
Abstract
A recent generation of family studies has revealed that autism can be predicted from an array of neurobehavioural susceptibilities that are appreciable before the syndrome is diagnosed, and that each may be traceable to partially-independent sets of genetic variation. Some of these liabilities are not necessarily specific to ASD—those that are non-specific could account for a significant share of the ‘missing heritability’ of autism, would (by definition) contribute to pleiotropy, and relate to so-called ‘co-morbidities’, which are inappropriately named if they actually contribute to (or exacerbate) the severity of autism itself. Linking genetic variants to these underlying traits rather than to a diagnosis of ‘autism’ may be more productive in devising personalized approaches to developmental intervention, especially if autism represents an epiphenomenon of earlier-interacting susceptibilities. In this article, the implications of conceptualizing autism as a syndrome of neurobehavioural degeneration is considered, predicated on the notion that it can arise from a critical co-aggregation of earlier-interacting neuropsychiatric liabilities, the phenotypic expression of which—importantly—can be moderated by sex.
Introduction
Twenty years ago (when I first entered the field of autism research), it was a time of heated controversy over the boundary between Autistic Disorder and Asperger Disorder, and the DSM IV criterion sets used to establish and distinguish these conditions—essentially social impairment, communicative impairment, and restricted interests/repetitive behaviours—were considered to be independent impairments, whose co-occurrence defined the syndrome, and each of which (it was hoped) might someday be resolved to a separate biology. In retrospect, there were many reasons—even at that time—to believe that this might not be the case, the most compelling of which was the fact that single, highly deleterious point mutations (e.g. in MECP2, TSC1, TSC2, FMR1, and, more recently, NF1) were capable of recapitulating autistic syndromes featuring symptoms from all three DSM-IV criterion sets. The goal of this article is to briefly synthesize a series of advances from family studies over the past two decades, which have contributed to a quantitative reconceptualization of autism—one that most recently capitalizes upon findings from prospective infant studies (see Jones & Klin, Citation2013; Ozonoff et al., Citation2014) that consider autism as a syndrome of developmental decompensation. It is now understood that such decompensation might be precipitated by a diverse array of contributing causes, some specific to autism, some not, a few potent enough to engender the syndrome ‘single-handedly’ (as in the above highly deleterious rare mutations) but in most individual cases influenced by a multiplicity of early developmental liabilities. Evidence for this can be derived from its ‘deconstruction’ into early developmental quanta that may more faithfully reflect the causal structure of the syndrome than is possible to derive from manifest symptoms (i.e. after the condition arises). Here, the current evidentiary basis for this view is summarized according to seven key advances from family and genetic research in autism.
Autism is predominantly influenced by additive genetic factors
Although it is common for autism to arise sporadically in families, even the sporadic cases are substantially influenced by the aggregation of recessive or additive genetic risk (Wellner et al., Citation2017), and there is overwhelming evidence for the influence of additive genetic risk accounting for a majority of the population-attributable-risk for autism (Constantino et al., Citation2013; Sandin et al., Citation2017). Given the profound effects of clinical autism spectrum disorders (ASD) in compromising reproduction, when autism does recur in families—which occurs in autism-affected families 20-times more commonly than in the general population—it does so on the basis of transmission of components of liability that can be carried sub-clinically by unaffected members of the family. Lower (sub-clinical) levels of autistic trait aggregation may have been preserved in the human gene pool on the basis of as-yet-unspecified aspects of selective advantage (e.g. attention to detail and/or motivation to sustain highly focused behaviours that enhance survival). The results from twin and family studies involving several million subjects worldwide (see Constantino, Citation2014; Constantino et al., Citation2013; Gaugler et al., Citation2014; Robinson et al., Citation2011; Sandin et al., Citation2017) have concluded that the vast majority (80–90%) of causal influences on the occurrence of autism are resolvable to genetic factors, and that these are predominantly additive (incremental—as opposed to highly penetrant single-gene mutations) in nature. Thus, common allelic variations, each presumably individually-preserved in the human population—and none of which singly can account for more than a tiny elevation in risk for ASD (odds ratio between 1.0 and 1.2)—are responsible for a majority of genetic liability for ASD on the basis of cumulative polygenic risk.
