
Abstract
Purpose: This review evaluates the role of dose rate on cell and molecular responses. It focuses on the influence of dose rate on key events in critical pathways in the development of cancer. This approach is similar to that used by the U.S. EPA and others to evaluate risk from chemicals. It provides a mechanistic method to account for the influence of the dose rate from low-LET radiation, especially in the low-dose region on cancer risk assessment. Molecular, cellular, and tissues changes are observed in many key events and change as a function of dose rate. The magnitude and direction of change can be used to help establish an appropriate dose rate effectiveness factor (DREF).
Conclusions: Extensive data on key events suggest that exposure to low dose-rates are less effective in producing changes than high dose rates. Most of these data at the molecular and cellular level support a large (2–30) DREF. In addition, some evidence suggests that doses delivered at a low dose rate decrease damage to levels below that observed in the controls. However, there are some data human and mechanistic data that support a dose-rate effectiveness factor of 1. In summary, a review of the available molecular, cellular and tissue data indicates that not only is dose rate an important variable in understanding radiation risk but it also supports the selection of a DREF greater than one as currently recommended by ICRP (Citation2007) and BEIR VII (NRC/NAS Citation2006).
Introduction
Regulatory bodies
Consideration of both dose rate (National Council for Radiation Protection and Measurements [NCRP] Citation1980, Citation1993; United Nations Scientific Committee on the Effects of Atomic Radiation [UNSCEAR] Citation1993) and total dose (National Research Council/National Academy of Sciences [NRC/NAS] 2006, UNSCEAR Citation2012) is important in estimating risk and setting radiation protection standards. One of the primary data sets used by regulatory agencies to estimate radiation risk for low LET-radiation is that developed for survivors of the Japan atomic bombs (Preston et al. Citation2007; Shimizu et al. Citation2010; Ozasa et al. Citation2012; Hsu et al. Citation2013). The exposures in this case were delivered over a very short time. Exposure to relatively high doses delivered at high dose rates such as those from the atomic bombs and also for some radiation therapy practices, are known to increase the frequency of cancer (Preston et al. Citation2007) and some non-cancer diseases such as cardiovascular disease (Little et al. Citation2008b), cataracts (Blakely Citation2012) and stroke (UNSCEAR Citation2008; Shimizu et al. Citation2010).
Using human data in the low-dose region (less than 100 mSv), it has been difficult to define the shape of the dose-response relationship. However, regulatory bodies, following the lead of major scientific organizations, have adopted an approach that assumes that when low LET radiation is delivered at a low total dose (less than 100 mSv) or low dose rate (less than 5 mSv/day) there is a decrease in the effectiveness of radiation for cancer induction. This has resulted in scientific review committees recommending a dose and dose-rate effectiveness factor (DDREF) of, for example, 1.5 (NRC/NAS Citation2006) or 2.0 (International Commission on Radiological Protection [ICRP] Citation2007). The French Academy suggested that at low doses and low dose rates the dose-rate effectiveness factor may be very high or even protective (Tubiana et al. Citation2005). The controversy continues as illustrated, for example, by the German Commission on Radiological Protection (Strahlenschutzkommission [SSK] Citation2014) proposal that the DDREF be abolished, i.e. set at 1.0. Other reviews suggest there is still evidence for a higher value (Morgan and Bair Citation2013).
Before beginning a review of the data pertinent to dose and dose rate responses, it is necessary to define the relevant terms. The use of the term dose and dose-rate effectiveness factor (DDREF) has been considered necessary, based on a range of biological studies and selected epidemiology studies, for converting cancer risks obtained at relatively high doses and dose rates to predict risks at low doses (<100 mGy) and low dose rates (<5 mGy per hour). The use of DDREF has been restricted to the development of low-dose/low dose-rate cancer risk estimates for calculating detriment values to be used in setting dose limits for radiation protection purposes. It is not a definitive, measured value but rather a derived one based upon a selected data set that varies from organization to organization making recommendations. For example, after considering various human and experimental information, a value of 2 was selected by ICRP in Publication 60 (ICRP Citation1991). A considerable amount of discussion has ensued since this time on what are the appropriate data sets upon which to base a selection of DDREF and the methods for calculating a specific value. The general conclusions at this time are that ICRP in its most recent set of Recommendations (ICRP Citation2007) retained a value of 2, BEIR VII using a Bayesian approach for data analysis selected a value of 1.5 (NRC/NAS Citation2006) and UNSCEAR most recently elected not to use a DDREF. A number of epidemiological studies for populations exposed at low dose rates have proposed values consistent with 2 and as low as 1 for DDREF conversion [reviewed, for example, in NCRP Report no. 171 (2012) pages 129–140].
However, it has been proposed by a number of sources that it is more appropriate and more correct based on the available literature to separately consider a low dose effectiveness factor (LDEF) and a dose-rate effectiveness factor (DREF) for risk estimate calculations (reviewed in Ruhm et al. Citation2015). There are international (e.g. ICRP, Multidisciplinary European Low Dose Initiative [MELODI], and UNSCEAR) and national organizations (e.g. Public Health England [PHE], NCRP, and Electric Power Research Institute [EPRI]) that are currently addressing this issue and the associated necessary data sets.
A LDEF is necessary when extrapolating from high to low dose effects for an adverse effect dose-response curve that is essentially linear-quadratic (LQ), The LDEF is calculated as the ratio of the slope of the linear extrapolation from a point on the LQ curve and the slope of the linear component of this LQ curve. Thus, for acceptance of this approach, the need is to establish if, for example, the dose-response for radiation-induced cancer (particularly that for the atomic bomb survivors) is described by an LQ curve. There has been an active discussion on this topic with opinions for and against an LQ for the all solid cancer for the cohort that survived the atomic bomb. While it is difficult to reach a definitive conclusion because of the uncertainties associated with effects at low doses, the recent report by Ozasa et al. (Citation2012) provides a convincing argument for their being no threshold for all solid cancer.
The DREF is calculated as the ratio of the slope of the dose response at low acute doses to that at low doses and low dose rates. For an LNT application, the slope for acute doses is described by the slope of the curve over the entire dose range of epidemiology assessment. If the dose response curve is best described by an LQ application, then the low dose slope is dominated by the linear component of the LQ curve. The greatest uncertainty in calculating a DREF arises from the relative lack of epidemiology data for low dose/low dose rate exposures. The data for occupational and environmental low dose-rate exposures of human populations together with the associated uncertainties were reviewed in NCRP Report 171 (NCRP Citation2012). The general conclusion was that a DDREF of 1 is feasible but that higher values cannot be excluded. Thus, to help reduce this uncertainty, additional reliance has to be placed on animal and cellular data. A concern is that there is a lack of direct association between the non-epidemiology data and human cancer induction. It might well be possible to strengthen this relationship through the design of research to develop data bases that more directly address this relationship (NCRP Citation2015). Given these uncertainties, the selection of a DREF for radiation protection purposes is somewhat subjective and values of 1, 1.5, 2 or greater can be defended. This topic will be discussed in great detail in the body of this review.
Manuscript goals
The authors of this manuscript are well aware of the controversy associated with the response in the low dose region, the large number of epidemiological studies conducted to help define the risk in the low dose region and the data bases that have been developed using animal studies to help address these issues. It is well established that the human data are the primary source for risk estimates from radiation exposure with animal studies providing additional supporting evidence. However, for many environmental stressors there are only limited human data. In these cases it has been necessary to use molecular, cellular and animal data as the primary source of information in setting regulatory standards (EPA Citation2005). To do this studies have focused on the key events in the critical pathways to the development of cancer (Adeleye et al. Citation2015; Preston Citation2015; Edwards et al. Citation2016). This particular approach, which is described in more detail below, has not been used until now for evaluation of the role of dose rate on risk from exposure to ionizing radiation. However, we have conducted an analysis using the human epidemiology data (Hoel Citation2015) and animal data (Brooks et al. Citation2009) to evaluate the role of dose and dose rate on risk. Additional research is currently underway to integrate the information at all levels of biological organization with a focus on the animal and epidemiological data. To accomplish the larger task, in this manuscript we provide a comprehensive review of the molecular and cellular studies that have been conducted in the low dose and dose-rate region.
