
Abstract
Background
Risankizumab is approved for treatment of moderate to severe plaque psoriasis. Availability of a patient-controlled single self-injection of risankizumab may improve adherence and long-term management of psoriasis.
Objective
To investigate efficacy, safety, and usability of a new risankizumab 150 mg/mL formulation administered as a single subcutaneous injection via prefilled syringe (PFS) or autoinjector (AI).
Methods
Efficacy, safety, usability, and acceptability of risankizumab 150 mg/mL PFS or AI were investigated in adults with moderate to severe psoriasis in two phase 3 studies. Study 1 was a multicenter, randomized, double-blinded, placebo-controlled study that investigated 150 mg/mL risankizumab PFS; study 2 was a multicenter, single-arm, open-label study that investigated 150 mg/mL risankizumab AI.
Results
At week 16, risankizumab 150 mg/mL demonstrated efficacy vs. placebo (Psoriasis Area and Severity Index ≥90% improvement (PASI 90), 62.9% vs. 3.8%; static Physician Global Assessment (sPGA) 0/1, 78.1% vs. 9.6%; both p< .001) in study 1; in study 2, PASI 90 and sPGA 0/1 were 66.7%, and 81.5%, respectively. All patients successfully self-administered study treatments via PFS or AI. Acceptability of self-injection was high in both studies. Efficacy and safety of risankizumab 150 mg/mL were comparable with results from previous risankizumab phase 3 studies using the 90 mg/mL formulation.
Conclusions
The efficacy, safety, and usability of 150 mg/mL risankizumab delivered as a single PFS or AI injection support use of this new formulation in patients with moderate to severe plaque psoriasis.
Clinical trials
NCT03875482 and NCT0387508
Introduction
Psoriasis is a chronic immune-mediated inflammatory disease associated with comorbidities and reduced quality of life (Citation1–4). Due to its chronic nature, therapies that provide long-term safety and efficacy are often needed to manage this disease (Citation5). Furthermore, adherence to therapy is important for favorable long-term treatment outcomes (Citation6). More specifically, injectable therapies that can be self-administered at home in a single injection can improve adherence and ease the management of disease (Citation7,Citation8).
Interleukin (IL)-23 inhibitors have demonstrated efficacy in clinical trials in patients with moderate to severe plaque psoriasis (Citation9–11). Risankizumab, a fully humanized monoclonal antibody that binds with high affinity to the human IL-23 p19 subunit (Citation12), has demonstrated superior efficacy in phase 3 clinical trials when compared with placebo, secukinumab, ustekinumab, or adalimumab for the treatment of patients with moderate to severe plaque psoriasis (Citation11,Citation13–15). In the pivotal phase 3 clinical trials, the risankizumab 150-mg dose was administered via two subcutaneous (SC) 75-mg injections of a 90-mg/mL formulation delivered with a prefilled syringe (PFS).
To allow a more patient-controlled self-injection experience, a new risankizumab formulation of 150 mg/mL that enables administration of the 150-mg dose with one SC injection was developed. A phase 1 pharmacokinetic (PK) study in healthy subjects demonstrated that the 150 mg/mL PFS is bioequivalent to the currently approved 90 mg/mL PFS, with comparable PK, immunogenicity, and safety profile (Citation16). In addition, the 150 mg/mL autoinjector (AI) showed bioequivalent exposure and comparable immunogenicity to the risankizumab 150 mg/mL PFS (Citation16). Here, we report efficacy, safety, tolerability, and usability of the single injection formulation of risankizumab 150 mg/mL administered via PFS or AI in adult patients with moderate to severe plaque psoriasis in two phase 3 clinical studies.
Materials and methods
Study designs
Study 1 (NCT03875482) was a multicenter, randomized, double-blinded, placebo-controlled, parallel-group study conducted at 38 sites in the United States, including Puerto Rico, that evaluated the efficacy and safety of risankizumab 150 mg/mL PFS in patients with moderate to severe plaque psoriasis. The study included a 30-day screening period, a treatment period with study drug self-administered at weeks 0, 4, and 16, and a subsequent follow-up safety call at approximately 20 weeks after the last dose of study drug; dosing on week 4 was self-administered at home after a study visit (Supplemental Figure 1A). Patients were randomized in a 2:1 ratio to risankizumab 150 mg or placebo.
