
Abstract
Speciation analysis of selenium metabolites in urine was performed using hyphenation of mixed ion-pair reversed-phase chromatography and inductively coupled plasma mass spectrometry. A chromatographic separation was performed with a C8 stationary phase and a mobile phase containing 2.5 mmol L−1 sodium butane-1-sulfonate, 8 mmol L−1 tetramethylammonium hydroxide, 4 mmol L−1 malonic acid, and 0.05% methanol, pH 3.0. Under this condition, the selenium species selenite, selenate, selenomethionine, selenoethionine, selenourea, trimethylselenonium, and Se-methylselenocysteine were successfully separated. Selenium determination was carried out by monitoring 80Se. The internal standard Ge was added into the mobile phase. The calibration was linear at least up to 100 μg L−1 Se for all species. The limit of detection was 0.4 μg L−1 Se. When higher (1400 W) ICP power was applied, the calibration based solely on selenate provided accurate results for all species. Stability tests revealed extremely short stability of selenite. After collection, the urine samples should be acidified to pH 3.0, stored below –5 °C, and analyzed before 12 h after collection. The method was used for the short-time (single capsule, one day observation, 5 persons) monitoring of the excretion of selenium after ingestion of Se-containing dietary supplements. When a formulation containing selenate was applied, almost 50% of Se was excreted during 24 h (mostly in the unchanged form of selenate) and the plasma Se remained unaltered. In the case of organically bound Se, only 8% of Se was lost by urination during 24 h as selenite, Se-methylselenocysteine, selenomethionine, and unknown species. Subsequently, a statistically significant increase of plasma Se was observed.
Keywords:
Introduction
Determination of selenium in environmental and biological systems is important, mainly due to the essentiality of this element and its coeval toxicity at higher concentration levels. This element takes part in several biological functions, such as antioxidative action, regulation of thyroid hormone metabolism, and cell growth. In the human body, selenium is also incorporated to the active site of selenoproteins, such as glutathione peroxidase (GPx) in organisms, and the prevention activity of some selenium-containing compounds against the development of diverse cancer types has been demonstrated.[Citation1] Trace amounts of Se are needed in the diet, but large amounts are poisonous (liver, skeletal, and cardiac muscle damage).[Citation2] Selenium deficiency leads to severe cardiomyopathy and increases the risk of carcinogenesis, the development of atherosclerosis, thyroid disorders, and immunodeficiency.
Although selenium is available in our food, its content in food varies widely among different world regions depending on the selenium content in the soil.[Citation3] Selenium deficiency should be considered in vegetarians, in malnourished people, in pregnant women, and in patients with diarrhea or chronic pancreatitis. Particularly, significant selenium deficiency occurs in patients in intensive care, for whom supplementation is recommended. To compensate for selenium deficiency, dietary supplements are being administered. Selenium is also involved in some parenteral nutrition formulae.
Selenium adsorbed in the human body is excreted mainly by urination,[Citation4] and consequently, the chemical speciation of selenium in urine carries important information about the state of selenium in the organism. Recently, the inductively coupled plasma mass spectrometry (ICP/MS) as a trace element specific detection method with superior analytical capabilities combined with a proper separation technique was successfully used. Several procedures based on high-performance liquid chromatography (HPLC) were reported for selenium speciation analysis. Linking HPLC and ICP/MS has received much attention due to the easy sample preparation and simple interface. Moreover, the use of HPLC enables the separation of many selenium species in a single run. Recently analyzed ones are the following:
Selenite (Se(IV)), selenate (Se(VI)), methylselenonic acid, selenourea (SeUr), trimethylselenonium ion (TMSe), selenocystine (SeCys), Se-methylselenocysteine (Se-MetSeCys), selenomethionine (SeMet), Se-methyselenomethionine (Se-MetSeMet), selenoadenosylmethionine, selenoethionine (SeEt), selenocystamine (SeCM), selenogammaaminoburyric acid, methylselenoglutathione, and selenosugars. Several modes of HPLC can be used; however, reversed-phase chromatography (RPC) is used most frequently. Basic papers in this field were published by Zheng [Citation5] who used the mixed ion-pair reagent butane-1-sulfonate and tetramethylammonium hydroxide for successful separation of the anionic, cationic, and neutral Se species. In addition, mobile phases based on perfluorinated carboxylic acids were used too. Ion exchange chromatography and gel permeation chromatography can also be advantageously used. Moreover, an electroseparation method as capillary electrochromatography capillary electrophoresis (CE) was recently used for the separation of Se species. Basic outline of separation method used for the speciation analysis of selenium is given in Table
Table 1. Short overview of separation techniques used for the speciation analysis of selenium in urine.
