
Abstract
Inherited epidermolysis bullosa is a heterogeneous group of hereditary skin diseases characterized by skin (mucosa) fragility, which leads to blistering. Junctional epidermolysis bullosa is associated with mutations in genes expressing proteins of the dermo-epidermal junction. Dupilumab, an antibody that directly targets interleukin (IL)-4 receptor alpha, may be an effective treatment for dystrophic epidermolysis bullosa. We describe a case of junctional epidermolysis bullosa that improved with dupilumab.
1. Introduction
Junctional epidermolysis bullosa (JEB) is characterized by extreme skin fragility, mechanically induced blistering, and chronic inflammation, in addition to gastrointestinal, renal, and respiratory complications (Citation1–3). A major hallmark of JEB is the recurrence of subepidermal blisters and mucosa caused by functional loss or complete absence of major structural proteins in the skin (Citation4). JEB is caused by mutations in the genes encoding components of the dermo-epidermal junction, including type XVII collagen (COL17A1), laminin 332 (LAMA3, LAMB3, and LAMC2), integrin α6β4 (ITGA6 and ITGB4), and integrin α3 (ITGA3). Intermediate JEB caused by mutations in the COL17A1 gene is characterized by blister formation and abnormalities of the hair and teeth (Citation5). Curative therapies targeting the genetic etiologies of JEB are not standard, and the management of JEB can be challenging (Citation6). Dupilumab, an anti-IL-4 receptor-alpha antagonist, is effective for moderate and severe atopic dermatitis by inhibiting IL-4 and IL-13 signalling (Citation7). Dupilumab might have a beneficial effect in dystrophic epidermolysis bullosa (Citation8–14). Here, we describe an adult patient with recalcitrant JEB intermediate who was successfully treated with dupilumab.
2. Case description
A 65-year-old man presented to the dermatology department of Huashan Hospital with a lifelong history of itching vesicles and bullae in his limbs and trunk. The condition was aggravated in the summer and relieved in the winter. The lesions had become more numerous, and the itching symptoms had become more severe in the past year, resulting in a poor quality of life. His average pruritus numeric rating scale (P-NRS) score in the last 7 days was 7 (maximum = 8) and his average visual analog scale in the last 7 days was 7.4 (maximum = 9). Physical examination revealed erythematous patches involving the friction areas of the trunk and upper and lower limbs covered by vesicles and bullae, and his toenails were dystrophic. Moreover, the patient suffered from alopecia and tooth loss. ( and ). A skin biopsy performed on an abdominal lesion showed a focal blister at the dermo-epidermal junction with perivascular mononuclear inflammatory infiltrates and a few eosinophilic infiltrates in the superficial and middle layers of the dermis. A direct immunofluorescence test of the perilesional skin showed negative results. There was an increased blood eosinophil count (2540 × 106/L, normal: 50–350 × 106/L) and serum IgE level (1495.20 ng/ml, normal: 0–240 ng/ml). The values of enzyme-linked immunosorbent assay-BP180, BP230, Dsg1, and Dsg3 were all negative, excluding concomitant bullous pemphigoid.
Figure 1. Clinical photographs. Intermediate junctional epidermolysis bullosa improved after 12 weeks of treatment with dupilumab (loading dose 600 mg s.c. with successive injections of 300 mg every other week)
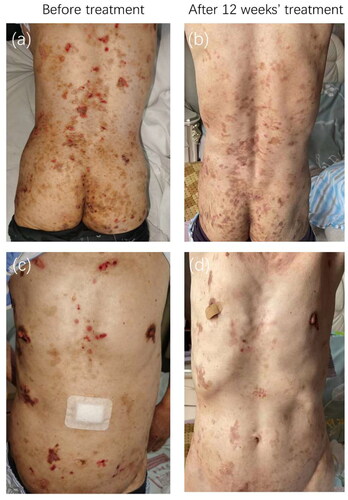
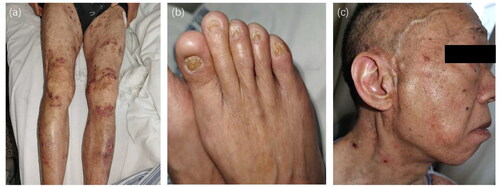
Figure 2. Other clinical features of junctional epidermolysis bullosa. (a) Erythematous patches covered by vesicles and bullae localized on the friction areas of lower limbs. (b) His toenails were dystrophic. (c) The patient suffered from alopecia.
