findings in the blank control group (**P < 0.01).
CK induced mitochondrial apoptosis in Bclaf1 knockout HCC cells
In SMMC-7721 cells, the apoptosis rates in the blank control, CK (40 µmol/L), sgRNA, and sgRNA + CK groups were 20.18 ± 1.43%, 32.86 ± 2.11%, 39.54 ± 1.29%, and 63.26 ± 1.59%, respectively, which were significantly higher than that in the control group (P < 0.01). The apoptosis rates of BEL-7404 cells in these groups were 15.72 ± 1.16%, 34.00 ± 1.43%, 43.86 ± 1.64%, and 56.30 ± 1.36% ((A)). Using the fluorescent probe JC-1, red fluorescence was significantly weakened and green fluorescence was enhanced after CK treatment in Bclaf1 stable knockout HCC cells compared with the findings in the control group, indicating that the mitochondrial membrane potential of HCC cells was further decreased after CK treatment and Bclaf1 knockout ((B)). After Bclaf1 depletion, apoptotic protein expression was examined in SMMC-7721 and BEL-7404 cells by western blotting. The results demonstrated that compared with the findings in the control group, the Bcl-2/Bax ratio was decreased by treatment, whereas Cyt-C and cleaved caspase 3 expression was upregulated (P < 0.01, (C)).
Figure 5. (A) Flow cytometry was used to detect the apoptosis of SMMC-7721 and BEL-7404 cells treated with CK (40 µmol/L) after Bclaf1 depletion compared with that in the blank control group (**P < 0.01). (B) The effect of CK on mitochondrial membrane potential in SMMC-7721 and BEL-7404 cells after Bclaf1 knockout was detected using a fluorescence probe (×400). (C) Western blotting was used to detect the expression of Cyt-C, cleaved caspase 3, Bcl-2, and Bax in SMMC-7721 and BEL-7404 treated with CK after Bclaf1 knockout in comparison with the findings in the blank control group (**P < 0.01; ginsenoside CK group compared with the sgRNA + ginsenoside CK group, ##P < 0.01).
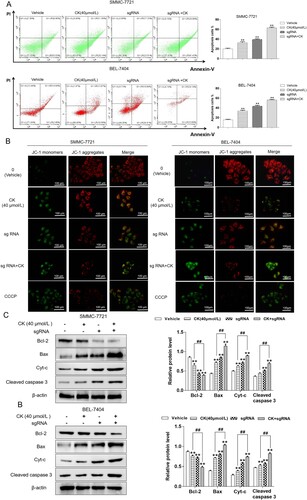
CK inhibited the ERK pathway in Bclaf1 knockout HCC cells
Western blotting was used to detect p-ERK expression in HCC cells with stable Bclaf1 knockout. Bclaf1 knockout enhanced the inhibitory effect of CK on p-ERK expression (P < 0.01). After Bclaf1 depletion, ERK expression decreased ((A)).
Figure 6. (A) Western blotting was used to detect the effect of CK on the expression of ERK and p-ERK in SMMC-7721 and BEL-7404 cells after Bclaf1 depletion compared with the findings in the blank control group (**P < 0.01; ginsenoside CK group compared with the sgRNA+ ginsenoside CK group, ##P < 0.01).