We discuss the influence of dose rate on key events and critical pathways at the cell and molecular levels as additional information on the mechanisms of action for low dose rate radiation. High doses of low LET radiation delivered at low dose rates markedly reduce the effectiveness of low LET radiation and thus, as noted above, it may be appropriate to separate the DREF from the DDREF (and LDEF) (Brooks et al. Citation2009). The current manuscript will thus, focus only on the molecular and cellular events and their influence on the dose rate effectiveness factor.
Dose rate and dose fractionation in medicine
It has long been accepted in the field of radiation biology that delivering high total radiation doses (greater than 1 Gy), at a low dose rate has a marked sparing effect on tissue reactions (formerly, deterministic effects. Without exception, when a high dose of radiation is delivered at a low dose rate (mGy/week or mGy/year) it has much less biological impact than when it is given at a high dose rate (Gy/min or Gy/sec). It has also been observed that giving a radiation dose as a series of fractions, with a time for ‘tissue recovery’ between exposures, decreases the impact on normal tissue in areas surrounding the tumor. These observations have been taken into the clinic. Radiation treatment for cancer is often delivered as dose fractions. With proper focusing of the radiation beam and fractionated treatment schedules, doses as high as 2.0 Gy per fraction can be delivered to tumor tissue (30–70 Gy total dose) maximizing the cancer cure rate and limiting normal tissue damage. The specific cancer risks from medical exposure, both diagnostic and therapeutic, have been estimated (NRC/NAS Citation2006; Brenner and Hall Citation2007; NCRP Citation2009). When these risks are weighed with an estimate of the measured benefits from these radiation procedures, the benefits have been shown to be much greater than the calculated risks (Zanzonico and Stabin Citation2014). Such information is critical when considering nuclear medicine procedures (Brooks and Dauer Citation2014).
Research on basic mechanisms of radiation responses to low dose and dose-rate exposures
Does low dose radiation exposure (less than 100 mGy) delivered at a low dose rate, result in a lower biological stochastic response (e.g. cancer induction) and consequently, a reduced risk than the same dose delivered at a high dose rate? This question has been the focus of much research over the past 20 plus years. This research includes, but is not limited to U.S. Department of Energy, Low Dose Radiation Research Program (http://lowdose.energy.gov), the extensive research programs in the European Union (MELODI, Epirad bio, Store and DoReMi) integrating low dose research (http://www.doremi-noe.net), the Japanese research IES (Institute for Environmental Sciences (http://www.ies.or.jp/index_e.html), and research conducted in Korea by the Korean Society for Radiation Bioscience (http://www.ksrb.kr/english/intro/intro_01.asp). All these organizations have invested and continue to invest considerable time and money to address this critical question.
The present manuscript applies the concepts of key events and Adverse Outcome Pathways (AOP), (or critical pathways) to help understand the effect of dose rate on biological responses. This approach makes it possible to evaluate the potential role of dose rate on molecular, cellular and tissue responses and provides a mechanistic and scientific basis for radiation protection standards. Using modern cell and molecular biological techniques recent research on the influence of dose rate is reviewed. In the same way the U.S. EPA has evaluated the effect of environmental chemicals on key events in critical pathways that result in cancer (EPA Citation2005; Simon et al. Citation2014; Preston Citation2015; Edwards et al. Citation2016). This approach is organized in a way that allows for an evaluation of the key events in critical pathways that result in cancer and their use in estimating risk at low exposure levels. For example, this concept has been refined and used to evaluate four categories of bioactive agents to define safe levels of intake for: food allergens, nutrients, pathogenic microorganisms and environmental chemicals (Julien et al. Citation2009). It has also been used in a number of additional adverse outcome/risk assessment situations (see for example, Organization for Economic Co-operation and Development [OECD] Citation2015). For the present review, the AOP/key event approach has been adapted for radiation exposure to investigate the influence of dose rate in the low dose range on adverse animal, cellular and molecular outcomes. It is important to stress that this same approach has been applied for chemical exposures and provides a framework for data collection and research planning (Edwards et al. Citation2016).
Hallmarks of cancer
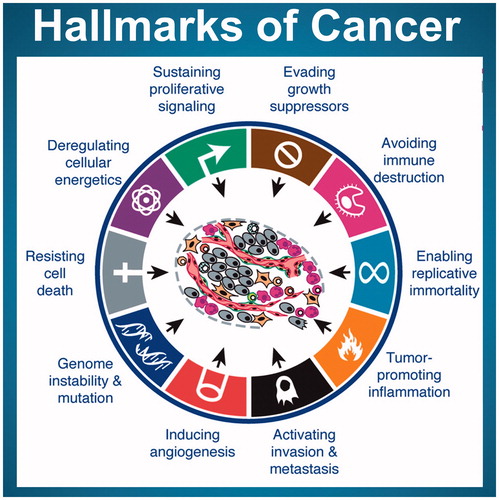
In order to be able to develop adverse outcome pathways (initially for cancer) and the associated key events, it is necessary to establish if there is a set of characteristics that can be used to define cancer in general, irrespective of the species, the site or the inducing agent. A set of such phenotypic outcomes (acquired characteristics) has been proposed by Hanahan and Weinberg in their so-called ‘Hallmarks of Cancer’ (Hanahan and Weinberg Citation2000). The original hallmarks were updated to include genomic instability, reprogramming of energy metabolism and evading immune destruction (Hanahan and Weinberg Citation2011).
These hallmarks are shown in (Hanahan and Weinberg Citation2011) and serve as the phenotypes that have to be produced by key events that would need to be induced by radiation for cancer to develop. Information is available on the radiation-induced dose and dose-rate response relationships for some of these ‘hallmarks’ (and underlying key events) but little is known about the radiation response of others (Boss et al, Citation2014). For the present review, we will consider the radiation responses of these underlying key events and discuss their dose-rate responsiveness. It is to be noted that for specific risk assessment considerations, the generic approach described here for cancer in general has to be modified to address specific adverse outcomes (for example, radiation-induced breast cancer or lung cancer) by determining the key events that are a feature of this specific outcome.
Figure 1. A graphic representation of the ‘Hallmarks of Cancer’ from Hanahan and Wienberg (Citation2011). These are the changes required for the production of cancer.
Critical pathways and key events for radiation-induced cancer
The key events in the critical pathways to radiation-induced cancer occur at all levels of biological organization. This manuscript will evaluate changes in key events measured at the molecular, cellular and tissue level after radiation exposure to low dose and dose rates. summarizes the types of key events that are applicable for cancer outcomes, in general, and not descriptive for any particular cancer. Where dose and dose-rate response data for a particular key event are available, this key event is shown in bold in . For the key events shown in red in , there is little information available from which direct comparisons can be drawn between the responses following low and high doses or low and high dose-rate radiation exposures. These key events will not be discussed further in this manuscript.
Table 1. Key events are listed at the molecular, cellular and tissue levels. The events in bold have data on the influence of dose rate and are discussed while those not in bold have little information and are not further discussed in the manuscript.
The AOP/key events approach described in is a generic one for illustrative purposes. In order to use the approach in a dose-response/risk assessment framework, it is necessary to adapt it for a specific cancer type, for example. Thus, the need will be to develop a detailed AOP for the specific example. The information on the key events identified (e.g. dose response, dose-rate response) can be used as input parameters for a biologically-based dose-response (BBDR) model (NCRP Citation2015). In fact, this approach has been applied in initial attempts to define low dose cancer outcomes in a qualitative manner. Moolgavkar and colleagues modified their two-stage clonal growth model for radiation exposures (Moolgavkar and Knudson Citation1981; Krewski et al. Citation2003). This approach has been further developed by a number of investigators to reduce uncertainty in the original approach (Little et al. Citation2002; Citation2008a; Little and Li Citation2007). Other approaches have been used that, in general, use a type of key event of approach with the aim of providing quantitative cancer risk estimates (Shuryak et al. Citation2010). In addition to a simplified model approach (such as with the two-stage carcinogenesis model), it is feasible to use more realistic models of carcinogenesis. For example, models that establish whether specific processes such as genomic instability or adaptive responses are predictive of the cancer response in specific studies (e.g. Eidemuller et al. Citation2011). Perhaps the most informative type of BBDR models are ones that are developed to examine the role of specific measured biological variables in the genesis of radiogenic cancer in a particular study. Examples of this informative approach have been reported by Heidenreich and Rosemann (Citation2012) and Heidenreich et al. (Citation2013). Further development of this latter type of model using key-event bioindicators as input parameters is encouraged. Thus, the AOP/key event approach is grounded in previous research for radiation exposures and a fairly extensive literature on risks from environmental chemicals.