The calibration techniques used in these studies were of various levels. Some papers described mere separation and identification of species without quantification. However, the very sophisticated technique of isotope dilution (ID) was used. Ohta [Citation16] used in-laboratory synthesized 76Se, 77Se, 78Se, 80Se, and 82Se labeled species for sample spiking. Such a method exhibits maximum accuracy; however, it is very laborious. Simpler ID was used by Jeong [Citation13] who worked with post-column mixing of the 78Se standard. In most other studies, the external calibration with multispecies calibration solutions was used. Although this procedure is very common and represents a standard approach, its weakness is that some standards may not be available and the quantification of unidentified species can be questionable.
The aim of the present work was to optimize and validate an effective and simple HPLC-ICP/MS method for the quantification of basic selenium species in human urine. Above all, our goal was to simplify the calibration procedure as much as possible, i.e., to restrict this process to the use of a single solution of a single species. Such an attitude facilitates easy quantification of all species regardless of whether the composition is unknown and/or the standard is unavailable. We verified the stability of the selenium species and determined the optimal condition for sample storage. To illustrate the capability of this method for real samples, the short study monitoring the excretion of Se by urine and its transfer to the serum and binding to main serum selenoprotein GPx after the ingestion of selenium-containing dietary supplements was performed.
Methodology
Instruments
ICP/MS measurements were performed with an ELAN DRC-e (Perkin-Elmer Concord, Canada) equipped with a concentric PTFE concentric nebulizer, a cyclonic glass spray chamber, and a high-efficiency quartz torch. The chromatographic system consisted of a high-pressure pump Series 200 (Perkin-Elmer, Shelton, USA), a degasser, a Rheodyne 9010 sampling valve (IDEX Health & Science LLC, Rohnert Park, CA, USA) equipped with a PEEK sample loop, and an analytical column PR-C8 (Purospher STAR-C8e, 250 4.6 mm, 5 μm, Merck, Darmstadt, Germany).
Other equipment included a PH 03 pH meter (Labio a.s., Prague, Czech Republic), Minipuls 3 (Gilson, Middleton, WI, USA) peristaltic pump, CAT S20 shaker (Ingenieurbüro M. Ziperer, Staufen, Germany), and 2–16 K model centrifuge (Sigma, Osterode am Harz, Germany).
Reagents
All commercial chemicals were used without further purification. The standards of the selenium species Se(VI) (min. 98%), Se(IV) (min. 99%), SeMet (min. 99%), Se-MetSeCys (min. 95%), and SeUr (min. 99%) were obtained from Sigma Aldrich (Steinheim, Germany). SeEt (min. 99%) was purchased from Santa Cruz Biotechnology (Dallas, USA). Trimethylselenium iodide was synthesized at the Department of Organic Chemistry of UCT, Prague, according to the procedure used earlier. [Citation20] Stock solutions containing 10 mg L−1 Se were prepared by dissolving individual species in water. Internal standard (IS) solutions (Ge, Rh, In) and calibration solutions for the total selenium concentration determination were prepared by diluting stock solutions containing 1000 mg L−1 of the specific element (CertiPur, Merck).
The mobile phase for the chromatographic separation consisted of 2.5 mmol L−1 sodium butane-1-sulfonate, 8 mmol L−1 tetramethylammonium hydroxide (all Sigma Alldrich), 4 mmol L−1 malonic acid, 0.05% (v/v) methanol, and 0.01% sodium azide. The pH of the solution was adjusted to 3.0 by adding small portions of hydrochloric acid (Suprapur, Merck). The mobile phase was spiked by the internal standard stock solutions to make a final concentration of 100 μg L−1.
Catalytic concentration of glutathione peroxidase (GPx) was determined using a reagent kit Ransel (Randox, Crumlin, UK).
Other reagents used were nitric acid (Suprapur, Merck), TritonTM X100 (Sigma Alldrich), and NaCl (Suprapur, Merck). Distilled and demineralized water (conductivity < 0.1 μS/cm) (Millipore, Bedford, MA, USA) was used to prepare all solutions.
Samples
Urine samples used for the method optimization were collected in PE bottles and were spiked by 50 μg L−1 Se of each species. Original concentration of individual specie in non-spiked samples were the following: Se(VI) 0–5.0 μg L−1 Se, Se(IV) 2–25 μg L−1 Se, Se-MetSeCys 0–8 μg L−1 Se, TMSe 0–5.0 μg L−1 Se, and SeMet 0–3.0 μg L−1 Se. Other selenium compounds (SeUr, SeEt) were below detection limits in all cases.