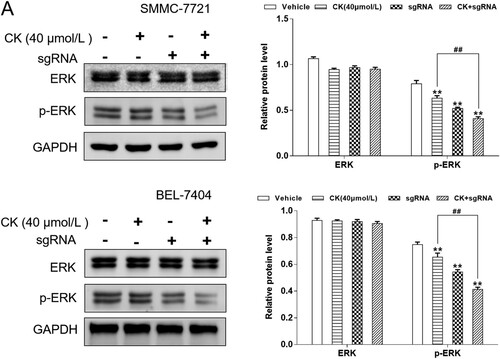
CK regulated mitochondrial apoptosis via the ERK pathway mediated by Bclaf1 in a nude mouse xenograft model of HCC
In vitro experiments, CK inhibited the proliferation of hepatoma cells, and in order to further explore the anti-hepatoma effect of CK in vivo, we established a nude mouse xenograft model of SMMC-7721. After CK (5, 10, and 20 mg/kg) administration for 15 days, the graft tumor volume was calculated, and recorded the weight of the tumor. The results showed that CK inhibite the tumor growth, compared with vehicle controls, tumor volume was significantly reduced following CK treatment (Fig.7A). The growth inhibition rate of the CK (5,10 mg/kg) group tumor was 46.69%, 55.72% respectively, after 20 mg/kg CK treatment, the tumor inhibition rate was 65.62% ((B)). There were no significant differences in weight of mice among the vehicle and 5 and 10 mg/kg CK-treated groups ((C)). Immunohistochemical analysis demonstrated that Bcl-2 and Bax were mainly expressed in the cytoplasm. CK inhibited the expression of Bcl-2 and promoted that of Bax, indicating that CK could induce mitochondrial apoptosis in transplanted tumor cells ((D)). In the xenograft tumor tissues of nude mice, Bclaf1 was expressed in both the cytoplasm and nucleus, and CK inhibited Bclaf1 expression ((E)). Compared with the findings in the blank control group, CK significantly inhibited the expression of p-ERK (P < 0.01, (F)).
Figure 7. (A) After CK (5, 10, 20 mg/kg) treatment, the nude micetumor volume (cm3), compared with blank control, **P < 0.01. (B) Tumor growth inhibition rate (%), compared with CK (5 mg/kg) group, **P < 0.01. (C) Weight of mice (g), compared with blank control, **P < 0.01. (D) The effects of CK on Bcl-2 and Bax in nude mice featuring SMMC-7721 xenografts were detected by immunohistochemistry (×200). (E) The effect of CK on Bclaf1 in nude mice featuring SMMC-7721 xenografts was detected by immunohistochemistry (×200). (F) ERK and p-ERK expression in tumor tissues was detected by western blotting compared with the findings in the blank control group (**P < 0.01).
Discussion
HCC is often diagnosed in intermediate and advanced stages because of its complex pathogenesis and insidious course. Although there are many treatments for HCC, the prognosis and quality of life of patients are poor. Therefore, it is crucial to develop effective and low-toxicity drugs for the treatment of liver cancer (Du et al., Citation2022). CK is an effective component of natural diol ginsenoside decomposition in vivo. As a potential anti-tumor drug with high efficiency and low-toxicity, CK can inhibit the proliferation of tumor cells, including liver cancer cells, but its mechanism remains unclear. In this study, the mechanism of the inhibitory effects of CK on HCC was deeply explored to provide an experimental and theoretical basis for the research and development of CK.
The CCK-8 assay demonstrated that CK could inhibit the proliferation of human HCC cells in a time- and concentration-dependent manner. Previous research groups showed that CK has no obvious toxicity to normal hepatocyte L02 and showed less L02 inhibition (Zhang et al., Citation2018). To further investigate the inhibitory effect of CK on HCC cell growth, we examined the effect of CK on the cell cycle of HCC cells and found that, as the CK concentration increased, the number of cells in G0/G1 phase gradually increased, whereas the numbers of cells in S and G2/M phases decreased. The aforementioned experimental results indicated that CK could inhibit the proliferation of HCC cells and arrest the cells in G0/G1 phase. When cells receive stimuli, such as tumor gene activation, DNA damage, and hypoxia, the mitochondrial apoptosis pathway can be activated, causing cell apoptosis. Bcl-2 family proteins play a key role in this pathway. The pro-apoptotic factor Bax can induce the release of mitochondrial apoptotic proteins, whereas anti-apoptotic factors such as Bcl-2 antagonize this process, which changes the mitochondrial membrane potential and opens mitochondrial membrane permeability transition pores. This promotes the release of pro-apoptotic proteins such as Cyt-c and activates caspase-3, ultimately causing apoptosis (Castillo Ferrer et al., Citation2021; Márquez-Jurado et al., Citation2018). The liver is an important organ in metabolism, which is closely related to mitochondria. The mitochondrial apoptosis pathway plays a crucial role in the occurrence and development of liver cancer. When cells undergo early apoptosis, phosphatidylserine in the cell membrane moves from the inner side to the outer side of the lipid membrane, and annexin V easily binds to phosphatidylserine. Then, cells in the early stage of apoptosis can be detected by fluorescein FITC labeling. Propidium iodide can penetrate