The AOP for a particular radiation-induced cancer, for example, will be characterized by a series of steps (key events). However, it is not necessary to be able to quantify all these steps to provide an estimate of risk at low doses. Of course, uncertainty will be reduced when more key events can be used as parameters in a BBDR model. Key events that are closest to the adverse outcome itself are likely to be more predictive of the adverse outcome and a suite of key events is likely to be more informative as regards risk than individual key events. Thus, the approach is a viable one based on current and predicted research capabilities and computational advances.
Influence of dose rate on dose to individual cells
Total radiation dose or ‘hits’ to an individual cell when exposed to low dose rates is limited by the time between cell divisions. This is a critical consideration when evaluating dose-rate effects on biological responses. Following acute radiation exposure the total energy is distributed over each and every cell present at the time of the exposure. Assuming a cell mass of 1 ng, it is possible to calculate that exposure to 0.1 Gy of 100 kV X-rays results in 100 ‘hits’ per ng or cell (Feinendegen and Graessle Citation2002). If the same exposure is protracted over one year, the hits per cell would depend on the cell turnover time of that particular cell. Each cell type has a unique cell cycle and turnover at different rates. For example, there are multiple types of cells in the bone marrow with many of them turning over in a day or two (Brooks and McClellan Citation1968), lung epithelial cells have an average cell turnover time of months (ICRP Citation2007; Brooks et al. Citation2009) and liver hepatocytes have a cell turnover time of years (NCRP Citation2001). The number of ‘hits’ per cell division in a rapidly dividing cell type at this dose is compared for a single acute exposure and one protracted over one year (). This shows individual cells receive different numbers of hits per cell cycle as a function of dose rate. For bone marrow exposed to 0.1 Gy/year and with an assumed cell turnover time of 3.6 days, the cells would over the year receive about 1.0 hit/cell cycle. For cell types with longer turnover times, like the liver, each cell could receive 100 hits/cell cycle when exposed to 0.1 Gy protracted over a year.
Figure 2. In this Figure the circles represent cells and the lines show energy deposition events within the cells. For an acute exposure of 0.1 Gy given acutely each cell has many energy deposition events. Protracting the dose over a year results in very few events in any one cell and little chance for the damage from one ionization to interact with that from a second.

In addition to the role of cell turnover on cellular dose, cells have multiple other mechanisms for repairing damage. At high dose rate some of these mechanisms may not be as effective as they are at low dose rates. Thus, at low dose rates the same total dose results in very different cellular doses and risks depending on the protective mechanisms and cell cycle of each individual cell type (Feinendegen et al. Citation2007).
Methods
Estimating dose rate effectiveness factor (DREF)
This section provides a description of the methods used to estimate a DREF from data available for the key events in the critical pathways to cancer. There is wide range of different types of data for these key events and so it is necessary to establish a set of methods to maximally use these data for estimating values for DREF. The research evaluated in this manuscript is represented by four major types of data.
In many of these research studies, either a single high or low dose exposure was used. These studies are not very useful for estimating the role of dose rate on key events.
Other research used a single high dose or a single low dose exposure both delivered at the same dose rate or at a single dose rate with a variable dose. Again these types of studies are not informative for calculating a DREF. However, such research demonstrated that the cell and molecular responses were different for high dose vs. low dose as well as for high dose rate vs. low dose rate. Such information provided mechanistic insight to the responses and suggests that the mechanisms of action differ as a function of both dose and dose rate. These studies also demonstrate that cell and molecular changes can be developed as biomarkers of exposure when physical dosimetry is not available.
Further studies used a single dose but with a variable dose rate. In these studies it was possible to use the ratio of the response to the high dose rate to that of the low dose rate to provide a single point estimate of the DREF. However, for many of these studies the endpoint responded to the high dose rate but not the low dose rate for which the response was not different from the controls. Thus, if the background response is subtracted from the radiation induced response from both high and low dose rate and if the net response for the low dose rate is undetectable (zero), then the ratio comparing high to low dose rate response approaches infinity. Such research, where there was no net response following low dose rate exposure, demonstrated that the relative responses were dependent on dose rate but could not be used to determine a numerical value but suggests a high value for DREF.
The most informative data sets are those that have complete dose response information obtained for different dose rates. If the dose responses were linear following both the high and low dose-rate exposure, a direct comparison of the slopes as a function of dose rate can be used to estimate DREF. For some endpoints, such as chromosome aberrations, the dose-response relationships for the low dose-rate exposure were linear and for the acute response were non-linear. There are different ways to estimate a DREF from such data. The method used in the present manuscript was to compare the response at a single dose (such as 1.0 Gy) delivered at different dose rates. This provides a single point estimate but one that changes depending on the specific dose selected. An alternative approach is to conduct curve fitting and compare the linear portion of the curves for the acute exposure with that for the chronic exposure. This method uses only the linear term so it often produces a DREF value of 1.
Results
Key events: molecular level
At the molecular level our review focuses on radiation-induced DNA damage and changes in gene expression in the low dose and dose-rate range. Information on changes in protein expression and post translational protein modifications are not currently well documented as a function of radiation dose or dose rate.
DNA damage, repair and signaling
High dose rate
The first key event and an important hallmark of cancer following exposure to ionizing radiation is the induction of changes in the DNA due to the deposition of energy in cells. This damage represents the primary cellular and molecular data that support the Linear-No-Threshold (LNT) hypothesis. The linear dose-response relationship for high dose-rate, acute radiation-induced DNA damage is well defined and covers a wide range of doses (Erixon and Cedervall Citation1995; Asaithamby and Chen Citation2009). This damage is indicated by double-strand breaks, evidenced by the presence of γH2AX. The use of γH2AX as a marker of DNA damage makes it possible to not only determine DNA damage from acute high doses, but extend the DNA damage dose-response relationship to very low doses and dose rates. This technique has become a useful tool (Rothkamm et al. Citation2007). At high dose rate there is a linear dose response for the induction of DNA alterations.
Research has also suggested that repair of DNA damage in the low dose region is limited (Rothkamm and Löbrich Citation2003; Löbrich et al. Citation2005). This was postulated to be related to the inability of low doses to induce a sufficient amount of DNA damage to stimulate the induction of DNA repair genes (Rothkamm and Löbrich Citation2003; Rothkamm et al. Citation2007; Grudzenski et al. Citation2010). These data suggest that the cellular response to double-strand breaks is substantially different for low vs. high doses of low LET radiation. If there is no DNA repair at low doses and if DNA damage is directly predictive of cancer, the cancer risk should increase linearly with dose. It has further been postulated, based on the possible lack of repair at low doses that the cancer risk at these low doses could be higher than that obtained by linear extrapolation from high doses. In addition, if the responses at low doses are independent of dose rate because of a lack of repair, then a DREF of one or less would be obtained.
Recent data, using ATM foci as an endpoint, demonstrated that DNA double-strand breaks induced by single acute low doses of radiation, in cells that do not have energy deposited in them (bystander cells) are persistent and have a longer life time than the DNA damage induced in cells by direct deposition of energy (Ojima et al. Citation2011). Such observations suggest that a greater risk for these cells to progress toward cancer. There are no data on the effect of dose rate in this model but these low dose data would not seem to support a DREF of greater than one.
Low dose rate
A major response to DNA damage is the induction of different signaling pathways (Lavin et al. Citation2005). These pathways trigger a range of responses some of which has been linked to genomic instability.
Studies on DNA damage suggest that dose rate has a marked influence on the response. Recent studies demonstrated that, when a total accumulated dose 400 times higher than natural background, was delivered at a low dose rate, it was not possible to detect an increase in DNA damage. DNA damage in these studies included base damage, micronuclei, or p53-inducible gene expression. However, when the same total accumulated dose was delivered at high dose rates, many types of DNA damage were readily measured (Olipitz et al. Citation2012).
Changes in mitochondrial DNA were not detected in animals living in the Chernobyl environment following the low dose-rate exposure. Changes in this biomarker could be readily detected following the same dose delivered as a single acute exposure (Wickliffe et al. Citation2002). The data from these studies indicate that repair does occur for both nuclear and mitochondrial DNA damage following low dose-rate exposure. A dose-rate effectiveness factor of infinity would be calculated for all these endpoints since at low dose rate the responses were not measurable.