Urine samples serving for the stability testing were stored in PP tubes. A total of 108 samples were prepared: 6 levels of the storing duration (0, 3, 7, 14, 35, and 75 days) × 3 levels of temperature (+25, –5, and –35 °C) × 2 levels of pH value (neutral and 3) × 3 repetitions. All samples were spiked by a mixture of Se(VI), Se(IV), Se-MetSeCys, TMSe, SeUr, SeEt, and SeMet, for which concentration of each species corresponded to 50 μg L−1 Se. A similar short-time test was carried out with samples containing only Se(IV) species with storing times of 0, 3, 6, 9, 12, 18, 24, and 48 h under the same conditions described above.
When the sorption of selenium from dietary supplements (DS) was investigated, urine samples were taken from five healthy volunteers (2 men and 3 women from 22 to 57 years old). The amount of DS containing 100 μg Se was administered, and the sampling times were just before administering (time 0) and 1, 3, 6, 9, 12, and 24 h after administering the DS. The urine samples were collected in PE bottles, and the volume of urine was measured to evaluate the total excreted selenium content. Experiments with two various DS were carried out with one-month delay.
Blood samples were taken by the venipuncture of probands. The study was approved by the local ethical committee and they gave their informed consent prior to entering the study. After coagulation of the blood and centrifugation (3000 rpm, 10 min), the portions of serum for the selenium determination were stored at 4 °C and the analysis was performed 12 h after collection. The portions of serum serving for the GPx determination were stored at –80 °C until analysis. The first blood sampling was carried out just before administering the DS, and the next sampling was completed 24 h after ingestion.
Two dietary supplements containing Se were used in our study. DS 1 was made by Nature’s Bounty (USA) and was declared as “Selenium Yeast.” DS 2 was produced by Pharma Nord (Denmark). This preparation was described as “Seleno Precise Yeast” containing SeMet and more than 30 others Se species.
Procedures
Speciation analysis of selenium
Samples (one piece of tablet) of DS were crushed in an agate mortar and leached by 50 ml of 0.3 mol L−1 hydrochloric acid. The mixture was shaken for 3 h and then centrifuged at 10,000 rpm for 5 min. Before analysis, urine samples were filtered through a 0.45-μm nylon filter (Whatman, United Kingdom). Calibration solutions with selenium concentrations of 100 μg L−1 were prepared by the dilution of a stock solution of Se(VI). The calibration solution was analyzed before the first urine sample and repeatedly after each three urine samples. Calibration curve was constructed as dependence of peak area calculated from relative signal (i.e., the signal of analyte divided by the signal of the internal standard, for selection of proper internal standard see Discussion section). The sample volume was 50 μl, and the mobile phase flow was 1 mL min−1. ICP/MS measurements were performed under the conditions summarized in Table . Selenium determination was carried out by monitoring 80Se isotope which provides the highest sensitivity. Spectral interferences due to forming of Ar2+ ion were fully suppressed using methane as reaction gas. For DRC optimization, see our previous paper.[Citation8]
Table 2. Operation conditions for ICP/MS and HPLC.
Determination of total selenium concentration in serum
A 1 ml of 1% Triton X-100 solution was pipetted into a 10-ml calibrated flask; a 1-ml plasma sample was added and diluted with approximately 5 ml of water and agitated; 2 ml of 0.14 mol L−1 HNO3 and 0.2 ml of a solution of 5 mg L−1 Ge (internal standard) were added and the whole solution was diluted to volume. Calibration solutions containing 0 and 50 μg L−1 Se were prepared by diluting the Se stock solution. They contained the same concentration of Triton X-100, HNO3, and Ge. Moreover, calibration solutions were spiked by a NaCl solution (final concentration: 0.9 g L−1) in order to minimize non-spectral interferences during ICP/MS measurement ). [Citation25] Details on the ICP/MS conditions are given in Table .
Catalytic concentration of glutathione peroxidase was determined using a reagent kit Ransel (Randox, Crumlin, UK). The method is based on conversion of reduced glutathione to its oxidized form. It is then reduced by glutathione reductase with a simultaneous oxidation of NADPH to NADP+; the reaction is followed spectrophotometrically as a decrease of absorbance at wavelength of 340 nm.
Results and discussion
Method optimization and validation
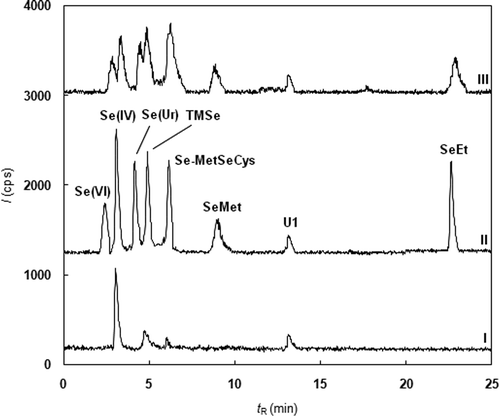
The aim of our work was to develop a robust method for the routine quantification of important selenium metabolites, such as Se(VI), Se(IV), Se-MetSeCys, SeMet, SeEt, SeUr, and TMSe. The chosen separation method of the selenium species was based on ion-pair reversed-phase chromatography originally described by Zheng [Citation5], who separated Se(VI), Se(IV), SeCys, SeUr, TMSe, SeMet, SeCM, and SeEt. The composition of the mobile phase was only slightly modified. The original pH value of the mobile phase (5.5) did not provide good separation of the inorganic selenium species. The decrease in pH to 3.0 provided the entire separation of all the tested species; see Figure for the chromatograms of spiked and unspiked human urine samples. In addition to the known species, one unidentified species (U1) was observed.