Study 2 (NCT03875508) was a multicenter, single-arm, open-label study conducted at 24 sites in the United States that evaluated usability, efficacy, safety, and tolerability of the risankizumab 150 mg/mL AI in patients with moderate to severe plaque psoriasis. The study included a 30-day screening period; a treatment period with study drug self-administered at weeks 0, 4, 16, and 28; and a subsequent follow-up safety call at approximately 20 weeks after the last dose of study drug. Dosing on weeks 4 and 16 was self-administered at home after a study visit (Supplemental Figure 1B).
All patients provided written informed consent, and the study protocol was approved by an institutional review board or independent ethics committee at each study site. Both studies were conducted in accordance with applicable regulations and the ethical principles of Good Clinical Practice as defined by the International Conference on Harmonisation and Declaration of Helsinki.
Participants
Adults (≥18 years old) from the United States, including Puerto Rico, with a diagnosis of moderate to severe plaque psoriasis for at least 6 months before the baseline visit were eligible for the studies. Eligible patients met the following disease activity criteria: ≥10% body surface area (BSA) psoriasis involvement, static Physician Global Assessment (sPGA) score of ≥3, and Psoriasis Area and Severity Index (PASI) ≥12. Patients were also required to have laboratory values meeting the following criteria during the screening period, prior to the first dose of study drug: serum aspartate transaminase <2 × upper limit of normal (ULN); serum alanine transaminase <2 × ULN; serum direct bilirubin ≤2.0 mg/dL (except for patients with isolated elevation of indirect bilirubin relating to a confirmed diagnosis of Gilbert syndrome); total white blood cell count >3000/μL; absolute neutrophil count >1500/μL; platelet count >100,000/μL; and hemoglobin >8 g/dL.
Patients with a history of erythrodermic psoriasis, generalized or localized pustular psoriasis, medication-induced or medication-exacerbated psoriasis, or new onset guttate psoriasis, active skin disease other than psoriasis that could interfere with the assessment of psoriasis, chronic infections, or documented active or suspected malignancy or history of any malignancy within the last 5 years (except for successfully treated non-melanoma skin cancer or localized carcinoma in situ of the cervix) were excluded from the studies. In addition, patients with previous exposure to risankizumab or concomitant use of systemic non-biologic or biologic therapy for psoriasis, topical psoriasis treatment, and phototherapy were also excluded.
Efficacy endpoints
Key efficacy endpoints (co-primary endpoints in study 1) were the proportion of patients achieving PASI 90 (defined as at least 90% improvement from baseline in PASI) at week 16, and proportion of patients achieving sPGA of clear (0) or almost clear (1) at week 16. Additional endpoints (ranked secondary endpoints in study 1) were proportion of patients achieving PASI 100 (defined as 100% improvement from baseline in PASI) at week 16 and proportion of patients achieving sPGA 0 at week 16; percent change from baseline in PASI at all visits was also assessed.
Usability and acceptability experience
In study 1, usability of the PFS for self-injection was assessed as the proportion of patients with an observer rating of successful patient self-administration, defined as any patients who successfully completed the sequence of three critical steps (‘select correct injection site’, ‘remove the needle cover’, and ‘slowly push plunger all the way in until all the liquid is injected and syringe is empty’) without errors to administer study drug, and measured at baseline and at week 16 by an observer at the study site. In study 2, usability of the AI for self-injection was assessed as the proportion of patients with an observer rating of successful self-administration, defined as any patients who successfully completed the sequence of 4 critical steps (‘chose an appropriate injection site’, ‘removed cap from AI’, ‘activated the injection’, and ‘performed a complete injection’) without errors in the administration of study drug via AI, and measured at baseline and at week 28 by an observer at the study site. In addition, potential hazards assessed by the observer based on a pre-defined possible use-related hazards checklist for self-administration with PFS or AI were measured at baseline and at week 16 (study 1) or at week 28 (study 2).