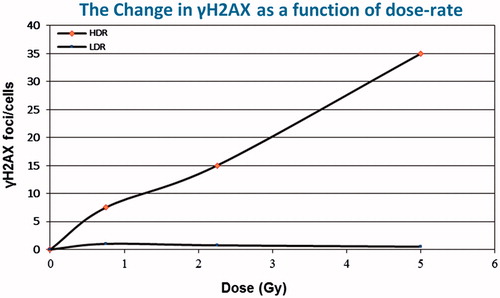
The influence of dose rate on DNA damage and repair was assessed using changes in the level of γH2AX foci/cell, thought to be a measure of DNA damage and repair. Additional data were included that measured changes in phosphorylation as a function of dose rate. The phosphorylation data were expressed as a ratio of the radiation response to that of the control so it is not possible to estimate a DREF. However, the frequency of γH2AX foci/cell provides a direct measure of the interaction between damage and repair. For this endpoint a complete dose response was generated for both high and low dose rate exposure, comparing the slopes of the lines provides a direct estimate of DREF. Exposure to high dose rates resulted in a linear increase in γH2AX over a wide range of doses. When the same dose was delivered at low dose rates, little increase in the frequency of γH2AX above background levels was observed. This observed difference was consistent up to a very high accumulated dose of 5.0 Gy. These results are shown in where γH2AX foci/cell is plotted against radiation dose delivered at either a high or low dose rate (Ishizaki et al. Citation2004). From such data a dose-rate effectiveness factor of about 30 can be calculated. After high doses delivered at a high dose rate, the interaction between damage and repair results in a linear increase in the frequency of γH2AX. Low dose rate exposures show that over the time required to deliver the dose much of the induced damage is repaired supporting a high dose-rate effectiveness factor, much greater than 1.
Figure 3. The influence of dose rate on the induction of γH2AX. This marker of DNA damage and repair is very sensitive to changes in dose rate. Comparing the slopes of the lines a DREF of about 30 can be calculated.
Studies have been conducted on the influence of aging on DNA repair since age seems to be an important factor in radiation-induced breast cancer. Human mammary epithelial cells were exposed to fractionated low doses (20 mGy), similar to those experienced in a mammogram and the frequency of γH2AX and micronuclei measured as a function of age in vitro (Hernandez et al. Citation2013). This study suggested that aging of cells in vitro decreased the ability of cells to repair DNA damage. Such studies are important and suggested that aging of cells even without shortening of telomeres can modify DNA repair. The major concerns about such studies are the relationship between aging in cell culture in an artificial environment which takes place in days and comparing it to aging in human populations over years. Since all the exposures in this study were given as single acute exposures it is not possible to relate these results to a dose rate effectiveness factor.
Radiation can cause multiple breaks in a very short region of the DNA. These damaged sites have been called locally multiply damaged sites (Ward Citation1994) and have been postulated to be very difficult to repair.
Recent publications suggest that DNA repair centers, radiation-induced foci (RFI), are formed following exposure to ionizing radiation. Research determined that formation of these centers was not linearly related to radiation dose. Following low doses of ionizing radiation (0.1 Gy) the number of repair centers per unit of dose was about four times higher than observed for high doses (2.0 Gy). From these studies the authors demonstrated a non-linear increase in repair foci at low dose rates and suggested that this would result in non-linear dose-response relationship with repair being more effective after low dose rates than at high (Neumaier et al. Citation2012). These studies are in marked contrast to the studies of (Rothkamm and Löbrich Citation2003; Grudzenski et al. Citation2010) who suggested, as noted above, that low doses of radiation do no induce genes needed for DNA repair and that little or no repair of DNA damage was detected following low dose exposures. Since both DNA repair and the production of RIFs decease rapidly as a function of time after induction, they would be expected to be much lower as a function of dose rate and may be of limited value in determining a DREF. Additional studies on the influence of dose rate on the formation of DNA repair centers are needed. For the induction of repair centers the DREF cannot be calculated, but the available data suggest that it would be much greater than 1.
Gene expression
High dose rate
Extensive advances in biological and physical sciences over the past 20 years have made it possible to measure radiation-induced biological changes in gene expression in the low dose and low dose-rate region. These advances have been spurred on by the sequencing of the genome and the development and use of gene expression arrays and other more informative methods such as RNA sequencing. Nevertheless, at the present time there are limited data on the effect of dose rate and total dose on biological responses using these newly developed techniques. This represents an important area for future research.
Using gene expression arrays it is now possible to measure radiation-induced changes in gene expression in thousands of genes at the same time (Yin et al. Citation2003). Following high dose-rate exposure, the pattern of gene expression changed markedly as a function of total dose. Research determined that changes in gene expression include, ‘… genes that respond only to low-dose exposures, genes that are unique to high-dose exposures, and genes that are modulated in their expression at both high and low doses’ (Coleman and Wyrobek Citation2006). The changes have been measured in human and mouse cells. A brief summary of these data are provided in .
Table 2. Biological pathways where changes in gene expression were measured as a function of radiation dose (Coleman and Wyrobek Citation2006). This table illustrates that many pathways respond to either high or low total doses suggesting unique mechanisms.
It is of interest to note from the table that some responses in human cells in vitro differ from those in mouse in vivo. Also of relevance to the present topic are the unique responses in the low dose region for genes responsible for heat shock proteins, immune response and protein synthesis. The data suggest that cell cycle genes and those involved in cell signaling are modified by both low and high doses in each of the systems tested. Such results make it difficult to use these data for human risk assessment. Extensive research on gene expression levels has been conducted and for the most part support the data presented in (Fornace et al. Citation1999; Amundson et al. Citation2003a, Citation2003b; Ding et al. Citation2005). Gene expression patterns change as a function of dose. Some of the genes involved in these radiation induced changes are very important in the critical pathways to cancer. A review of the changes in gene expression as a function of dose suggests that they reflect a change in the mechanisms of action (Dauer et al. Citation2010). Such studies demonstrate that the responses to this key event (gene expression) in the critical cancer pathways change as a function of radiation dose. High doses trigger pathways that have been postulated to be damaging while the molecular pathways upregulated by low doses have been considered to be protective.
Such information provides the basis for postulating that the cellular responses to radiation-induced damage are different following low doses than after high doses. These mechanistic studies have been expanded to evaluate further the high and low dose-rate radiation responses that can be used to predict cancer outcome based on mechanistic understanding. Even though such data cannot be used to calculate a dose or dose-rate effectiveness factor, they do indicate that it is significantly greater than one.
Radiation alters gene expression patterns and induces signaling processes. Such signaling processes initiated by DNA damage are an important factor in determining radiation responses. These responses are involved in activation of many genes associated with stress following radiation exposure (Amundson et al. Citation2003a). Many of these stress genes are associated with transcription factor p53, ‘one of the key elements in cellular response, which can regulate nearly 100 genes that have already been identified’ (Fornace et al. Citation1999).
Low dose rate
Studies on gene expression using low dose-rate exposures have also been conducted and illustrate that different genes are activated as a function of dose rate, time after exposure and tissue type (Amundson et al. Citation2003b). All of these variables become important in understanding the risks associated with low dose-rate exposures.
A number of studies have been conducted using gene expression as a biomarker of radiation exposure (Paul and Amundson Citation2008; Paul et al. Citation2015). The data were derived as a function of both dose and dose rate. It was determined that gene expression is a useful biomarker and can be used to estimate radiation dose following exposures at both high and low dose rates.
Human exposures (ex vivo exposures)
Human blood was exposed to either high or low doses of low LET radiation and expression measured in genes transcriptionally regulated through DNA damage by the tumor suppressor p53 (Manning et al. Citation2013). Individual variation was noted among samples. It was also determined that the dose-response relationship following high doses was best fit by a polynomial expression while the dose-response from low doses was found to be linear. It was also determined that some of the p53-regulated genes were responsive following high or low doses delivered at either low or high dose rates (Ghandhi et al. Citation2015). Such studies suggest that there are minor differences in the response to these informative genes and seem to support a DREF that is not different from 1. The major impact of these studies is to demonstrate that changes in gene expression may be useful as biomarkers of exposure and can detect differences in gene expression induced as a function of both radiation dose and dose rate. Additional research is needed to determine how and if these biomarkers can be used to estimate DREF.