Figure 1. Chromatogram of selenium species in urine (I-blank urine, II-urine spiked by 50 µg⋅L–1 Se of Se(IV), tR = 2.57 min; Se(VI), tR = 2.98 min; SeUr, tR = 3.85 min; TMSe, tR = 4.12 min; Se-MetSeCys, tR = 5.85; SeMet, tR = 8.52 min; SeEt, tR = 22.25 min; III-decrease in separation efficiency caused by the post-column mixing of the eluent with IS solution).
In order to evolve a precise and simple calibration procedure, several basic problems should be solved:
| • | the selection of proper IS and the method of mixing with the sample, | ||||
| • | minimization of the number of calibration solutions (i.e., checking the linearity testing), | ||||
| • | minimization of the number of calibrants (i.e., checking the sensitivity of the determination of an individual species). | ||||
The application of IS is necessary for suppressing some undesirable effects (matrix effect, long-term plasma instability, etc.). There are two ways to add the IS solution to the sample in chromatographic separation. First, post-column mixing of the column eluent with the IS solution delivered by a peristaltic pump was tested. Unfortunately, mixing of the two flows by a T piece caused peak broadening and, consequently, the rapid decrease in separation efficiency, see Figure . Due to this reason, the internal standard at a concentration level 100 μg L−1 was added directly into the mobile phase. Six isotopes, 74Ge, 73Ge, 72Ge, 103Rh, 115In, and 38Ar (present in plasma), were tested. Samples of unspiked urine and of urine spiked by Se(VI) at 50 μg L−1 (n = 5) were analyzed, and a water solution containing 100 μg L−1 Se in the Se(VI) form was used for the calibration. The recovery was evaluated by Student’s t-test (p = 0.05), and only the 74Ge (recovery 101%) and 73Ge (recovery 102%) isotopes provided accurate results, see Table . Finally, 74Ge was chosen as the internal standard because the less abundant 73Ge had low signal.
Table 3. Accuracy of the determination of Se(VI) for different IS (added 50 μg L−1 Se).
The linearity of the calibration was tested individually for each selenium species. Water solutions containing 0, 10, 20, 50, and 100 μg L−1 Se (n = 2) were analyzed, and Mandel’s test (p = 0.05) [Citation22] proved the linearity in the case of all species. Consequently, the mixed calibration solution containing all seven analyzed species at a level corresponding to 100 μg L−1 Se was used for the analyses of unspiked and spiked (all Se species, 50 μg L−1 Se in each) urine samples (n = 5). Student’s t-test (p = 0.05) confirmed the accuracy of the results for all tested species (recovery 102–107%); see Table .
Table 4. Accuracy of the selenium species determination using multispecies calibration solutions (added 50 μg L−1 Se).
Although the above-described calibration led to satisfactory results, the abbreviated calibration based on a solution of a single Se species was also tested. A water solution of Se(VI) was chosen for this purpose because this species is very stable, and it is easily available. The same samples as described above were analyzed using this simplified calibration. The first set of measurements was performed using a routine ICP/MS power of 1100 W. The results showed (Table ) that this type of calibration was not suitable for some species (Se-MetSeCys, SeMet, and SeUr). The difference between the expected and the obtained concentration was higher than 40% in some cases. Different sensitivity of the individual species is caused by the different energy needed for atomization. It could be expected that this effect will be suppressed by higher plasma power. The next experiments were conducted with the highest value recommended by the plasma torch producer, i.e., 1400 W. The results (see Table ) indicate that the effect of various sensitivity was rapidly suppressed, and recoveries of individual species were lying within the range 102–105%. It is obvious that the accuracy of determination based on the single-species calibration can be met for all species, assuming that higher plasma power is applied. The value of 1400 W was used for all the following measurements.
Table 5. Accuracy of the selenium species determination using single-species calibration solutions containing Se(VI) (added 50 μg L−1 Se).
The limit of detection (LOD) was estimated as three times the standard deviation of the signals of solutions that contained Se(VI) at low concentration level (approximately 1 μg L−1 Se) (n = 5). Analogously, the limit of quantification (LOQ) was estimated as ten times the standard deviation of this solution (n = 5). The final value of the LOD and LOQ was 0.4 and 1.20 μg L−1 Se, respectively.