Familial autism exhibits a unitary symptom structure
The characterizing features that define the autistic syndrome are tightly inter-correlated, by definition, among clinically-affected individuals (Constantino et al., Citation2006), and they are either partly (Ronald, Larsson, Anckarsater, & Lichtenstein, Citation2011) or largely (Frazier, Ratliff, Gruber, Zhang, & Law, Citation2014) inter-correlated in the general population as well, depending on the method of ascertainment of traits. This has profound implications for the biology of autism because it suggests that symptoms as disparate as social-communicative impairment, restriction in range of interests, repetitive behaviour, and coordination difficulties with actions like riding a bicycle or catching a ball (Hilton, Zhang, Whilte, Klohr, & Constantino, Citation2012; Moruzzi, Ogliari, Ronald, Happe, & Battaglia, Citation2011)—whether mild or severe—can be collectively generated from biological liabilities whose genetic influences either directly overlap or which interact early in the development of autistic traits. It is possible that our field will continue to struggle in the search for a neural signature for autism until a lesion can be ‘placed’ (as neurologists might say) within circuits that are either jointly affected by these liabilities (see Mahajan, Dirlikov, Crocetti, & Mostofsky, Citation2016), or whose compromise could give rise to many or all of these inter-correlated symptoms. It is important to reflect on the fact that autism is rarely recapitulated by severe head trauma, hypoxia, or encephalitis in the way that intellectual disability commonly results from such global insults to the brain. Thus, the specificity inherent in coordinated impairment in a set of competencies that ‘travel together’ (at least to some degree) suggests either that autism might represent a convergent path of decompensation in the setting of these interacting liabilities, or that the various symptom clusters arise secondarily from a parsimonious biological insult.
Sub-clinical characteristics of the autistic syndrome aggregate in the unaffected first degree relatives of individuals with ASD, and have the same factor structure as autism
When standardized quantitative assessment methods are implemented in the study of families affected by ASD, sub-clinical autistic symptoms and traits are observed among first degree relatives (including siblings and parents) with a frequency an order of magnitude higher than observed in the general population (Constantino, Zhang, Frazier, Abbacchi, & Law, Citation2010; Frazier et al., Citation2015; Lyall et al., Citation2014; Page et al., Citation2016), and these traits exhibit the same symptom structure as autism itself. Furthermore, in very large genetic-epidemiologic studies it has been confirmed that genetic susceptibilities to these sub-clinical syndromes observed in the general population exhibit near-complete overlap with the genetic underpinnings of clinical-level autistic syndromes (Robinson et al., Citation2011). This strongly suggests that variation in social communication in all people is related to the biological underpinnings of ASD itself (see Constantino, Citation2011; Constantino & Todd, Citation2003). A recent extension of this principle is that rare deleterious single-gene mutations can be superimposed upon additive (polygenic) ASD risks that run in a family to compound the severity of affectation in an individual and ‘push’ him/her across a threshold for clinical-level affectation (Wellner et al. Citation2017; Yuen et al., Citation2015). Conversely, the level of familial ‘background’ genetic risk for ASD–which may soon be specifiable on the basis of molecular genetic profiles (so-called ‘polygenic’ risk scores), but can also be phenotypically indexed by the level of sub-clinical affectation of parents—may play a major role in ‘variability-of-expression’ of a rare mutation. For example, Moreno-De-Luca et al. (Citation2015) showed that the effect of a germline (de novo) 16p11.2 deletion on a child was far from absolute, rather reflected a predictable ‘shift’ (on the order of 2SD) against what would be expected on the basis of the average trait burden of his/her parents. Thus, children with de novo deletions of 16p11.2 whose parents fell within the upper quartile of the general population distribution for autistic-like traits (i.e. still entirely within normal limits) were much more likely to be affected by clinical-level ASD than deletion-affected counterparts whose parents fell within the lower quartile of the general population distribution for these traits. The notion of a highly-penetrant mutation inducing a predictable shift against a measurable genetic background (rather than an absolute effect) represents an important new way of thinking about the effect of disease variants in clinical populations (Finucane, Challman, Martin, & Ledbetter, Citation2016). The fact that background variation within the range of normality can exert such profound effects on the phenotypic expression of a superimposed ‘deleterious’ mutation suggests the opportunity for disease prediction and possibly the discovery of disease modifiers that can be simulated in novel therapies.