Human exposures (in vivo exposures)
Following low dose-rate exposure of workers, many of the genes activated are involved in normal physiological processes that have been shown to protect the body from harm, such as ubiquitin cycle, DNA repair, cell cycle and proliferation and stress response genes (Fachin et al. Citation2009). Medical workers exposed to low and fractionated doses also showed changes in gene expression which were related to important biological processes including DNA packaging and mitochondrial electron transport. In the higher exposed group, medical workers showed ‘a significant modulation of ion homeostasis and programmed cell death as well.’ These changes suggest different mechanisms at low doses from those occurring at high doses (Morandi et al. Citation2009). These unique mechanisms of action at different dose levels for low dose-rate radiation exposure support the non-linear dose-response data and the increased repair observations in humans and suggest the a dose-rate effectiveness factor could be greater than one. How such responses influence risk for cancer is difficult to determine and requires additional research. An accurate estimate of DREF for these low dose and dose rate exposures cannot be calculated from the data available.
Paul et al. (Citation2015) conducted a study to determine if it was possible to develop a molecular biomarker which could help determine the exposure dose rate by comparing the changes in gene expression as a function of dose rate. The high dose rate was 1.03 Gy/min and the low dose rate 3.09 mGy/min (6-h total dose 1.1 Gy, 12-h total dose 2.2 Gy, 24-h 4.4 Gy). These low dose-rate exposures were delivered to the whole animal and changes in gene expression were measured in blood samples. With this protocol and given the slow turnover of blood cells, almost every individual cell would receive the total dose regardless of dose rate. This is valuable information. However, it is difficult to extrapolate effects from these short-time dose protractions to real world long-time exposures to very low dose-rates. For example, for fallout, internally deposited radioactive materials and other protracted exposures, the dose is usually received over weeks, months or years. Additional research on gene expression is needed for these very long exposure times.
All these studies support the observation that there are unique genes up- and down-regulated as a function of dose rate. The mRNA metabolic and processing genes as well as IgA production genes are among those modified only by low dose-rate exposures. Using rather large total doses, both low dose- rate and high dose-rate exposures resulted in an enhanced p53 signaling pathway. Not surprisingly, genes in this pathway were also up-regulated in response to acute exposures (Paul et al. Citation2015). This approach and these data are useful in developing a biomarker of dose-rate effects but it is not possible to use this information to estimate a DREF.
Key events: cellular
At the cellular level, induced key events along the critical pathway to cancer following low dose-rate exposure include mutations, chromosome aberrations, cell killing, cell cycle alterations and cell transformation. Dose-response data are available as a function of dose and dose rate for several of these endpoints.
Radiation-induced mutations
High dose rate
Radiation is an effective cell killer which also produces chromosome damage. Because of its ability to kill cells, radiation is widely used in cancer therapy. Since it is such an effective cell killer, radiation is not a strong inducer of recoverable mutations. Studies on the A-bomb survivors did not report an increase in mutations transmitted to offspring (Neel Citation1998). Can we consider mutations as a key event in the production of radiation-induced cancer? For example, if one conducts a radiation-induced mutation study on somatic cells at medium to high doses, it is necessary to seed a large number of cells since the radiation kills most of these cells and produces few mutations in the surviving cells (Hsie et al. Citation1978). Thus, radiation-induced mutations are expressed as mutations per surviving cell. Radiation-induced somatic cell mutations have been characterized as mostly being large deletions and rearrangements (Jostes et al. Citation1994). DNA strand breaks induced by ionizing radiation in general are not predictive of the production of many of the types of mutations that are important in cancer induction (in particular small base changes and small deletions). Many chemicals produce base changes and rearrangements important in cancer induction at chemical doses where there is relatively little cell killing. Thus, mutated cells induced by some chemicals are available to progress along the cancer pathway. However, the picture is more complex since many agents classified as human carcinogens have been shown to be non-mutagens which questions the role of directly-induced mutations in many types of cancer (International Agency for Research on Cancer [IARC] Citation2012).
Low dose rates
A number of techniques have been developed to detect mutations induced by radiation in germ cells that can be transmitted to the offspring (Russell and Matter Citation1980; Brooks Citation1986). Early research on transmitted germ cell mutations suggested that large radiation doses were necessary to produce a detectable number of mutagenic changes in offspring (Russell et al. Citation1958; Russell Citation1968). When the radiation was delivered at a low dose rate, the frequency of transmitted germ cell mutations decreased markedly, especially for female mice (Russell et al. Citation1959). This research suggested a dose-rate effectiveness factor of about three. The frequency of mutations induced by internally deposited radioactive materials in mice was similar to that produced by low dose-rate whole body exposures (NCRP Citation1987), and so a similar DREF would be predicted. Further evaluation of these data determined that the major influence of dose rate was a reduction in the frequency of large lesions such as deletions and rearrangements (the major types of genetic damage produced by ionizing radiation) (Russell and Hunsicker Citation2012), again resulting in a dose-rate effectiveness factor of about 3.
An informative method for measuring mutations in mice following low doses and dose rates was recently developed based on the frequency of recombinational events with the pKZi recombinational mutation (Sykes et al. Citation2006). This assay measured the impact of priming low doses of ionizing radiation followed by a high challenge dose as was used in previous adaptive response studies (Wolff Citation1995). These studies used a priming dose of either 0.01 or 10 mGy and a challenge dose of 1000 mGy (1.0 Gy). The mutation frequency induced by the large challenge dose was depressed by a low priming dose to levels at or below the spontaneous mutation frequency (Hooker et al. Citation2004; Day et al. Citation2007).
Concerns about this particular assay have been expressed since it was postulated to be unique to blood lymphocytes (Zeng et al. Citation2006). These cells are part of the immune system and it is feasible that the observed adaptive response may simply have been a reflection of the spontaneous rearrangements that occur and may not play a role in cancer induction. To address these concerns, additional research was conducted which measured mutation frequency in prostate (Hooker et al. Citation2004) and spleen (Day et al. Citation2007) using four different strains of mice. These studies indicated that the somatic tissues showed the same radiation-induced decrease in the frequency of mutations as was observed for lymphocytes, namely the frequency of mutations following low levels of radiation exposure were not significantly different or below those observed in the tissues of non-exposed animals. These studies resulted in very interesting non-linear, protective dose-response relationships (J-shaped curve). However, the phenomenon does require additional study. If radiation-induced mutations are a key event in the progression to cancer then these particular results could suggest that the LNT model was not valid for this endpoint with the potential need to assign a negative or protective DREF.
Chromosome aberrations
High dose rate
Radiation-induced chromosome aberrations change as a function of dose, dose rate radiation energy and radiation type (Bender et al. Citation1988; Guerrero-Carbajal et al. Citation2003). The role of total dose and dose rate on the induction of chromosome aberrations has been reviewed a number of times (for example, Brooks Citation1980; Lucas et al. Citation1999, Citation2004). Chromosome aberration frequency has long been used as a biomarker of radiation exposure (Bender et al. Citation1988). Following acute exposures, non-stable aberrations such as dicentrics provide the best early biomarker of radiation dose. With the development of chromosome painting technology, it became possible to detect both asymmetrical and symmetrical translocations more accurately than by traditional staining methods (Edwards et al. Citation2005). Symmetrical translocations survive cell division and thus are more stable than asymmetrical aberrations. This makes them particularly valuable for detecting changes induced by low dose-rate exposures (Hsieh et al. Citation1999).
Even though chromosome damage provides a useful measure of radiation dose, does an increase in the frequency of chromosome aberrations always predict an increased risk? Some animal studies suggest that they do not (Bao et al. Citation1997; Brooks et al. Citation2003). There are disconnects between the frequency of chromosome aberrations and the induction of cancer. For example, following inhalation of radon, rats develop a high frequency of deep lung cancers, yet no cancers of the trachea have been detected in these same animals. The frequency of chromosome aberrations induced by radon exposure, measured as micronuclei, is the same in the trachea and the deep lung epithelial cells and fibroblasts (Bao et al. Citation1997; Brooks et al. Citation1997). Aberration frequency per se in these tissues does not predict cancer: the same amount of chromosome damage results in a different frequency of cancer. The aberrations predict the dose to the tissues but not the cancer frequency. There are also disconnects between aberration frequency and radon induced lung cancer in different species. Rats have a high frequency of radon-induced lung cancer and Syrian hamsters or Chinese hamsters exposed to similar doses of radon have a very low frequency. All three species have the same frequency of chromosome aberrations induced by radon or gamma rays in the lung cells (Khan et al. Citation1995; Bao et al. Citation1997). Thus, aberration frequency was very effective in predicting past exposure and dose, but was not a useful predictor of radiation-induced cancer in different tissues or species.