Stability of selenium species
The accuracy of selenium speciation analysis in urine is affected not only by the measurement steps but also by sample handling and storage. Although the stability study of selenium compounds in water has been reported earlier, [Citation23,24] data on the stability of Se compounds in urine are very scarce. In view of the complex composition and physicochemical properties of urine, the stability of Se species can be different from that in aqueous solution. Recently, Juresa [Citation25] studied the stability of selenosugars in urine. They found that samples cooled immediately after collection and stored at 4 °C for up to two weeks were stable with no appreciable loss of selenosugar. For longer term storage, urine samples should be kept at –80 °C or at –20 °C after lyophilization. Unfortunately, no attention was put on the stability of other selenium species. A more comprehensive study was conducted by Zheng [Citation7] who investigated the stability of Se(VI), SeUr, TMSe, SeMet, and SeEt. Based on this study, it is considered that urine sample storage at –20 °C within one month is safe for speciation analysis. The important selenium species Se(IV) was unfortunately omitted from the study due to the incomplete chromatographic separation of Se(IV) and Cl– ions. Because the ICP/MS detection was performed on 77Se, the determination of Se(IV) was complicated by polyatomic interferences from ArCl+. In our study, we worked with 80Se in DRC mode, which is why Se(IV) could also be included in the study.
To assess the stability of selenium compounds Se(VI), Se(IV), Se-MetSeCys, SeEt, SeMet, SeUr, and TMSe during 10 weeks of storage, urine samples spiked by a species mixture (100 μg L−1 Se each) were kept in polyethylene tubes at three different temperatures (25, –5, and –35 °C). The first series of samples was stored without the addition of a stabilizing reagent and the next was acidified by the addition of 1 mol L−1 HCl with a final pH value of 3.0.
As shown in Figure (A), unsatisfactory results were obtained when the urine samples were stored at room temperature 25 °C. Only two species, Se(VI) and TMSe, were stable during the entire storage time. When the sample was acidified (Figure (B)), only the stability of Se-MetSeCys and SeMet was slightly prolonged. The losses of other species were greater than 50% after only three days of storage. However, the concentration of SeUr increased for the last sampling point (70 days), most likely due to the conversion of the other species.
Figure 2. Stability of selenium species in urine (A – urine stored at 25 °C; B – acidified urine stored at 25 °C; C – urine stored at –5 °C; D – acidified urine stored at –5 °C; E – urine stored at –35 °C; F – acidified urine stored at –35 °C).
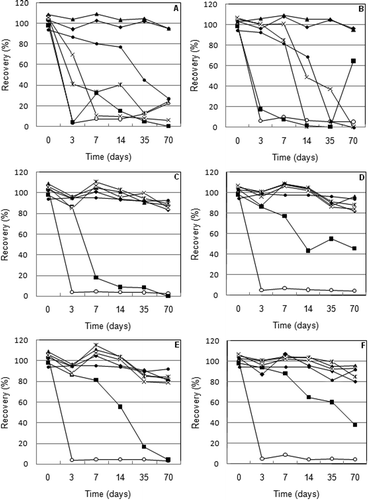
Better results were obtained at –5 °C (Figure (C) and (D)). No significant decrease of the recovery of Se(VI), Se-MetSeCys, TMSe, SeEt, and SeMet was observed; however, the stability of Se(IV) was invariably low. After only three days, almost all the Se(IV) disappeared in both types (acidified and un-acidified) of samples. The effect of acidifying the sample was significant only in the case of long-term storage of SeUr. The recovery of this species in un-acidified urine was less than 20% after seven days, while the recovery in the acidified urine exceeded 80% at the same sampling time. Moreover, the losses of this compound in acidified urine did not increase over 50% during all tested times.
The best stability of the Se species was reached at –35 °C (Figure (E) and (F)). The stability of SeUr was confirmed for three days (acidified urine), which is satisfactory for the collection of real samples for routine analysis. However, the stability of Se(IV) was very low even under these conditions. The loss of this species exceeded 90% in all cases. Because the decrease in the concentration of some species has not been accompanied by an increase in the concentration of other species, it can be assumed that during storage no interconversion of species occurred. Decrease in the concentration of species can be caused by their decomposition to volatile (H2Se, dimethylselenide, dimethyldiselenide) or insoluble selenium compounds (elemental selenium). Recently, formation of dimethyldiselenide during long-term storage of solution of selenosugars in urine was observed.[Citation25]
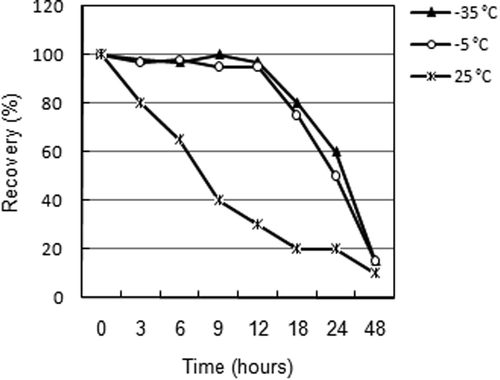
Due to the unsatisfactory stability of Se(IV) during the long-term test, a new short-term stability test (up to 20 h) covering only Se(IV) in acidified urine was performed. It is obvious (Figure ) that the stability of this species at 25 °C is very short; therefore, it is necessary to transport the urine samples frozen at least at –5 °C. Despite this measure, the samples must be analyzed no more than 12 h after collection because the loss increases rapidly even at –35 °C.