the cell membrane and stain the nucleus, thus distinguishing the stage of apoptosis. To explore the mechanism of action of the anti-tumor growth of CK, in this study, the apoptosis rates of SMMC-7721 and BEL-7404 cells after CK treatment was detected by phosphatidylserine effusion analysis. The results illustrated that as the drug concentration increased, the apoptosis rate gradually increased, and the number of viable cells gradually decreased. To explore whether CK can induce liver cancer cell apoptosis through the mitochondrial apoptosis pathway, the mitochondrial membrane potential of SMMC-7721 and BEL-7404 cells was detected using a fluorescence probe. The results indicated that the mitochondrial membrane potential in the cells decreased significantly after CK treatment, and the permeability of the mitochondrial outer membrane increased. The Bcl-2/Bax ratio is a key factor of mitochondrial apoptosis. The content of Bcl-2 and Bax in cells was detected by western blotting. As the drug concentration increased, the expression Bax increased, and that of Bcl-2 decreased, resulting in a gradually decreasing Bcl-2/Bax ratio. Mitochondrial membrane permeability transition pores were opened, and the expression of Cyt-C and cleaved caspase 3 was detected by western blotting. CK promoted the expression of Cyt-C and cleaved caspase 3, indicating that CK could induce mitochondrial apoptosis in liver cancer cells.
ERK is ubiquitous in a variety of tissues, and it is believed to bind MEK1, phosphorylate threonine or tyrosine, and then undergo degradation. Monophosphorylated ERK will recombine with activated MEK1 to perform double phosphorylation and participate in cell proliferation and differentiation. ERK activation can activate the signal transduction pathways of growth and nutrient factors in the body and participate in the completion of substance metabolism in the body (Guo et al., Citation2020; Salaroglio et al., Citation2019). In this experiment, the effect of CK on the ERK pathway in SMMC-7721 and BEL-7404 cells was explored. The results demonstrated that 20, 40, and 60 µmol/L CK could downregulate the expression of p-ERK, indicating that CK inhibits the ERK pathway. Concentration dependence was also observed. To further explore the effect of drugs on the ERK pathway, the ERK inhibitor U0126 was used. Compared with the findings in the control group, the inhibitory effect of CK was strengthened (P < 0.01), and the Bcl-2/Bax ratio was further decreased. These results indicated that ginsenoside CK could induce mitochondrial apoptosis in human HCC cells by inhibiting the ERK pathway.
The activation of tumor-related oncogenes, expression of transcription factors, and changes in signaling pathways are closely related to the development of cancer cells. Bclaf1 is involved in several physiological and pathological processes in cells. In bladder cancer tissues, the expression of Bclaf1 is significantly increased, and it has been observed that it can induce autophagy in tumor cells, which is extremely important for maintaining the homeostasis of the cellular environment and overcoming the malignant development of tumors (Yu et al., Citation2022). In this study, the expression and localization of Bclaf1 in HCC cells were detected by immunofluorescence analysis. The results demonstrated that Bclaf1 was expressed in both the cytoplasm and nucleus, and Bclaf1 expression was significantly lower in the CK groups than in the control group. After treatment with different concentrations of CK (20, 40, or 60 µmol/L), Bclaf1 expression in SMMC-7721 and BEL-7404 cells decreased gradually compared with that in the control group (P < 0.01), indicating that CK could inhibit the expression of Bclaf1. To explore whether CK can induce mitochondrial apoptosis of SMMC-7721 and BEL-7404 cells through Bclaf1, liver cancer cells with stable Bclaf1 knockout were treated, and mitochondrial membrane potential was detected. The results demonstrated that compared with findings in the control group, mitochondrial membrane potential was decreased significantly by CK treatment, and the green monomer content was significantly increased. After Bclaf1 depletion, the expression of the mitochondrial apoptosis-related proteins Cyt-C and cleaved caspase 3 was significantly increased (P < 0.01), and the Bcl-2/Bax was gradually decreased, indicating that CK could induce mitochondrial apoptosis in HCC cells through Bclaf1. In exploring whether CK can regulate the ERK pathway through Bclaf1, p-ERK expression was significantly downregulated after Bclaf1 knockout cells were treated with CK compared with the findings in the control group. The aforementioned in vitro experiments indicated that CK could inhibit the ERK pathway through Bclaf1. Thus, mitochondrial apoptosis was induced in hepatocellular carcinoma cells.