Early neurodevelopmental impairments that (a) are non-specific to ASD and (b) do not aggregate in the unaffected relatives of ASD probands nevertheless account for ASD recurrence in multiplex families
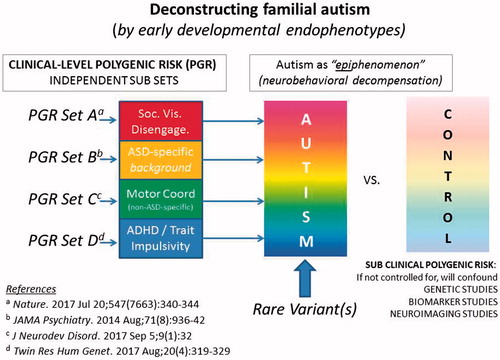
In a recent contemporaneous analysis of symptom burden of non-ASD-specific neurodevelopmental traits and ASD recurrence in the siblings of affected probands (Mous, Jiang, Agrawal, & Constantino, Citation2017) it was observed that Attention Deficit Hyperactivity Disorder (ADHD) symptoms and motor coordination impairment jointly accounted for a large share of the variance (over 50%) in both categorical ASD recurrence and quantitative trait severity of recurrent autistic syndromes in families affected by autism. These findings in a clinical family cohort confirmed observations from general population studies (Lichtenstein, Carlstrom, Rastam, Gillberg, & Anckarsater, Citation2010), indicating substantial genetic overlap between the characterizing symptoms of autism and those of both inattention/hyperactivity and developmental coordination disorder. Impairments in attention and motor co-ordination are strongly genetically-influenced, but not specific to autism, and may relate to autism by amplifying the effect of ‘ASD-specific’ factors early in development, as presented in the graphic abstract for this article. Although such traits are sometimes referred to as ‘co-morbidities’, these and other research findings (e.g. Floris et al., Citation2016) have suggested that they may be part-and-parcel of the autistic syndrome. Rather than being discarded (or mathematically regressed away in research studies—see Constantino & Frazier, Citation2013) it may be extremely important to include characterization of these traits as key components of autistic syndromes, of comparable relevance to traits that have traditionally been viewed as more ‘specific’ to the measurement of autistic severity.
This particular role of non-ASD-specific risk factors could actually explain elements of ‘missing heritability’ for autism, and may help resolve apparent discrepancies between genetic epidemiologic (population-based) and molecular genetic (case-control) studies in estimating the extent of genetic overlap between autism and other neuropsychiatric impairments (such as ADHD). Estimations of molecular genetic overlap have historically depended upon case-control study designs that have not accounted for sub-clinical cross-trait aggregation among controls. Beyond the issue of genetic overlap, non-ASD-specific factors have important implications for the phenomenology of infant development and the clarification of early liabilities that might contribute to the development of autism as depicted schematically in . In recent studies of the early development of ASD among high-risk infant siblings of children with ASD, trajectories of delayed motor development have been shown to predict later ASD diagnosis (Estes et al., Citation2015; Landa, Gross, Stuart, & Bauman, Citation2012; Ozonoff et al., Citation2014). Similarly, early abnormalities in visual social engagement predict ASD among high-risk infant siblings (see below). Given the high prevalence of motor impairments and attention/hyperactivity problems in ASD, either or both might serve as important early targets for intervention, both with respect to reducing non-specific risk and to reducing so-called “comorbidity” in affected children. For example, studies have shown that developmental therapies targeted to the acquisition of motor skills may have broad-ranging positive effects on the development of executive functioning, reduce symptoms of inattention/hyperactivity, and improve social behaviour (Diamond & Lee, Citation2011; Kamp, Sperlich, & Holmberg, Citation2014).
Figure 1. A developmental deconstruction of familial autism. Under this model, attempts to fractionate autism according to the traditional symptom triad (rather than by the orthogonal axes of neuropsychiatric liability indexed by A, B, C, and D) erode statistical power to link the autistic syndrome with genetic variants and other biological markers.