Figure 3. Short-term stability of Se(IV) in urine.
Example of method application: excretion of selenium obtained from the dietary supplements
Dietary supplements are being administered to compensate for selenium deficiency in food. The real efficiency of these products to human health is frequently discussed. In our test, two randomly chosen supplements were observed in a short-term test.
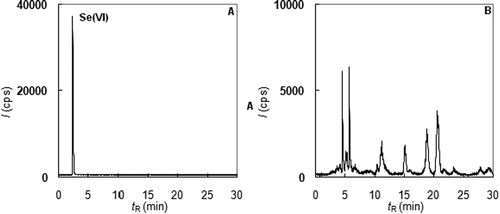
The declared form of selenium was the same in both formulations: “Selenium bound to yeast.” However, speciation analyses of the samples dissolved in diluted HCl gave inconsistent results. Although the total content of Se declared by the producer was proven (100 μg Se/tablet for DS 1 and 50 μg Se/tablet for DS 2), the profiles of both chromatograms were very different. The analysis of DS 1 showed (Figure (A)) that, despite the declaration by the producer, purely inorganic Se(VI) was present. However, product DS 2 (Figure (B)) contained more than 15 organic selenium species, and it was possible to expect that the retention of Se from this product should be higher.
Figure 4. Speciation analysis of selenium in dietary supplements (A – extract of DS 1, B – extract of DS 2).
The next part of the study covered an investigation of Se excretion by urination and the alternation of plasma Se levels after one-shot administration of DS containing 100 μg Se. Urine from five volunteers was regularly collected for 24 h after the administration, while blood samples were taken twice: just before and 24 h after the administration. All urine samples were analyzed immediately after collection (no longer than 3 h), and the total excreted amount of selenium was evaluated. The results confirmed different dynamics of excretion of inorganic and organic bound Se. Almost 50% of the ingested selenium from DS 1 was excreted by urine during 24 h (see Table ). In the case of DS 2, a maximum of 8% of this element was excreted during this period; see Table . This result indicates that a greater portion of selenium from of DS 2 could be incorporated to organisms. This effect was reported previously by Pedrero [Citation4], who reported that the bioavailability of inorganic species is less than organic forms of selenium.
Table 6. Course of Se excretion after the administration of DS 1 (100 μg Se). Contents of unreported species were under LOQ.
Table 7. Course of Se excretion after the administration of DS 2 (100 μg Se). Contents of unreported species were under LOQ.
In view of the Se speciation, a major portion of the selenium from DS 1 was excreted in the unchanged form as Se(VI) (Figure (A)). Only a small portion of Se was altered by the human metabolism and was excreted as organic species, namely as U1 found in urine samples collected 3 h after the administration. This result confirms that the absorption of Se(VI) is very low, and its usage in the diet supplements is questionable. In the case of DS 2 (Figure (B)), the situation was very different. This supplement did not contain any inorganic species; in excreted urine, only a minor increase in Se(IV) after 3 h was observed. The species U1 was a major excreted selenium metabolite. Moreover, two other species, Se-MetSeCys and SeMet, were also found in all samples collected from 1 to 9 h after the administration. This result provides convincing evidence that the selenium from DS 2 was better integrated into the metabolic processes.
Figure 5. Examples of selenium speciation in urine after the administration of the dietary supplements (A – DS 1, B – DS 2, I-before ingestion, II-an hour after ingestion, III-three hours after ingestion, IV-six hours after ingestion).
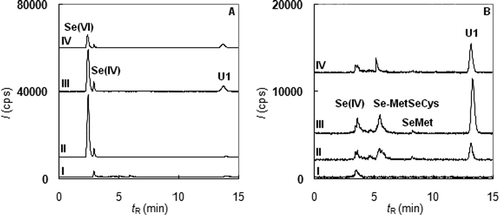
Previous results were also confirmed by the analysis of blood serum. The found concentrations of total selenium before and after ingestion were compared by Student’s paired-matched test. The results showed (Table ) that the difference in concentration after the application DS 1 was not statistically significant. Although this short-term investigation has less explanatory power, this result implies that dietary supplements containing inorganic Se are not beneficial for human health. In the case of DS 2, the statistical test confirmed the increase in serum Se even after unrepeated ingestion of DS. However, the increase in plasma Se was not followed by an increase in GPx. Student’s paired-matched test (see Table ) showed that the GPx activity was not altered by the administration of both products. It was most likely caused by the short-term investigation and unrepeated ingestion of the dietary supplements. It would be necessary to carry out more long-term experiments to trace the path of Se in the body.