Patient rating of acceptability of their experience with the PFS or AI using the Self-Injection Assessment Questionnaire (SIAQ) was assessed at study visits in both studies. SIAQ measures overall patient experience with SC self-injection and includes two parts: the PRE module completed by the patient immediately before baseline self-injection and the POST module completed by the patient 20–40 min after each self-injection at all visits with study drug dosing. The PRE module included three domains: ‘feelings about injections’, ‘self-confidence’, and ‘satisfaction with self-injection’. The POST module included six domains: ‘feelings about injections’, ‘self-image’, ‘self-confidence’, ‘injection-site pain’, and ‘reactions during and after the injection’, ‘ease of use’, and ‘satisfaction with self-injection’ (Citation17). Patients rated each item of the SIAQ, with ratings that varied from ‘not at all’ to ‘extremely’; these ratings were later transformed to scores ranging from 0 (worst experience) to 10 (best experience) (Supplemental Table 1). Transformed scores were averaged to compute a domain score. Higher domain scores indicated higher acceptability by patients to use the PFS or AI.
Safety
Safety evaluations included adverse event (AE) monitoring, physical examinations, vital sign measurements, and clinical laboratory testing (hematology and chemistry). Treatment-emergent AEs were defined as any event with an onset that is after the first dose of study drug and with an onset date within 20 weeks (140 days) after the last dose of study drug.
Statistical analysis
In study 1, the intent-to-treat (ITT) population included all randomized patients; patients were analyzed according to treatment as randomized. The safety analysis population consisted of all patients who received at least one dose of study drug; patients were analyzed according to the first dose of study drug (risankizumab or placebo) received. In study 2, the ITT population, which included all patients who received at least one dose of study drug, was analyzed for efficacy, usability, and safety.
In study 1, overall type-I error was controlled by simultaneously testing co-primary endpoints, followed by the ranked secondary endpoints, in a hierarchical order. Comparison of the primary and secondary efficacy endpoints were made between the risankizumab and the placebo treatment groups using the Chi-square test. In study 2, no statistical tests were performed.
For analysis of both studies, categorical efficacy variables were analyzed using non-responder imputation (NRI) to handle missing data; mixed-effect model repeat measurements (MMRMs) of additional efficacy endpoints was used for continuous variables. As-observed and modified NRI (mNRI) were conducted as sensitivity analyses for efficacy endpoints (co-primary and ranked secondary efficacy endpoints in study 1) for the two studies. For mNRI, a patient was considered as a non-responder for the visit if the patient did not have an evaluation and discontinued study drug due to lack of efficacy or due to an AE of worsening of psoriasis during the visit window. Other missing values were excluded from this sensitivity analysis. Usability endpoints were analyzed as-observed cases for both studies. Of note, the NRI approach was coupled with multiple imputation to mitigate the impact of missing data due to COVID-19 for key efficacy endpoints in study 2.
Sample size
Study 1 was designed to enroll approximately 150 patients. Assuming the response rates at week 16 to be 74% for the risankizumab arm and 3% for the placebo arm for PASI 90, and 85% for the risankizumab arm and 7% for the placebo arm for sPGA 0/1, the study provided more than 95% power for each of the two co-primary efficacy endpoints (an overall power of more than 90%).
Study 2 was designed to enroll approximately 100 patients receiving risankizumab. Assuming the point estimate is 90% for the proportion of patients with an observer rating of successful self-administration at week 28, the current sample size of 100 patients provided a 95% confidence interval of ±5.9% around a point estimate. In addition, assuming comparable response rates as the phase 3 pivotal studies, with the sample size of 100, the PASI 90 rate at week 16 was estimated with a 95% confidence interval of ±8.6%, when the point estimate is 74%; and the sPGA 0/1 rate at week 16 was estimated with a 95% confidence interval of ±7.0%, when the point estimate is 85%.