Impairments in social visual engagement in infant males appear ‘necessary but not sufficient’ to predict the development of autism
Long before infants walk or talk, they explore the world by looking, normally giving preferential attention to social stimuli including faces, face-like stimuli, and biological motion. This capacity—social visual engagement—shapes typical infant development from birth, and is pathognomonically impaired in children affected by autism. Recently (Constantino et al., Citation2017), using methodologies that had identified predictors of risk for autism identifiable between the ages of 2–6 months (Jones & Klin, Citation2013), we showed that variation in viewing of social scenes—including levels of preferential attention and the timing, direction, and targeting of individual eye movements—is under stringent genetic control in infants in the general population, with effects appreciable at a moment-to-moment level of analysis (tens of milliseconds time scale) and directly traceable to the active seeking of social information. Moreover, the measures that were most highly heritable, preferential attention to eye and mouth regions of the face, were those that most clearly distinguished typically-developing children from those with autism. These results implicated deficits in social visual engagement as a neurodevelopmental endophenotype—not only for autism, but for population-wide variation in social-information-seeking. While most infants who went on to be affected by autism exhibited marked deficiencies in this highly-heritable developmental competency, a small proportion of infants in the general population with the same deficiency nevertheless developed typically, suggesting that such deficits may be necessary but not sufficient to cause clinical autistic syndromes. Thus, just as occurs for highly-penetrant mutations such as the 16p11.2 deletion—these inherited deficits may superimpose upon ASD-specific background genetic liabilities in the development of autism. This would explain why they are even more powerful predictors of autism in the setting of familial risk (i.e. among infant siblings of ASD-affected probands) than they are in the context of unselected screening in the general population. Other recently-identified predictors of ASD within infant sibling research designs—including brain imaging signatures which are promising biomarkers (see Hazlett et al., Citation2017)—may similarly predict autism more faithfully in the context of known familial background risk (i.e. when a relative or an older sibling is affected) than among all infants in the population.
The known molecular genetic causes of autistic syndromes are markedly heterogeneous, and most cases of autism are caused by combinations of genetic liabilities, each of which may be traceable to effects on developmental endophenotypes
The above considerations consequently bring us back to a more sophisticated understanding of how molecular genetic variants might relate to the development of ASD and other complex neuropsychiatric syndromes. It is now known that autism is associated with a diversity of rare and common genetic variants involving hundreds (Sanders et al., Citation2015), if not thousands (Boyle, Li, & Pritchard, Citation2017) of different genes. It is important to note that most of the molecular genetic variants that have been statistically associated with autism are autosomal (i.e. not on sex chromosomes), indicating that the universally-observed sex ratio in autism arises from mechanisms other than sex linkage. The inference from a vast amount of family recurrence and molecular genetic data that has accumulated to date is that most—but not all—females are relatively ‘protected’ from most—but not all—inherited forms of autism (Constantino, Citation2016; Constantino et al., Citation2010; Jacquemont et al., Citation2014; Palmer et al., Citation2017). This suggests at least one highly parsimonious resiliency factor that operates against numerous genetic liabilities to ASD.
In the same manner that a protective factor such as sex may mitigate a diversity of pathways to ASD risk, targeted biological therapies may someday prove capable of modifying syndrome severity across a multitude of genetic pathways (as has been the case for the effect of stimulant medication in ADHD syndromes, which are as genetically diverse as ASD). This would be especially true if the genetic causes of autism converge upon a discrete set of fundamental variations in early social and behavioural development. The specification of developmental endophenotypes through which independent or partially-overlapping sets of genetic liability operate, stands to contribute to a reconceptualization of autism (depicted in the graphic abstract), as a convergent pathway of developmental decompensation (a ‘behavioural version’ of neurodegeneration) engendered by a critical accumulation of inherited developmental liabilities—each component of which constitutes a potential target for a next generation of early preventive intervention.
Developmental deconstruction is a ‘flip side’ of pleiotropy
It has long been known that specific genetic variants associated with autism are also associated with epilepsy, intellectual disability, ADHD, or schizophrenia across different families. A reconceptualization of autism via developmental deconstruction—in which some of its contributing components are specific to autism, but others are non-specific—provides a direct way of accounting for pleiotropy in genetic studies. Recently, analyses of large twin and family data sets in which general psychiatric symptoms were uniformly ascertained has revealed a remarkably homologous ‘phenotypic pleiotropy’ of a small number of highly-inherited, highly-influential symptom clusters, accounting for causation across diverse syndromes of distress psychopathology, including disruptive behaviour disorders, anxiety disorders, depressive syndromes, and substance use disorders (Lahey, Krueger, Rathouz, Waldman, & Zaid, Citation2017). One of the most influential of these general psychopathology determinants is a domain empirically-derivable through factor analysis of comprehensive data collections and perhaps best captured by the descriptor ‘emotion dysregulation’—which encompasses a parsimonious and highly pleiotropic predictor of all of these disorders. Other lines of investigation have identified additional independent developmental liabilities (see Beauchaine & Constantino Citation2017) that represent other parsimonious ‘building blocks’ of psychopathology that offer the prospect of a new taxonomy for neuropsychiatric impairment predicated on advanced understanding of its developmental causal structure, rather than on manifestations of symptoms after a syndrome of impairment or decompensation emerges.