Our research group established a xenograft tumor model of SMMC-7721 cells in nude mice and tested the inhibitory effect of CK on xenograft tumors in nude mice. The results illustrated that CK inhibited tumor growth. However, the effect of CK on the mitochondrial apoptosis pathway has not been clarified in the xenograft tumor tissues of nude mice. Immunohistochemistry demonstrated that the mitochondrial apoptosis-related genes Bcl-2 and Bax were mainly distributed in the cytoplasm. After CK treatment, Bclaf1 and Bcl-2 were downregulated, and the expression of Bax was increased. These results indicated that CK could induce mitochondrial apoptosis and inhibit the expression of Bclaf1 in the xenograft tumor tissues of nude mice. The expression of ERK and p-ERK was detected, the results illustrated that p-ERK expression decreased as the CK concentration increased above 5 mg/kg (P < 0.01), indicating that CK could inhibit the ERK signaling pathway in xenograft tumor tissues in nude mice.
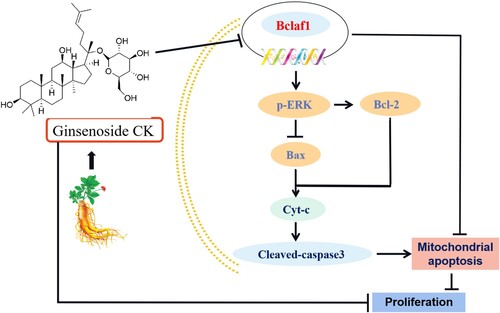
Based on these findings, it can be concluded that CK induces mitochondrial apoptosis in human liver cancer cells by inhibiting the ERK pathway via Bclaf1 ().
Figure 8. CK induces mitochondrial apoptosis in human liver cancer cells by inhibiting the ERK pathway via Bclaf1.
Ethics statement
The experiment procedures were approved by the Institutional Animal Care and Use Committee of Yanbian University (Resolution number, 201501022).
Acknowledgements
XZ and JC designed the study; JC and MS performed the research, analyzed data, and wrote the initial draft of the paper; XC contributed to refining the ideas, carrying out additional analyses and finalizing this paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data underlying this study are presented in the article. Further inquiries can be directed to the corresponding author.