Reviewing the data on dose-rate effects demonstrated that, if the cell cycle is short relative to the exposure time, the asymmetrical translocations (dicentrics and rings) are lost rapidly from the population. However, there is evidence that a high proportion of symmetrical (reciprocal) translocations is able to survive through cell division and can modify the genotype and phenotype of the cells. Thus, they are considered to be important in cancer development (Lucas et al. Citation2004) and a key event (Preston Citation2015). After high dose rates, chromosome aberrations are produced as a non-linear function of radiation dose (Bender et al. Citation1988). This has been postulated to be related to ‘hits’ and the requirement for multiple hits to produce complex exchange types of chromosome aberrations. This non-linear function makes it impossible to derive a single value for a DREF. To estimate a DREF, the response after a single dose is often used, such as the response following exposure to 1 Gy. Using this method to calculate DREF from chromosome aberrations, dose rate effectiveness factors ranging from 1.0–3.0 have been found.
To illustrate the role of cell turnover time on the frequency of radiation-induced chromosome aberrations induced by low and high dose rates, research was conducted using rapidly dividing bone marrow cells, lung fibroblasts with intermediate cell cycle times and slowly dividing liver epithelial cells.
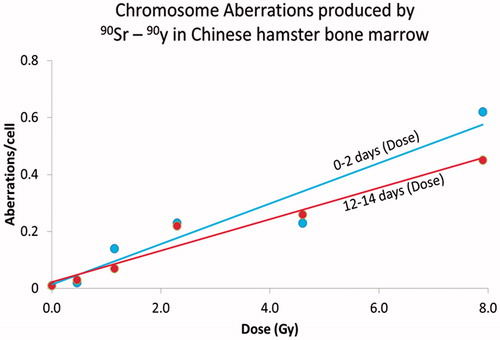
For rapidly dividing bone marrow cells the rate of formation of chromosome aberrations at low dose rates is in equilibrium with the rate of loss of aberrations through cell division. This is especially true for non-stable aberrations such as chromatid aberrations, deletions, dicentrics and rings. Since 90Sr delivers the dose to the bone marrow at a low and rather constant dose rate the amount of radiation-induced damage induced by 90Sr increased as a function of dose rate or dose delivered per cell cycle and reached equilibrium within two days. At this time, the loss through cell division of cells containing chromosome aberrations was equal to the frequency of aberrations produced during each cell cycle. To illustrate this, shows the frequency of aberrations plotted as a function of dose delivered either over the first 2 days, (days 0–2) or over the last two days (days 12–14) prior to aberration scoring (Brooks and McClellan Citation1968, Citation1969). The slope of the dose-response relationships using these metrics of dose are the same. This demonstrates that the rate of formation of aberrations over the early two day time period was the same and the frequency of aberrations induced over the last 2 days. This demonstrates that aberration frequency is dependent on dose-rate or dose per cell cycle and not total dose. If the dose-response for the total dose were used the slope of the dose response relationship over the 14-day time interval would be much lower since the bone marrow tissue had a much higher total dose over 14 days than over 2 days.
Figure 4. The influence of dose rate on the frequency of chromosome damage in the rapidly dividing bone marrow cells in Chinese hamsters exposed to 90Sr-90Y. In this Figure the chromosome aberrations frequency is related to either the dose delivered over the first 2 days (0–2 days) or the dose delivered over the last 2 days of the study (12–14 days). The frequency of aberrations increases as the same function of dose over these selected time periods. If the total dose were to be used on the x-axis the slope over the total time would be much less than that observed over the first 2 days of the study. This demonstrates that the frequency of aberrations increased as a function of dose per cell cycle or rate not total dose.
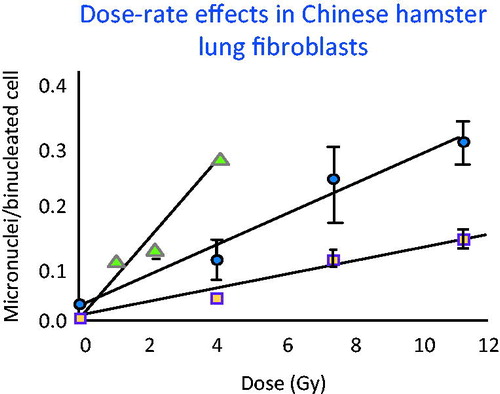
Dose rate effects were evaluated in lung fibroblasts which represent a cell type that has an intermediate cell turnover time of the order of more than 30 days. As shown in , when the slope of the dose-response for the acute exposure (triangles) was compared to that for the 4-h exposure (circles), the DREF was about 2.5. Comparing the acute exposure (triangles) to the 67-h exposure (squares), the DREF increased to 6.1 (Brooks et al. Citation1995). This demonstrates that even when individual cells are exposed to low dose rates and all the energy is deposited in a single cell cycle, the low dose-rate exposures are still less effective in producing cellular and molecular damage than exposures to high dose rate. This influence of dose rate would seem to be related to changes in cellular, sub-cellular and molecular repair.
Figure 5. Frequency of micronuclei were measured in lung cells exposed over a range of different times, dose rates and total doses. The dose rate had a marked influence on the frequency of aberrations even when the cell cycle time was long relative to the exposure time. These data made it possible to calculate DREF as a function of dose rate by comparing the slope of the acute exposures (solid triangles) with that of the protracted exposures (solid circles 4-h exposure) DREF of 2.5 and (squares 67-h exposure) DREF of 6.1.
The dose rate for most environmental exposures is very low (Rogers et al. Citation2001). To measure the response as a function of dose it is necessary to evaluate cells that have very long cell cycle times like blood lymphocytes (peripheral ones divide infrequently) or liver (where the turnover time is measured in years) (Brooks Citation1980). Research demonstrated that the distribution and retained pattern for 144Ce-144Pr (Sturbaum et al. Citation1970) in the livers of Chinese hamsters was similar to that seen in humans (and dogs) making this a useful model for study of chromosome aberration induction in liver. Chinese hamsters were injected with 144Ce-144Pr, a beta gamma emitter (Brooks et al. Citation1972), and the induced chromosome aberration frequency in the liver was compared with the frequency induced following either acute (Brooks et al. Citation1971a) or chronic external exposure to gamma rays from 60Co (Brooks et al. Citation1971b). This experimental design made it possible to study the influence of dose and dose rate on the induction of chromosome aberrations. shows that the slope of the dose-response relationship for the induction of aberrations in the liver produced by either internally deposited 144Ce-144Pr or external protracted 60Co exposure are linear and with the same slope. As with studies conducted in vitro, the acute 60Co exposure resulted in a non-linear dose response relationship. This makes it impossible to derive a single value for a DREF. High doses of low LET radiation delivered at a high dose rate result in many more chromosome aberrations that the same dose delivered at a low dose rate. Thus, after high doses it is not possible to estimate a DREF since it changes with dose, but the data do demonstrate that it would be higher than one.
Figure 6. The induction of chromosome aberrations in the slowly dividing liver cells of the Chinese hamster. The cells were exposed to either internally deposited 144Ce -144Pr or external 60Co gamma rays delivered acutely or over a protracted period of time. The DREF changes as a function of total dose. The protracted exposure resulted in linear dose response relationship and the acute exposure was linear-quadratic. At high doses, greater than 3 Gy there was a large DREF. The DREF in the low dose region was 1.
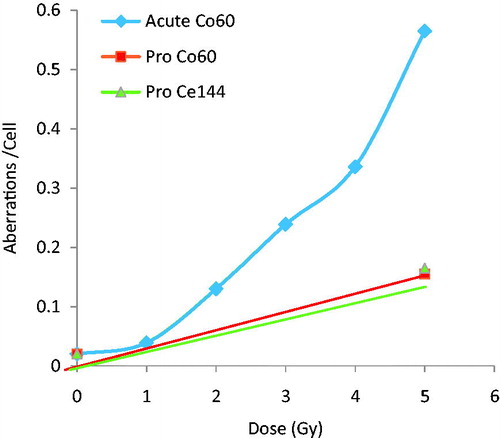
The non-linear nature of the acute dose-response for chromosome aberrations in the liver makes it impossible to derive a single DREF the factor. A DREF can be estimated at a single dose level. When comparing doses greater than 3 Gy, the DREF is very large. However, for doses less than 3 Gy there where the linear portion of the dose-response is driving the response no significant difference between acute and chronic exposures were detected, suggesting a DREF in the low dose region of one for liver chromosome aberrations.