Table 8. Alterations of the blood serum Se after the administration of dietary supplements.
Table 9. Alterations of GPx activity after the administration of dietary supplements.
Conclusion
In this study, we performed a validation of the quantification step for a method of speciation analysis of Se in human urine. The study has shown that the commonly accepted idea, that the sensitivity of ICP/MS detection is the same for all analyzed species of one element, may not be valid and that the proper conditions of detection should be optimized. Therefore, we managed to simplify the calibration procedure for using a single species allowing the determination of all selenium compounds in urine, even those whose standards are not available. The ability of the method to monitor changes in the speciation of selenium metabolites was demonstrated using short studies concerning the absorption of selenium from selenium-containing dietary supplements. The result of this study, i.e., the small resorption and rapid excretion of inorganically bound selenium through urination, is not surprising. However, the main objective was to show that this method, despite the fact that samples must be analyzed almost immediately, is suitable for the routine analysis of samples from bulk studies. The analysis of selenium metabolites allows us to look for connections between the metabolism of selenium and some internal diseases, which is a challenge for our future work.
Notes on contributors
Štěpán Eichler holds MSc degree in analytical chemistry at the University of Chemistry and Technology Prague, Czech Republic. He is now working as a PhD student at the University of Chemistry and Technology Prague, Atomic Spectroscopy Research Group, Faculty of Chemical Engineering.
Antonín Kaňa is an assistant professor at the University of Chemistry and Technology Prague, Czech Republic. His current research interest is the speciation analysis of trace elements in biological samples.
Marta Kalousová is professor of medical chemistry and biochemistry at the Institute of Medical Biochemistry and Laboratory Diagnostics of the First Faculty of Medicine, Charles University and General University Hospital in Prague. She is a specialist in clinical chemistry and internal medicine – nephrology.
Magda Vosmanská is an assistant professor at the University of Chemistry and Technology Prague, Czech Republic. Her current research interest is the analysis of trace elements and minerals in biological and environmental samples.
Milan Korotvička is the head of the Department of Clinical Biochemistry and Hematology, Psychiatric Hospital in Dobřany and the postgraduate student at the Institute of Clinical Biochemistry and Hematology of the Faculty of Medicine, Charles University, and University Hospital in Pilsen. He is specialized in study of metabolic changes in patients with kidney diseases.
Tomáš Zima is professor of medical chemistry and biochemistry and specialist in clinical chemistry and internal medicine – nephrology. He is the head of the Institute of Medical Biochemistry and Laboratory Diagnostics of the First Faculty of Medicine, Charles University and General University Hospital in Prague and Rector of The Charles University.
Oto Mestek is professor at the University of Chemistry and Technology Prague, Czech Republic. He is the head of Atomic Spectroscopy Research Group, Faculty of Chemical Engineering. His research group carries out research related to speciation analysis of trace elements in environmental and biological samples. He published over 100 scientific papers.
Acknowledgments
Financial support from specific university research MSMT No. 20/2013 is greatly acknowledged.
Funding
This work was financially supported by specific university research [MSMT No. 20/2013] and study was also supported by research projects [PRVOUK - P25/LF1/2] and [MH CZ - DRO VFN64165].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Tapiero H, Townsend DM, Tew KD. The antioxidant role of selenium and seleno-compounds. Biomed. Pharmacother. 2003;57:134–144.10.1016/S0753-3322(03)00035-0
- Linder MC. Nutrition and metabolism of the trace elements. In: Linder MC, editor. Nutritional biochemistry and metabolism with clinical application. 2nd ed. Englewood Cliffs: Prentice-Hall; 1991. p. 215–276.
- National Research Council. Dietary reference intakes: applications in dietary assessment. Washington, (DC): The National Academies Press; 2002.
- Pedrero Z, Madrid Y. Novel approaches for selenium speciation in foodstuff and biological specimen: a review. Anal. Chim. Acta. 2002;634:135–152.