Conclusions
Stepping back and taking stock of the accumulated results of a generation of family studies reveals that autism may be predicted from an array of neurobehavioural susceptibilities, many appreciable before the syndrome is diagnosed, and each potentially traceable to specific sets of genetic influence, as depicted in . Some of these liabilities are not necessarily specific to ASD. Background ASD susceptibilities that are inherited but non-specific (‘BASINS’, see Mous et al., Citation2017) may contribute to additive genetic liability, could account for a significant share of the ‘missing heritability’ of autism, and may be responsible for so-called ‘co-morbidities’, which are inappropriately named if they actually contribute to (or exacerbate) the severity of autism itself. Future biomarker and molecular genetic studies should strongly consider cross-trait ascertainment, particularly among controls, as a means of capturing ‘missing heritability’ that might be tagged to biological signatures and/or genetic factors that are not necessarily specific to ASD but, nevertheless, (paradoxically) contribute to its severity. Early interventions capable of improving or resolving early non-specific developmental liabilities that may contribute risk for ASD can be directly tested for their ability to ameliorate ASD severity, particularly among infants known to be at elevated risk.
In this article the implications of conceptualizing autism as a specific syndrome of neurobehavioural degeneration have been considered, predicated on the possibility that the syndrome is principally engendered by a critical co-aggregation of more fundamental contributing, measurable neuropsychiatric liabilities that conspire to confer accumulated susceptibility to the disorder within an individual, the phenotypic expression of which—importantly—can be moderated by sex. Linking genetic variants to the respective underlying traits that contribute to the development of autistic syndromes—rather than to a diagnosis of ‘autism’—may be more productive in devising personalized approaches to developmental intervention, especially if autism represents an epiphenomenon of earlier-interacting susceptibilities.
Disclosure statement
Dr Constantino receives royalties from Western Psychological Services for the commercial distribution of the Social Responsiveness Scale-2.
Additional information
Funding
References
- Beauchaine, T. P., & Constantino, J. N. (2017). Redefining the endophenotype concept to accommodate transdiagnostic vulnerabilities and etiological complexity. Biomarkers in Medicine. Advance online publication. doi:10.2217/bmm-2017-0002
- Boyle, E. A., Li, Y. I., & Pritchard, J. K. (2017). An expanded view of complex traits: From polygenic to omnigenic. Cell, 169, 1177–1186. doi:10.1016/j.cell.2017.05.038
- Constantino, J. N. (2011). The quantitative nature of autistic social impairment. Pediatric Research, 69, 55R–62R. doi:10.1203/PDR.0b013e318212ec6e
- Constantino, J. N. (2014). Recurrence rates in autism spectrum disorders. JAMA, 312, 1154–1155. doi:10.1001/jama.2014.9841
- Constantino, J. N. (2016). Data from the baby siblings research consortium confirm and specify the nature of the female protective effect in autism: A commentary on Messinger et al. Molecular Autism, 7, 32. doi:10.1186/s13229-016-0092-x
- Constantino, J. N., & Frazier, T. W. (2013). Commentary: The observed association between autistic severity measured by the social responsiveness scale (SRS) and general psychopathology-a response to Hus et al. (2013). J Child Psychol Psychiatry, 54, 695–697. doi:10.1111/jcpp.12064
- Constantino, J. N., & Todd, R. D. (2003). Autistic traits in the general population: A twin study. Archives of General Psychiatry, 60, 524–530. doi:10.1001/archpsyc.60.5.524