Results
Patient disposition and baseline characteristics
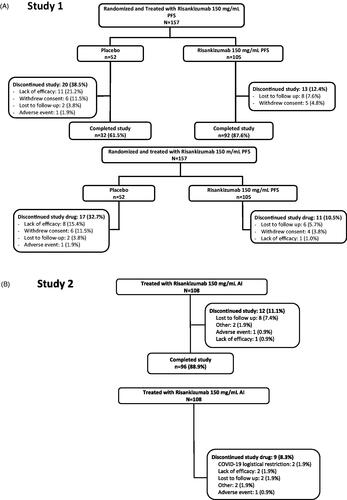
In study 1, a total of 157 patients were randomized to risankizumab 150 mg/mL PFS (n = 105) or placebo (n = 52) and received treatment. Of these, 124 completed the study (risankizumab, n = 92 (87.6%); placebo, n = 32 (61.5%)). The most common primary reasons for study discontinuation were lack of efficacy (11 patients (7.0%); risankizumab, n = 0; placebo, n = 11), withdrawal of consent (11 patients (7.0%); risankizumab, n = 5; placebo n = 6), and lost to follow-up (10 patients (6.4%); risankizumab, n = 8; placebo n = 2). In study 2, 108 patients were enrolled to the study and received risankizumab 150 mg/mL formulation via AI; of these, 96 (88.9%) completed the study. The most common reason for discontinuation was lost to follow-up (eight patients (7.4%); ).
Figure 1. Patient disposition and study drug discontinuation in (A) study 1 and (B) study 2. AI: autoinjector; PFS: prefilled syringe.
Baseline demographic and disease characteristics, such as duration of psoriasis, baseline PASI, BSA involvement, and prior treatment with biologic therapy, were similar in the two treatment arms in study 1 and also when comparing study 1 with study 2 ().
Table 1. Patient demographics and baseline disease characteristics in study 1 and 2.
Efficacy
Significantly greater proportions of patients receiving risankizumab 150 mg/mL PFS met the co-primary endpoints of PASI 90 (62.9%) and sPGA 0/1 (78.1%) at week 16 vs. placebo (3.8% and 9.6%, respectively; both p < .001, NRI analysis) in study 1. Similarly, significantly greater proportions of patients receiving risankizumab 150 mg/mL PFS achieved PASI 100 (38.1%) and sPGA 0 (39.0%) at week 16 vs. placebo (1.9% and 1.9%, respectively; both p < .001, NRI analysis, ). The sensitivity analysis supported these results (PASI 90 was 67.3% and sPGA 0/1 was 83.7% for mNRI and as-observed case analyses in the risankizumab arm).
Figure 2. Proportion of patients achieving PASI 90, PASI 100, sPGA 0/1, and sPGA 0 at week 16 with risankizumab and placebo (study 1) and OL risankizumab (study 2). NRI analysis. AI: autoinjector; NRI: non-responder imputation; OL: open-label; PASI: Psoriasis Area Severity Index; PFS: prefilled syringe; sPGA: static physician’s global assessment.
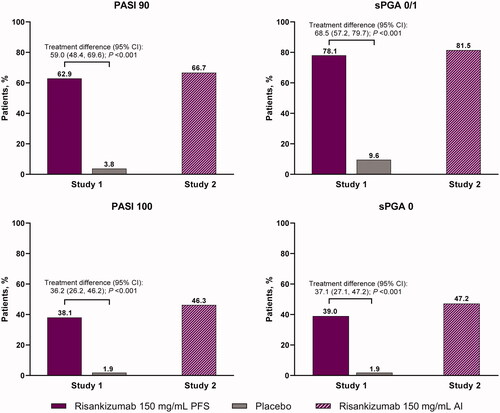
These results were also consistent with risankizumab 150 mg/mL AI in study 2 in which 66.7% (72/108) of patients achieved PASI 90, 81.5% (88/108) achieved sPGA 0/1, 46.3% (50/108) achieved PASI 100, and 47.2% (51/108) achieved sPGA 0 at week 16 (NRI analysis, ). Compared with the NRI analyses, the proportions of patients achieving PASI 90 and sPGA 0/1 were numerically similar or higher for mNRI and as-observed case analyses (67.9% and 83.0%, respectively). Of note, 84.4% of patients achieved PASI 90 and 90.9% of patients achieved sPGA clear or almost clear at week 28 in study 2 (NRI analysis, Supplemental Figure 2). The percentage change from baseline in PASI for studies 1 and 2 is shown in .