- Zheng J, Ohata M, Furuta N, et al. Speciation of selenium compounds with ion-pair reversed-phase liquid chromatography using inductively coupled plasma mass spectrometry as element specific detection. J. Chromatogr. A. 2000;874:55–64.10.1016/S0021-9673(00)00077-7
- Zheng J, Ohata M, Furuta N. Reversed-phase liquid chromatography with mixed ion-pair reagents coupled with ICP-MS for the direct speciation analysis of selenium compounds in human urine. J. Anal. At. Spectrom. 2002;17:730–735.10.1039/b202531k
- Zheng J, Shibata Y, Tanaka A. Study of the stability of selenium compounds in human urine and determination by mixed ion-pair reversed phase chromatography with ICP-MS detection. Anal. Bional. Chem. 2002;374:348–353.10.1007/s00216-002-1425-5
- Pan F, Tyson JF, Uden PC. Simultaneous speciation of arsenic and selenium in human urine by high-performance liquid chromatography inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2007;22:931–937.10.1039/b703713a
- Eichler S, Mestek O. Validation of determination and speciation analysis of selenium in urine by HPLC and inductively-coupled plasma mass spectrometry. Chem. Listy. 2011;105:200–206.
- Jäger T, Drexler H, Göen T. Ion pairing and ion exchange chromatography coupled to ICP-MS to determine selenium species in human urine. J. Anal. At. Spectrom. 2013;28:1402–1409.10.1039/c3ja50083g
- Gammelgaard B, Bendahl L, Sidenius U, et al. Selenium speciation in urine by ion-pairing chromatography with perfluorinated carboxylic acids and ICP-MS detection. J. Anal. At. Spectrom. 2002;17:570–575.10.1039/b202256g
- Wrobel K, Wrobel K, Kannamkumarath SS, et al. Identification of selenium species in urine by ion-pairing HPLC-ICP-MS using laboratory-synthesized standards. Anal. Bioanal. Chem. 2003;377:670–674.10.1007/s00216-003-2147-z
- Jeong JS, Lee J, Pak YN. Quantitative speciation of selenium in human blood serum and urine with AE- RP- and AF-HPLC-ICP/MS. Bull. Korean Chem. Soc. 2013;34:3817–3824.10.5012/bkcs.2013.34.12.3817
- Gammelgaard B, Jøns O. Determination of selenite and selenate in human urine by ion chromatography and inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2000;15:945–949.10.1039/b003637o
- Lu Y, Rumpler A, Francesconi KA, et al. Quantitative selenium speciation in human urine by using liquid chromatography–electrospray tandem mass spectrometry. Anal. Chim. Acta. 2012;731:49–59.10.1016/j.aca.2012.04.016
- Ohta Y, Suzuki N, Kobayashi Y, et al. Rapid speciation and quantification of selenium compounds by HPLC-ICP MS using multiple standards labelled with different isotopes. Isotopes Environ. Health Stud. 2011;47:330–340.10.1080/10256016.2011.587609
- Chatterjee A, Tao H, Shibata Y, et al. Determination of selenium compounds in urine by high-performance liquid chromatography–inductively coupled plasma mass spectrometry. J. Chromatogr. A. 2003;997:249–257.10.1016/S0021-9673(03)00434-5
- Lin SY, Wang GR, Huang QP, et al. Speciation of selenium compounds by open tubular capillary electrochromatography-inductively coupled plasma mass spectrometry. Electrophoresis. 2006;27:4257–4265.10.1002/(ISSN)1522-2683
- Bendahl L, Gammelgaard B, Jøns O, et al. Interfacing capillary electrophoresis with inductively coupled plasma mass spectrometry by direct injection nebulization for selenium speciation. J. Anal. At. Spectrom. 2001;16:38–42.10.1039/b007137o
- Zhao QX, Chen YW, Montaut S, et al. Synthesis, identification and chemical features of high-purity trimethylselenonium iodide. J. Sulfur Chem. 2010;31:373–385.10.1080/17415993.2010.516435
- Zima T, Mestek O, Němeček K, et al. Trace elements in hemodialysis and continuous ambulatory peritoneal dialysis patients. Blood Purif. 1998;16:253–260.10.1159/000014342
- Funk W, Dammann V, Donnevert G, et al. Quality assurance in analytical chemistry: applications in environmental, food, and materials analysis, biotechnology, and medical engineering. 2nd ed. Weinheim: Wiley-VCH; 2007.
- Cobo MG, Palacios MA, Cámara C, et al. Effect of physicochemical parameters on trace inorganic selenium stability. Anal. Chim. Acta. 1994;286:371–379.10.1016/0003-2670(94)85083-6
- Olivas RM, Donard OFX, Cámara C, et al. Analytical techniques applied to the speciation of selenium in environmental matrices. Anal. Chim. Acta. 1994;286:357–370.10.1016/0003-2670(94)85082-8
- Juresa D, Darrouzès J, Kienzl N, et al. An HPLC/ICPMS study of the stability of selenosugars in human urine: implications for quantification, sample handling, and storage. J. Anal. At. Spectrom. 2006;21:684–690.10.1039/b602976k