Exome sequencing of peripheral blood revealed two mutations (c.2039-6G > A and c.C1177T:p. Q393X) in the COL17A1 gene. Mutation c.2039-6G > A was likely pathogenic and Mutation c.C1177T:p. Q393X was pathogenic. The diagnosis of JEB was made based on the presence of typical clinical features and medical history, consistent histopathological findings, negative direct immunofluorescence assessments, and compound heterozygous mutations in COL17A1. The patient responded poorly to therapy with oral antihistamines and topical corticosteroid ointments. Owing to the patient’s persistent itching, off-label dupilumab therapy was initiated (600 mg subcutaneous loading dose followed by 300 mg every 2 weeks), and a significant reduction in itching was observed after 12 weeks of treatment. The body surface area (BSA) of the bulla decreased from 50% to 10%. The P-NRS score improved from 8/10 to 2/10. The VAS of itch improved from 9/10 to 1/10. Blood eosinophil count (500 × 106/L, normal: 50–350 × 106/L) and serum IgE level decreased (600.80 ng/ml, normal: 0-240 ng/ml). Erythema, crusts, and hyperpigmentation improved leaving atrophic plaques (). The patient discontinued dupilumab after 12 weeks because of the cost, and no relapse occurred six months after the discontinuation. No side effects occurred.
3. Discussion
JEB is a rare inherited genetic disorder with few treatment options beyond symptomatic, palliative care. One on-label drug for EB is available in Europe; Oleogel-S10 (birch triterpenes) accelerates wound healing in EB and is well-tolerated (Citation15). In an analysis of the genotype–phenotype correlation in 441 patients with epidermolysis bullosa from China, only one COL17A1 pathogenic mutation was found in four (four families; 28.6%) cases (Citation16). A genotypic and phenotypic analysis of 34 cases of inherited junctional epidermolysis bullosa caused by COL17A1 mutations in European and Mediterranean populations identified 39 mutations in COL17A1; 24 mutations were novel (Citation17).
Pruritus occurred in 85% of the EB patients, with substantial differences across the subtypes (EBS 74%, JEB 100%, DEB 93%). Itch, in all its dimensions, was most pronounced in patients with JEB and DEB, and less prominent in patients with EBS (Citation18). Inflammation is a prominent feature of EB skin. In response to mechanical injury, keratinocytes release various proinflammatory cytokines. These may be released into the systemic circulation and may mediate the recruitment of inflammatory cells and development of T helper (Th)17 and Th2 cells at the injury site (Citation19). In RDEB, gene expression of the itch-related mediators IL13RA1 and IL4R is dysregulated in comparison with that in aged-matched healthy individuals (Citation20). Biological agents are gaining usage in EB treatment with some encouraging results. Recently, several cases of DEB pruriginosa were successfully treated with dupilumab (Citation8,Citation9,Citation11,Citation12,Citation21). Dupilumab can block the downstream signaling of Th2 cytokines, IL-4 and IL-13 (Citation7). Immune markers related to general inflammation (MMP12), Th2 (CCL13/CCL17), Th17/Th22 (IL-12B, CXCL1, S100A12). and innate immunity (IL-6, IL-8, IL-17C) decrease after dupilumab administration (Citation22). Dupilumab might decrease the levels of IL-8 through blocking IL-4 in the reported JEB patient. The blister fluids of JEB patients contain increased levels of MMP-9 and CXCL8/IL-8 and may contribute to blister formation (Citation23); inhibition of these two factors is a potential mechanism by which dupilumab may improve JEB.
Author contributions
L Zhang, S Wang, Q Chen, and L Xiang designed the study and performed the acquisition, analysis, and interpretation of data. L Zhang and S Wang wrote the manuscript. L Zhang, S Wang, Q Chen, and L Xiang performed critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patient for granting permission to publish this information.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings are available from the corresponding author, upon reasonable request.
Additional information
Funding
References
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):1–4. doi: 10.1111/bjd.18921.
- Cifuentes L, Kiritsi D, Chen W, et al. A case of junctional epidermolysis bullosa with prurigo-like lesions and reduction of collagen XVII and filaggrin. Br J Dermatol. 2013;169(1):195–198. doi: 10.1111/bjd.12241.
- Bauer JW, Lanschuetzer C. Type XVII collagen gene mutations in junctional epidermolysis bullosa and prospects for gene therapy. Clin Exp Dermatol. 2003; 28(1):53–60. doi: 10.1046/j.1365-2230.2003.01192.x.