Table 2. Percentage change from baseline in PASI over time.
Usability and acceptability
In study 1, all patients in the risankizumab and placebo groups successfully self-administered study treatments, defined as successfully completing all three critical steps in the instructions for use at baseline and week 16. Similarly, in study 2, all patients successfully self-administered study treatments, defined as successfully completing all four critical steps in the instructions for use at baseline and week 28. In study 1, two observed use hazards were reported in the placebo and risankizumab groups at baseline (one each, delayed administration due to needle cover removal difficulties); no observed use hazards were reported at week 16. No reported use hazards were observed at baseline and at week 28 in study 2 (Supplemental Table 2).
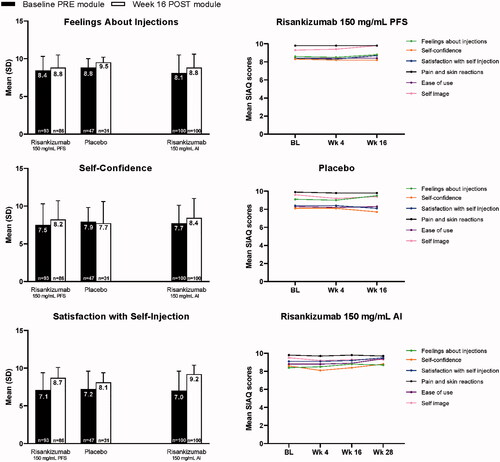
Mean domain scores for acceptability of self-injection experience were high at baseline (PRE module) and numerically higher at week 16 (POST module) for both the risankizumab and placebo groups for PFS in study 1. Similar results were seen for risankizumab AI in study 2, comparing PRE module and week 28 (POST module).
Domains with shared questions for the PRE and POST modules are reported in . In the risankizumab groups in studies 1 and 2, the mean scores for ‘feelings about injection’ domain were 8.4 and 8.1, for the baseline PRE module and 8.8 and 8.8 for the POST module at week 16 (study 1) or week 28 (study 2); for ‘self-confidence’ domain the mean scores were 7.5 and 7.7 for PRE module and 8.2 and 8.4 for POST module at week 16 (study 1) or week 28 (study 2); and for ‘satisfaction with self-injection’ domain the mean scores were 7.1 and 7.0 for PRE module and 8.7 and 9.2 for POST module at week 16 (study 1) or week 28 (study 2). Similar results were observed in the placebo group in study 1. POST module mean SIAQ scores were high at baseline and remained high or improved over time among all patients in both studies.
Figure 3. Mean SIAQ baseline PRE module domain scores and week 16 POST module domain scores and SIAQ POST module domain scores over time with risankizumab and placebo (study 1) and OL risankizumab (study 2). Observed case analysis. AI: autoinjector; BL: baseline; OL: open-label; PFS: prefilled syringe; SIAQ: Self-Injection Assessment Questionnaire.
Responses to individual POST module items are summarized in Supplemental Table 3. The majority of patients achieved the highest two response categories in all of the POST module individual SIAQ questions. ‘Satisfaction with self-injection’ domain represented by the question ‘overall, how satisfied are you with current way of taking your medication?’ showed the greatest increase in patients responding in the top two categories (i.e. satisfied and very satisfied) in the risankizumab groups, increasing from 64.5% of patients in study 1 and 57.0% of patients in study 2 in the PRE module at baseline to 93.0% with the PFS (study 1) and to 97.6% with the AI (study 2) in the last POST module assessment (week 16 for study 1 and week 28 for study 2).