Additional information
Funding
References
- Aouacheria, A., Baghdiguian, S., Lamb, H. M., Huska, J. D., Pineda, F. J., & Hardwick, J. M. (2017). Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochemistry International, 109, 141–161. https://doi.org/10.1016/j.neuint.2017.04.009
- Castillo Ferrer, C., Berthenet, K., & Ichim, G. (2021). Apoptosis - Fueling the oncogenic fire. The FEBS Journal, 288(15), 4445–4463. https://doi.org/10.1111/febs.15624
- Chidambaranathan-Reghupaty, S., Fisher, P. B., & Sarkar, D. (2021). Hepatocellular carcinoma (HCC): epidemiology, etiology and molecular classification. Advances in Cancer Research, 149, 1–61. https://doi.org/10.1016/bs.acr.2020.10.001
- Chu, Q., Gu, X., Zheng, Q., Wang, J., & Zhu, H. (2021). Mitochondrial mechanisms of apoptosis and necroptosis in liver diseases. Analytical Cellular Pathology, 2021, 1–9. https://doi.org/10.1155/2021/8900122
- Dong, L., Gopalan, V., Holland, O., & Neuzil, J. (2020). Mitocans revisited: Mitochondrial targeting as efficient anti-cancer therapy. International Journal of Molecular Sciences, 21(21), 7941. https://doi.org/10.3390/ijms21217941
- Du, D., Liu, C., Qin, M., Zhang, X., Xi, T., Yuan, S., Hao, H., & Xiong, J. (2022). Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharmaceutica Sinica B, 12(2), 558–580. https://doi.org/10.1016/j.apsb.2021.09.019
- Fan, Y., Xue, H., & Zheng, H. (2022). Systemic therapy for hepatocellular carcinoma: Current updates and outlook. Journal of Hepatocellular Carcinoma, 9, 233–263. https://doi.org/10.2147/JHC.S358082
- Guo, Y. J., Pan, W. W., Liu, S. B., Shen, Z. F., Xu, Y., & Hu, L. L. (2020). ERK/MAPK signalling pathway and tumorigenesis. Experimental and Therapeutic Medicine, 19(3), 1997–2007. https://doi.org/10.3892/etm.2020.8454
- Jiang, T., Liu, B., Wu, D., & Zhang, F. (2020). BCLAF1 induces cisplatin resistance in lung cancer cells. Oncology Letters, 20(5), 227. https://doi.org/10.3892/ol.2020.12090
- Liu, J., Wang, Y., Yu, Z., Lv, G., Huang, X., Lin, H., Ma, C., Lin, Z., & Qu, P. (2022). Functional mechanism of ginsenoside compound K on tumor growth and metastasis. Integrative Cancer Therapies, 21, 1–13. https://doi.org/10.1177/15347354221101203
- Márquez-Jurado, S., Díaz-Colunga, J., das Neves, R. P., Martinez-Lorente, A., Almazán, F., Guantes, R., & Iborra, F. J. (2018). Mitochondrial levels determine variability in cell death by modulating apoptotic gene expression. Nature Communications, 9(1), 389. https://doi.org/10.1038/s41467-017-02787-4
- Salaroglio, I. C., Mungo, E., Gazzano, E., Kopecka, J., & Riganti, C. (2019). ERK is a pivotal player of chemo-immune-resistance in cancer. International Journal of Molecular Sciences, 20(10), 2505. https://doi.org/10.3390/ijms20102505
- Tian, C., Liu, Y., Li, Z., Zhu, P., & Zhao, M. (2022). Mitochondria related cell death modalities and disease. Frontiers in Cell and Developmental Biology, 10, 832356. https://doi.org/10.3389/fcell.2022.832356
- Wen, Y., Zhou, X., Lu, M., He, M., Tian, Y., Liu, L., Wang, M., Tan, W., Deng, Y., Yang, X., Mayer, M. P., Zou, F., & Chen, X. (2019). Bclaf1 promotes angiogenesis by regulating HIF-1α transcription in hepatocellular carcinoma. Oncogene, 38(11), 1845–1859. https://doi.org/10.1038/s41388-018-0552-1
- Yu, Z., Zhu, J., Wang, H., Li, H., & Jin, X. (2022). Function of BCLAF1 in human disease. Oncology Letters, 23(2), 58. https://doi.org/10.3892/ol.2021.13176
- Zhang, S., Zhang, M., Chen, J., Zhao, J., Su, J., & Zhang, X. (2020). Ginsenoside compound K regulates HIF-1α-mediated glycolysis through Bclaf1 to inhibit the proliferation of human liver cancer cells. Frontiers in Pharmacology, 11, 583334. https://doi.org/10.3389/fphar.2020.583334
- Zhang, X., Zhang, S., Sun, Q., Jiao, W., Yan, Y., & Zhang, X. (2018). Compound K induces endoplasmic reticulum stress and apoptosis in human liver cancer cells by regulating STAT3. Molecules, 23(6), 1482. https://doi.org/10.3390/molecules23061482