- Constantino, J. N., Kennon-McGill, S., Weichselbaum, C., Marrus, N., Haider, A., Glowinski, A. L., … Jones, W. (2017). Infant viewing of social scenes is under genetic control and is atypical in autism. Nature, 547, 340–344. doi:10.1038/nature22999
- Constantino, J. N., Lajonchere, C., Lutz, M., Gray, T., Abbacchi, A., McKenna, K., … Todd, R. D. (2006). Autistic social impairment in the siblings of children with pervasive developmental disorders. American Journal of Psychiatry, 163, 264–296. doi:10.1176/appi.ajp.163.2.294
- Constantino, J. N., Todorov, A., Hilton, C., Law, P., Zhang, Y., Molloy, E., …Geschwind, D. (2013). Autism recurrence in half siblings: Strong support for genetic mechanisms of transmission in ASD. Mol Psychiatry, 18, 137–138. doi:10.1038/mp.2012.9
- Constantino, J. N., Zhang, Y., Frazier, T., Abbacchi, A. M., & Law, P. (2010). Sibling recurrence and the genetic epidemiology of autism. The American Journal of Psychiatry, 167, 1349–1356. doi:10.1176/appi.ajp.2010.09101470
- Diamond, A., & Lee, K. (2011). Interventions shown to aid executive function development in children 4–12 years old. Science (New York, N.Y.), 333, 959–964. doi:10.1126/science.1204529
- Estes, A., Zwaigenbaum, L., Gu, H., St. John, T., Paterson, S., Elison, J. T. … IBIS Network. (2015). Behavioral, cognitive, and adaptive development in infants with autism spectrum disorder in the first 2 years of life. Journal of Neurodevelopmental Disorders, 7, 24. doi:10.1186/s11689-015-9117-6
- Finucane, B., Challman, T. D., Martin, C. L., & Ledbetter, D. H. (2016). Shift happens: Family background influences clinical variability in genetic neurodevelopmental disorders. Genetics in Medicine, 18, 302–304. doi:10.1038/gim.2015.92
- Floris, D. L., Barber, A. D., Nebel, M. B., Martinelli, M., Lai, M. C., Crocetti, D., … Mostofsky, S. H. (2016). Atypical lateralization of motor circuit functional connectivity in children with autism is associated with motor deficits. Mol Autism, 7, 35. doi:10.1186/s13229-016-0096-6
- Frazier, T. W., Ratliff, K. R., Gruber, C., Zhang, Y., & Law, P. A. (2014). Confirmatory factor analytic structure and measurement invariance of quantitative autistic traits measured by the social responsiveness scale-2. Autism, 18, 31–44. doi:10.1177/1362361313500382
- Frazier, T. W., Youngstrom, E. A., Hardan, A. Y., Georgiades, S., Constantino, J. N., & Eng, C. (2015). Quantitative autism symptom patterns recapitulate differential mechanisms of genetic transmission in single and multiple incidence families. Molecular Autism, 6, 58. doi:10.1186/s13229-015-0050-z
- Gaugler, T., Klei, L., Sanders, S. J., Bodea, C. A., Goldberg, A. P., Lee, A. B., … Buxbaum, J. D. (2014). Most genetic risk for autism resides with common variation. Nature Genetics, 46, 881–885. doi:10.1038/ng.3039
- Hazlett, H. C., Gu, H., Munsell, B. C., Kim, S. H., Styner, M., Wolff, J. J. … For the IBIS Network. (2017). Early brain development in infants at high risk for autism spectrum disorder. Nature, 542, 348–351. doi:10.1038/nature21369
- Hilton, C. L., Zhang, Y., Whilte, M. R., Klohr, C. L., & Constantino, J. (2012). Motor impairment in sibling pairs concordant and discordant for autism spectrum disorders. Autism: The International Journal of Research and Practice, 16, 430–441. doi:10.1177/1362361311423018
- Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., … Eichler, E. E. (2014). A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. American Journal of Human Genetics, 94, 415–425. doi:10.1016/j.ajhg.2014.02.001
- Jones, W., & Klin, A. (2013). Attention to eyes is present but in decline in 2-6-month-old infants later diagnosed with autism. Nature, 504, 427–431. doi:10.1038/nature12715
- Kamp, C. F., Sperlich, B., & Holmberg, H.-C. (2014). Exercise reduces the symptoms of attention-deficit/hyperactivity disorder and improves social behaviour, motor skills, strength and neuropsychological parameters. Acta Paediatrica, 103, 709–714. doi:10.1111/apa.12628