Safety
Adverse events for studies 1 and 2 are reported in . In study 1, the occurrence of treatment-emergent AEs (21.0% vs. 21.2%), serious AEs (1.0% vs. 0%), and infections (5.7% vs. 5.8%) was similar between risankizumab 150 mg/mL and placebo PFS groups, respectively. The most common AEs in the risankizumab group were nausea (4 (3.8%)) and nasal congestion (3 (2.9%)). All other AEs were reported in <2% patients in the risankizumab group. One mild injection site reaction was reported in the risankizumab 150 mg/mL PFS group. A serious treatment-emergent AE was reported in one patient (grade 3 acute pancreatitis, in a patient with a medical history of chronic pancreatitis, considered not related to study drug). In study 2, AEs were observed in 43.5% of patients, serious AEs in 5.6% of patients, infections in 27.8%, and serious infections in 1.9% of patients receiving risankizumab 150 mg/mL AI. Most AEs were mild or moderate and not related to study drug. The most common AEs were nasopharyngitis (8 (7.4%)), upper respiratory tract infection (6 (5.6%)), and gastroenteritis (4 (3.7%)); all other AEs occurred in <3% of patients. Seven serious treatment-emergent AEs were reported in six patients; appendicitis and prostatitis occurring in the same patient, and pyelonephritis, pyrexia, thermal burn, lumbar spinal stenosis, and atrial fibrillation.
Table 3. Treatment-emergent adverse events in study 1 and 2.
No deaths, malignancies, serious hypersensitivity reactions, active tuberculosis, or adjudicated major adverse cardiovascular events were observed in either study. There were two AEs of COVID-19: one in each study; both were non-serious.
Discussion
The two reported clinical studies were conducted to demonstrate efficacy and safety as well as to evaluate usability and acceptability of self-administration of risankizumab 150 mg/mL formulation delivered either via PFS or AI in patients with moderate to severe plaque psoriasis. The placebo-controlled study using the PFS (study 1) met the efficacy endpoints with significant improvements in PASI and sPGA observed with risankizumab 150 mg/mL PFS vs. placebo as early as week 4, with these benefits continuing to improve over time. A similar improvement was observed for risankizumab 150 mg/mL AI in the open-label study (study 2). Efficacy results for both studies were consistent with the week 16 results observed in the pivotal clinical studies investigating risankizumab in adult patients with moderate to severe plaque psoriasis administered as two SC 75-mg injections of a 90 mg/mL formulation delivered via PFS (Citation11,Citation13–15).
All patients successfully self-administered the study treatments as demonstrated by performing the critical steps pre-determined in both studies in the instructions for use, and by showing high compliance with the remaining defined steps. Nearly, all patients had no use hazards with either the PFS (with the exception of two patients who had difficulty removing the needle cover at the baseline visit) or AI in the two studies and were able to administer the full volume of the injection.
These studies collected patient-reported assessments of their perceptions about and experiences with PFS and AI through use of the SIAQ. SIAQ is a reliable and valid tool that is used in many clinical studies to evaluate the patient experience and acceptability of self-injection (Citation18–23). The results of the current studies demonstrated that patients generally found both PFS and AI to be easy to use. Patient confidence and satisfaction with self-injection of risankizumab was high from baseline through week 16 (for study 1) or week 28 (for study 2). Patient acceptability of self-injection of risankizumab 150 mg/mL formulation was rated high for the three common domains of the PRE and POST modules of SIAQ (feelings about self-injections, self-confidence, and satisfaction with self-injection) at baseline and at the final assessments, and for all the remaining POST modules. This demonstrates that self-injection was acceptable to patients before they administered the self-injection, and remained highly acceptable, and in some cases, even improved up to weeks 16 and 28. When the questions of the domains were analyzed individually, ‘satisfaction with self-injection’ was the domain where the proportion of patients either ‘satisfied’ or ‘very satisfied’ increased the most between the PRE module and POST module at the last assessment, indicating that the patient experience might have an impact on improved adherence to treatment.