Dose rate also had a marked effect on the frequency of chromosome aberrations when human lymphocytes were exposed to external gamma ray sources. When the dose was protracted over a period of 20 h, the aberration frequency decreased and suggested a DREF of 2 (Purrott and Reeder Citation1976).
Using modern molecular chromosome painting techniques, it is possible to paint each chromosome a unique color. This made it possible to accurately detect symmetrical chromosome exchanges and to evaluate the role of dose rate on these important aberrations (Lucas et al. Citation2004). Since these aberrations have been shown to be important in the production of leukemia (Rowley Citation1980) and are biomarkers of this disease (Brooks Citation1999), they are postulated to be a key event in the critical pathway for the induction of cancer. Fitting the data for acute and chronic radiation exposure for the induction of simple exchanges resulted in a non-linear fit following acute exposure and a linear fit for the low dose-rate exposure. By comparing the ratio of simple exchanges for high and low dose-rate exposure, at a single dose value (1.0 Gy) a DREF of 2.0 was derived. Using exchange breakpoints per cell as the measure of chromosome damage the ratio was estimated to be about 3.0. These are very important data and demonstrate that the DREF for this key event is greater than 1.
The dose-response relationship for the induction of micronuclei in cultured cells following exposure to high dose rates and for a range of different radiation types can be described as linear or linear-quadratic (Heddle et al. Citation1991; Mill et al. Citation1996). However, when the dose rate is low, induced micronuclei do not follow linear kinetics. In fact, studies in vitro using normal human fibroblasts demonstrated that following an acute exposure to a low total dose (10 cGy), the frequency of micronuclei/cell was higher than the control values. However, when the same dose was protracted over 48 h, the level of micronuclei observed in the irradiated cells was lower than observed in control cells (deToledo et al. 2006). These data are shown in where frequency of micronuclei per 100 cells induced by a low dose (10 cGy) is related to radiation exposure time and thus, dose rate.
Figure 7. Micronuclei frequency measured as a function of exposure time. Of special note is that the frequency of micronuclei in these cultured cells was lower in the cells exposed to 10 cGy over 48 h than observed in the controls. Such data suggest induction of a protective response that may require a negative term in modeling risk.
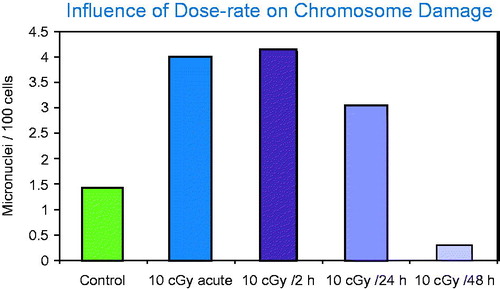
The major point of interest is that when the radiation exposure was protracted over a 48-h time period the frequency of micronuclei was less than observed in the control cells. The role of cell cycle and the elimination of damaged cells could be a competing factor in this study.
Bank voles were measured for the induction of micronuclei in the zones of high radiation following Chernobyl. In spite of having calculated doses that were greater than 1.0 Gy, delivered at a low dose rate, there was no detectable increase in the frequency of micronuclei (Rogers and Baker Citation2000).
Data on micronuclei support the concept that radiation adaptive protection for the induction of chromosome aberrations exists in the low dose and low dose-rate region (deToledo et al. 2006; Dauer et al. Citation2010; Feinendegen et al. Citation2011). Adaptive protection results in non-linear dose-response relationships which suggest that risks for cell and molecular damage from low dose-rate exposures are considerably lower than predicted by the LNT models and that there is therefore the need for a dose-rate effectiveness factor greater than 1. If the induction of micronuclei following low dose rate represents a critical event in the pathway to cancer, the observed radiation-induced decrease in micronuclei below the level seen in the control cells may suggest the need for a protective factor. Contrary to common risk assessment practices, it has in fact even been suggested that models include a negative DREF which would suggest protection at low dose rates (Scott Citation2004, Citation2007)
Cell killing
High dose rate
Cell killing has two major impacts on the tissues. First, the loss of damaged cells may decrease the risk for cancer and second, the loss of cells from a tissue can stimulate cell proliferation that has been shown to play an important role in cancer progression. It has been recently postulated that errors occur during normal cell division in stem cells that result in changes in the key events on the critical pathways which produces cancer (Tomasetti and Vogelstein Citation2015). This suggests that ‘bad luck’ may be the major cause of spontaneous cancer and that exposure to environmental stress plays a minor role. The number of cell divisions in each tissue type is highly variable and seems to provide a useful indicator of cancer risk. Since radiation exposure is effective in killing cells and this loss results in an increase in cell turnover in the exposed cells this would result in more cell divisions and may increase the risk for cancer. At low dose rates, the effectiveness for cell killing is decreased. However, exposure to very high total doses delivered at low dose rates results in marked cell killing. This loss of cells can produce an increase cell turnover and may be an important mechanism in radiation-induced cancer.
Low dose rate
Studies conducted some years ago showed that following low doses of low-LET radiation, cell killing plateaued in the low dose region of the dose-response curve and then increased as an exponential function of dose. Decreasing the dose rate also resulted in a marked decrease in cell killing. Using the colony-forming assay, extensive cell killing studies have been conducted in human cells, and in summary demonstrate a dose rate effectiveness factor of between 1.5 and 10. The range of DREF values is dependent on the genetic background of the human cells (Hall Citation2000). When the dose rate is protracted over many cell divisions, either in vitro or in vivo, the cell population sets up a new equilibrium and the cells continue to divide. This was demonstrated with Chinese hamster cells grown in culture and exposed in a low dose rate radiation field (Bedford and Mitchell Citation1973), and results in a DREF up to 10.
Cell killing and colony formation: radiation hypersensitivity and induced radiation resistance
With the development of techniques that make it possible to measure cell killing in the low dose region, the fine structure of the dose-response curve for cell killing in the low dose region was defined. Following low doses of radiation, it was determined that cell killing increased rapidly as a function of very low dose (radiation hypersensitivity). As the dose increased, the cells became radiation resistant (induced radiation resistance). Thus, there was fine structure in the dose-response relationship for cell killing that had not been appreciated in the past. Cells in the G2 stage of the cell cycle were most sensitive to low dose hypersensitivity followed by radiation-induced resistance (Marples et al. Citation2003). The literature on low dose hypersensitivity and radiation-induced resistance has been comprehensively reviewed (Joiner et al Citation2001; Marples and Collis Citation2008). These observations are very important for describing dose-response relationships in the low dose region. If low doses of radiation disproportionately increase cell killing, this treatment could be eliminating cells from the population that may be at higher risk for the induction of cell transformation and decrease risk. The other side of this coin is that radiation-induced resistance to cell killing may allow transformed cells to survive and increase risk. An important part of cell killing in the process of cancer seems to be the compensatory cell proliferation to replace the cells lost. After exposures to very low doses or dose rates the amount of induced cell proliferation will be limited, bringing into question the role that it plays as a key event in a critical pathway to cancer. From these low dose cell killing data, it is not appropriate to estimate a DREF.
Apoptosis
The process of apoptosis or programmed cell death has long been recognized as playing a critical role during embryonic development. As cells differentiate and form organs, many cells are programmed to die. During the early days of radiation biology, it was not widely recognized that radiation-induced cellular damage could lead to apoptosis. Cells were thought to be killed by radiation through either the process of mitotic death or necrosis. Radiation-induced apoptosis had in the past been called interphase death. Lymphocytes seemed to be one of the major cell types to undergo this type of death. Studies have been conducted to determine the dose-response relationships that exist for the induction of apoptosis. These suggested that for non-genotoxic insults a threshold existed below which apoptosis could not be observed in human lymphocytes. It was suggested that ‘Applications of non-genotoxic carcinogens at doses too low to interfere with life-death decisions of cells or for time periods too short to cause irreversible transitions in cell populations may therefore be considered below the biological threshold for a carcinogenic effect’ (Schulte-Hermann et al. Citation2000). Since radiation is classified as a genotoxic carcinogen, it was suggested that such responses do not apply to ionizing radiation. Studies using radiation demonstrated that many systems are very sensitive to radiation-induced apoptosis, and that no threshold could be detected below which there was no response. It was also determined that, at very low doses, the induction of apoptosis may be protective relative to the induction of cell transformation and other endpoints on the critical pathways to cancer (Mendonca et al. Citation1999).