- Lahey, B. B., Krueger, R. F., Rathouz, P. J., Waldman, I. D., & Zaid, D. H. (2017). A hierarchical causal taxonomy of psychopathology across the life span. Psychological Bulletin, 143, 142–186. doi:10.1037/bul0000069
- Landa, R. J., Gross, A. L., Stuart, E. A., & Bauman, M. (2012). Latent class analysis of early developmental trajectory in baby siblings of children with autism. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 53, 986–996. doi:10.1111/j.1469-7610.2012.02558.x
- Lichtenstein, P., Carlstrom, E., Rastam, M., Gillberg, C., & Anckarsater, H. (2010). The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. American Journal of Psychiatry, 167, 1357–1363. doi:10.1176/appi.ajp.2010.10020223
- Lyall, K., Constantino, J. N., Weisskopf, M. G., Roberts, A. L., Ascherio, A., & Santangelo, S. L. (2014). Parental social responsiveness and risk of autism spectrum disorder in offspring. JAMA Psychiatry, 71, 936–942. doi:10.1001/jamapsychiatry.2014.476
- Mahajan, R., Dirlikov, B., Crocetti, D., & Mostofsky, S. H. (2016). Motor circuit anatomy in children with autism spectrum disorder with or without attention deficit hyperactivity disorder. Autism Research, 9, 67–81. doi:10.1002/aur.1497
- Moreno-De-Luca, A., Evans, D. W., Boomer, K. B., Hanson, E., Bernier, R., Goin-Kochel, R. P., … Ledbetter, D. H. (2015). The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry, 72, 119–126. doi:10.1001/jamapsychiatry.2014.2147
- Moruzzi, S., Ogliari, A., Ronald, A., Happe, F., & Battaglia, M. (2011). The nature of covariation between autistic traits and clumsiness: a twin study in a general population sample. Journal of Autism and Developmental Disorders, 41, 1665–1674. doi:10.1007/s10803-011-1199-8
- Mous, S. E., Jiang, A., Agrawal, A., & Constantino, J. N. (2017). Attention and motor deficits index non-specific background liabilities that predict autism recurrence in siblings. Journal of Neurodevelopmental Disorders, 9, 32. doi:10.1186/s11689-017-9212-y
- Ozonoff, S., Young, G. S., Belding, A., Hill, M., Hill, A., Hutman, T., … Iosif, A.-M. (2014). The broader autism phenotype in infancy: When does it emerge? Journal of the American Academy of Child and Adolescent Psychiatry, 53, 398–407.e2. doi:10.1016/j.jaac.2013.12.020
- Page, J., Constantino, J. N., Zambrana, K., Martin, E., Tunc, I., Zhang, Y., … Messinger, D. (2016). Quantitative autistic trait measurements index background genetic risk for ASD in Hispanic families. Molecular Autism, 7, 39. doi:10.1186/s13229-016-0100-1
- Palmer, N., Beam, A., Agniel, D., Eran, A., Manrai, A., Spettell, C., … Kohane, I. (2017). Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatrics, 171, 1107–1112.
- Robinson, E., Koenen, K. C., McCormick, M. C., Munir, K., Hallett, V., Happé, F., … Ronald, A. (2011). Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5%, 2.5% and 1%). Archives of General Psychiatry, 68, 1113–1121. doi:10.1001/archgenpsychiatry.2011.119
- Ronald, A., Larsson, H., Anckarsater, H., & Lichtenstein, P. (2011). A twin study of autism symptoms in Sweden. Molecular Psychiatry, 16, 1039–1047. doi:10.1038/mp.2010.82
- Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., … State, M. W. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron, 87, 1215–1233. doi:10.1016/j.neuron.2015.09.016
- Sandin, S., Lichtenstein, P., Kuja-Halkola, R., Hultman, C., Larsson, H., & Reichenberg, A. (2017). The heritability of autism spectrum disorder. JAMA, 318, 1182–1184. doi:10.1001/jama.2017.12141
- Wellner, D. J., Wigdor, E. M., Ripke, S., Walters, R. K., Kosmicki, J. A., Grove, J., … Robinson, E. B. (2017). Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nature Genetics, 49, 978–985. doi:10.1038/ng.3863
- Yuen, R. K., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., … Scherer, S. W. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nature Medicine, 21, 185–191. doi:10.1038/nm.3792