No new safety findings were observed, and the risankizumab 150 mg/mL safety profile was consistent with risankizumab phase 3 studies using risankizumab 90 mg/mL PFS (Citation11,Citation13–15). No deaths, malignancies, serious hypersensitivity reactions, active tuberculosis, or adjudicated major adverse cardiovascular events were observed in either study. Only two patients discontinued the study drug due to COVID-19 logistical restrictions, and there were only two AEs of COVID-19 reported.
There were some limitations to this analysis. The two clinical studies were relatively small and conducted in a single country (the United States, including Puerto Rico), and did not have a similar study design. The placebo-controlled study (study 1) was not followed by an open-label extension, so the use of placebo, even when the randomization was 2:1, might have contributed to higher rates of discontinuation. Study 2 had an open-label design. The higher proportion of treatment-emergent AEs in study 2 might be partly explained by the longer study duration in comparison with study 1. However, it is important to note that the proportion of patients with treatment-emergent AEs in both studies was similar or even lower than in phase 3 clinical studies (Citation11,Citation14,Citation15). The occurrence of the COVID-19 pandemic when the two studies were ending had some impact on study discontinuations or missing assessments. Nevertheless, it did not affect the studies outcome or the interpretation of the results or overall conclusions.
In summary, superiority of risankizumab 150 mg/mL PFS vs. placebo was demonstrated in all co-primary and ranked secondary efficacy endpoints. The efficacy and safety profile of the risankizumab 150 mg/mL AI was consistent with the 150 mg/mL PFS, and with the 90 mg/mL formulation used in the pivotal phase 3 clinical studies. Usability and positive patient acceptability of the PFS and the AI as shown here support the use of these devices in patients with moderate to severe plaque psoriasis.
Supplemental Material
Download PDF (379.8 KB)Acknowledgements
AbbVie and the authors thank the patients who participated in the trials and all trial investigators for their contributions. Medical writing support was provided by Maria Hovenden, PhD, Jennifer Venzie, PhD, and Janet Matsuura, PhD, of ICON (North Wales, PA). All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
Disclosure statement
Andrew Blauvelt has served as a scientific adviser and/or clinical study investigator for AbbVie, Aligos, Almirall, Amgen, Arcutis, Arena, Athenex, Boehringer Ingelheim, Bristol-Myers Squibb, Dermavant, Eli Lilly and Company, Evommune, Forte, Galderma, Incyte, Janssen, Leo, Novartis, Pfizer, Rapt, Regeneron, Sanofi Genzyme, Sun Pharma, and UCB Pharma. Kenneth B. Gordon has received honoraria and/or research support from AbbVie, Amgen, Arcutis, Arena Pharma, Bristol Myers Squibb, Dermavant, Dermira, Incyte, Janssen, Kyowa Hakko Kirin, LEO Pharma, Novartis, Pfizer, Sanofi Genzyme, Sun Pharma, and UCB. Patricia Lee does not have any conflicts of interest, but her spouse is a speaker for AbbVie. Jerry Bagel has received research funds payable to Psoriasis Treatment Center from AbbVie, Amgen, Arcutis Biotherapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Celgene Corporation, Corrona LLC, Dermavant Sciences Ltd, Dermira, UCB, Eli Lilly and Company, Glenmark Pharmaceuticals Ltd, Janssen Biotech, Kadmon Corporation, Leo Pharma, Lycera Corp, Menlo Therapeutics, Novartis, Pfizer, Regeneron Pharmaceuticals, Sun Pharma, Taro Pharmaceutical Industries Ltd, and Ortho Dermatologics; consultant fees from AbbVie, Amgen, Celgene Corporation, Bristol-Myers Squibb, Eli Lilly and Company, Janssen Biotech, Novartis, Sun Pharmaceutical Industries Ltd, UCB; and fees for speaking from AbbVie, Celgene Corporation, Eli Lilly, Janssen Biotech, and Novartis. Howard Sofen has served as a scientific adviser and/or clinical study investigator for AbbVie, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Eli Lilly, Incyte, Janssen, Leo, Novartis, Pfizer, Sanofi Genzyme, Sun Pharma, and UCB. Benjamin Lockshin has served as a speaker, consultant and/or clinical study investigator for AbbVie, Bristol Myers Squibb, Celgene, Corrona registry, Eli Lilly, Incyte, Novartis, Regeneron, Sanofi Genzyme, Sun Pharma, and UCB. Ahmed M. Soliman, Ziqian Geng, Tianyu Zhan, and Gabriela Alperovich are employees of AbbVie Inc. and may hold stock or stock options. Linda Stein Gold has served as a scientific adviser, speaker and/or clinical study investigator for AbbVie, Almirall, Arcutis, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, Galderma, Incyte, Leo, Novartis, Ortho Derm, Pfizer, Regeneron, Sanofi Genzyme, Sun Pharma, and UCB Pharma.