- Condrat I, He Y, Cosgarea R, et al. Junctional epidermolysis bullosa: allelic heterogeneity and mutation stratification for precision medicine. Front Med (Lausanne). 2018;5:363. doi: 10.3389/fmed.2018.00363.
- McGrath JA, Gatalica B, Christiano AM, et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet. 1995;11(1):83–86. doi: 10.1038/ng0995-83.
- Keith AR, Twaroski K, Ebens CL, et al. Leading edge: emerging drug, cell, and gene therapies for junctional epidermolysis bullosa. Expert Opin Biol Ther. 2020;20(8):911–923. doi: 10.1080/14712598.2020.1740678.
- Beck LA, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–139. doi: 10.1056/NEJMoa1314768.
- Wang Y, Zhou M, Zhang L, et al. Amelioration of dystrophic epidermolysis bullosa pruriginosa symptoms with dupilumab: a case report. Dermatol Ther. 2021;34(6):e15130. Novdoi: 10.1111/dth.15130.
- Caroppo F, Milan E, Giulioni E, et al. A case of dystrophic epidermolysis bullosa pruriginosa treated with dupilumab. J Eur Acad Dermatol Venereol. 2022;36(5):e365–e367. doi: 10.1111/jdv.17887.
- Wu XG, Yan S, Jiang JQ, et al. Successful treatment of epidermolysis bulls parmigiano by unplumbed. J Dermatol. 2023;50(6):837–842. doi: 10.1111/1346-8138.16729.
- Yu L, Wang JB, Bian L, et al. Dystrophic epidermolysis bullosa pruriginosa: successfully treated with dupilumab. Dermatitis. 2023;34(1):58–59. doi: 10.1089/DERM.0000000000000954.
- Shehadeh W, Sarig O, Bar J, et al. Treatment of epidermolysis bullosa pruriginosa-associated pruritus with dupilumab. Br J Dermatol. 2020;182(6):1495–1497. doi: 10.1111/bjd.18855.
- Zhou AG, Little AJ, Antaya RJ. Epidermolysis bullosa pruriginosa treated with dupilumab. Pediatr Dermatol. 2021; 38(2):526–527. doi: 10.1111/pde.14493.
- Darbord D, Hickman G, Pironon N, et al. Dystrophic epidermolysis bullosa pruriginosa: a new case series of a rare phenotype unveils skewed Th2 immunity. J Eur Acad Dermatol Venereol. 2022;36(1):133–143. doi: 10.1111/jdv.17671.
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes) for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188(1):12–21. doi: 10.1093/bjd/ljac001.
- Chen F, Wei R, Deng D, et al. Genotype and phenotype correlations in 441 patients with epidermolysis bullosa from China. J Eur Acad Dermatol Venereol. 2023;37(2):411–419. doi: 10.1111/jdv.18692.
- Herisse AL, Charlesworth A, Bellon N, et al. Genotypic and phenotypic analysis of 34 cases of inherited junctional epidermolysis bullosa caused by COL17A1 mutations. Br J Dermatol. 2021;184(5):960–962. doi: 10.1111/bjd.19752.
- Snauwaert JJ, Yuen WY, Jonkman MF, et al. Burden of itch in epidermolysis bullosa. Br J Dermatol. 2014;171(1):73–78. doi: 10.1111/bjd.12885.
- Papanikolaou M, Onoufriadis A, Mellerio JE, et al. Prevalence, pathophysiology and management of itch in epidermolysis bullosa. Br J Dermatol. 2021;184(5):816–825. doi: 10.1111/bjd.19496.
- Breitenbach JS, Rinnerthaler M, Trost A, et al. Transcriptome and ultrastructural changes in dystrophic epidermolysis bullosa resemble skin aging. Aging (Albany NY). 2015;7(6):389–411. doi: 10.18632/aging.100755.
- Clawson RC, Duran SF, Pariser RJ. Epidermolysis bullosa pruriginosa responding to dupilumab. JAAD Case Rep. 2021; 16:69–71. doi: 10.1016/j.jdcr.2021.07.036.
- He H, Olesen CM, Pavel AB, et al. Tape-strip proteomic profiling of atopic dermatitis on dupilumab identifies minimally invasive biomarkers. Front Immunol. 2020;11:1768. doi: 10.3389/fimmu.2020.01768.
- Lettner T, Lang R, Bauer JW, et al. Increased levels of matrix metalloproteinase-9 and interleukin-8 in blister fluids of dystrophic and junctional epidermolysis bullosa patients. J Eur Acad Dermatol Venereol. 2015;29(2):396–398. doi: 10.1111/jdv.12399.