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g. protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Additional information
Funding
Related Research Data
References
- Baeta IG, Bittencourt FV, Gontijo B, et al. Comorbidities and cardiovascular risk factors in patients with psoriasis. An Bras Dermatol. 2014;89(5):735–744.
- Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–390.
- Augustin M, Radtke MA. Quality of life in psoriasis patients. Expert Rev Pharmacoecon Outcomes Res. 2014;14(4):559–568.
- Gelfand JM, Feldman SR, Stern RS, et al. Determinants of quality of life in patients with psoriasis: a study from the US population. J Am Acad Dermatol. 2004;51(5):704–708.
- Gisondi P, Del Giglio M, Girolomoni G. Treatment approaches to moderate to severe psoriasis. Int J Mol Sci. 2017;18(11):2427.
- Augustin M, Holland B, Dartsch D, et al. Adherence in the treatment of psoriasis: a systematic review. Dermatology. 2011;222(4):363–374.
- Yelamos O, Ros S, Puig L. Improving patient outcomes in psoriasis: strategies to ensure treatment adherence. Psoriasis. 2015;5:109–115.
- Domanska B, Stumpp O, Poon S, et al. Using patient feedback to optimize the design of a certolizumab pegol electromechanical self-injection device: insights from human factors studies. Adv Ther. 2018;35(1):100–115.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–288.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–661.
- Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7(4):778–791.
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(6):649–658.
- Warren RB, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br J Dermatol. 2021;184(1):50–59.
- Reich K, Gooderham M, Thaci D, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394(10198):576–586.
- Lon H-K, Cheng L, Nudurupati S, et al. Pharmacokinetic comparability of risankizumab formulations in prefilled syringe and auto-injector for subcutaneous injection. Clin Ther. 2021;43(3):629–636.
- Keininger D, Coteur G. Assessment of self-injection experience in patients with rheumatoid arthritis: psychometric validation of the Self-Injection Assessment Questionnaire (SIAQ). Health Qual Life Outcomes. 2011;9:2.
- Bailey K, Mountian I, Bruggraber R, et al. Patient Satisfaction with CIMZIA(®) (Certolizumab Pegol) AutoClicks(®) in the UK. Adv Ther. 2020;37(4):1522–1535.
- Lacour J-P, Paul C, Jazayeri S, et al. Secukinumab administration by autoinjector maintains reduction of plaque psoriasis severity over 52 weeks: results of the randomized controlled JUNCTURE trial. J Eur Acad Dermatol Venereol. 2017;31(5):847–856.
- Paul C, Lacour JP, Tedremets L, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29(6):1082–1090.
- Blauvelt A, Prinz JC, Gottlieb AB, et al. Secukinumab administration by pre-filled syringe: efficacy, safety and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol. 2015;172(2):484–493.
- Ferris LK, Ott E, Jiang J, et al. Efficacy and safety of guselkumab, administered with a novel patient-controlled injector (One-Press), for moderate-to-severe psoriasis: results from the phase 3 ORION study. J Dermatolog Treat. 2020;31(2):152–159.
- Nash P, Mease PJ, McInnes IB, et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: results from a randomized, placebo-controlled trial (FUTURE 3). Arthritis Res Ther. 2018;20(1):47.