An important outcome of apoptosis being induced is the possibility that low doses of radiation can trigger biochemical and signaling pathways in bystander cells that result in selective apoptosis and differentially killing of cells that are transformed. There are a large number of transformed cells in the body at all times. If low dose radiation exposure increases the frequency of selective apoptosis of transformed cells, a key event, then low doses of radiation would reduce the number of existing transformed cells at a greater rate than the radiation exposure produces them. This would result in a large DREF. Such a result suggests that total cancer risk would be reduced by low doses of radiation (Bauer Citation2007; Portess et al. Citation2007). Selective apoptosis would help explain why low doses of radiation have been shown in some studies to reduce both the cell transformation (Redpath Citation2006) and mutation frequency (Sykes et al. Citation2006).
The role of programmed cell death in radiation oncology has been reviewed (Meyn et al. Citation2009). Apoptosis may become a key pathway as the science of low dose radiation biology moves forward and new mechanistic information is used to inform radiation protection and risk assessment. If selective apoptosis is a key event in the pathway to cancer, then this process could be postulated to reduce risk following low doses of ionizing radiation delivered at either a high or low dose rate. Currently there are limited data on the role of dose rate on the induction of selective apoptosis. Such information is needed to calculate a reliable DREF for this key event.
Cell transformation
High dose rate
A number of different systems have been developed for following the progression of cells from the normal state to a ‘transformed’ state. Cells in culture have already acquired many of the hallmarks of cancer and require only a few additional changes to become ‘transformed.’ Cell transformation has been suggested to be a critical step as the cells progress from normal to acquiring the characteristics needed to develop cancer. In the field of radiation biology, primary cell transformation systems used to study the final steps in the progression from normal to cancer cells include a human hybrid cell system that has a high spontaneous frequency of cell transformation (Redpath Citation2007) and the mouse embryo C3H 10T1/2 cell system (Azzam et al. Citation1996). With these tools, it was possible to expose the cells to graded acute doses of radiation and carefully measure the frequency of cell transformation. Many studies were conducted to measure cell transformation and demonstrated that low doses of ionizing radiation delivered at a high dose rate decreased the spontaneous frequency of cell transformation below that observed in control cells receiving no radiation exposure (Azzam et al. Citation1996; Redpath et al. Citation2003; Redpath Citation2007). An example of the cell transformation responses following high dose-rate exposure in the low dose region is illustrated in (Redpath et al. Citation2001).
Figure 8. The frequency of transformed cells decreases below the level observed in the controls following acute low doses of radiation. After this initial decrease the frequency then increases in a rather linear manner with dose resulting in a J-shaped dose response relationship. These data support adaptive protection in the low dose region.
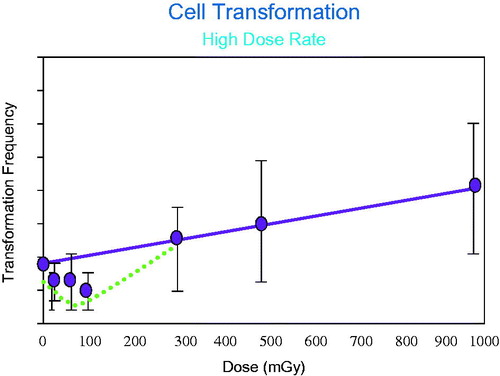
This decrease in cell transformation frequency following low dose exposures, below the background has been demonstrated in other cell transformation systems (Azzam et al. Citation2002; Redpath Citation2007). Extensive studies have been conducted to determine the role of exposure variables on the induction of cell transformation that decreases the frequency to levels below the spontaneous frequency. It is important to determine if the types of radiation used in diagnostic procedures would induce a decrease in cell transformation, since it has been demonstrated that there is an increase in use and in the associated collective dose from of such diagnostic procedures, this increased medical exposure has almost doubled the ‘background’ radiation dose (Brenner and Elliston Citation2004; Mettler et al. Citation2008). It has been postulated that this increased exposure will result in an increase in total radiation-induced cancers in the population (Brenner and Hall Citation2007). Research on the induction of cell transformation has been conducted with diagnostic energy X-rays, (Redpath et al. Citation2003), photons (Redpath Citation2006) and mammographic energy X-rays (Ko et al. Citation2006). All of these radiation types delivered at a low total dose induced a decrease in the frequency of cell transformation below the background level. The results of these types of studies were reviewed and it was concluded that, following low dose exposures, the low photon energies used in medical imaging all produced a reduction in the frequency of cell transformation (Ko et al. Citation2006; Redpath Citation2006). Use of this cell system as an indicator of cancer risk has been reviewed (Redpath Citation2007). If cell transformation in vitro represents a key event in the pathway as cells progress toward radiation-induced cancer, then such cellular studies suggest that a negative or protective value may be required in risk models (Scott Citation2004, Citation2007). The role of in vitro studies on cell transformation in estimating cancer risk remains an area of controversy and requires additional research (Morgan and Bair Citation2013).
Cell transformation
Low dose rate
It is important to determine the role of dose rate on the induction of cell transformation. Dose rate has a marked impact on cell transformation frequency (Elmore et al. Citation2006). Research has demonstrated that by changing dose rate, the relationship between the control values and the exposed ones could be varied. It was of interest to note that the levels of transformation in the controls varied across experiments, with a range between 3 and 5. This makes it essential to compare the response following exposure to the background for that specific experiment. It was demonstrated that for the highest two dose rates used in these studies the data fit equally well to a LNT model and a threshold model. At the lowest two dose rates used (0.47 or 0.19 mGy/min), the level of transformation in the exposed cells was lower than observed in the controls over the dose range of the study (up to 1000 mGy or 1 Gy). The most extreme indication of the potential for a protective adaptive response was observed after exposure to 0.47 mGy/min. These data are shown in . Thus, exposure to low dose rate resulted in a decrease in the frequency of cell transformation below the background level over a much wider dose range (up to 1.0 Gy) than was observed for high dose-rate exposures (Redpath Citation2007).
Figure 9. When cell transformation is measured following low dose rate exposures the dose region where adaptive protection is observed is much larger than observed following acute exposures. The Figure illustrates that this dose rate (0.47 Gy/min) again suggests adaptive protection response even after a total dose of 1000 mGy (1 Gy).
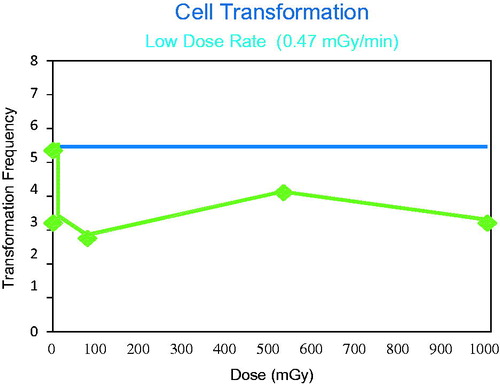
When the dose rate was decreased, the response in the exposed cells was dependent on both the total dose and the dose rate. If low dose and dose rate exposures decrease cell transformation, and if these are important key events in the critical pathways to cancer, the possibility of a decrease in cancer risk in the low dose region must be considered and a negative DREF used. However, since both high and low dose rate exposures result in non-linear dose response relationships for cell transformation it is not possible to calculate a single DREF. One approach to estimating a DREF from such data would be to compare the response at 1.0 Gy for the two different dose rates, independent of the fact that for the low dose rate exposure there is a response less than that observed in the control. This approach results in a DREF of about 3.
Key events: tissue level
Genomic instability
Solid cancers contain multiple genetic changes. Such changes reflect the loss of genetic stability of the cells. This loss of genomic stability is one of the hallmarks of the cancer process (Hanahan and Weinberg Citation2011) and is critical as the cells take on a cancer phenotype. More data are needed to determine if genomic instability is actually induced by an agent (such as radiation) that ‘caused’ the cancer or is simply a reflection of the cancer process where cells have escaped genetic control present in normal tissues. Radiation exposure of cells in culture to either high or low dose rate can induce a low level of genomic instability (Limoli et al. Citation1999). Following acute high dose radiation exposure, cells can make multiple, apparently normal cell divisions, then a fraction of the irradiated cells appear to suffer some loss of control of their genome. The implication of such delayed changes was unappreciated until quite recently. This ‘genomic instability’, or loss of genetic control, results in multiple genetic changes in the cells. Genomic instability has been defined as the increased rate of acquisition of genetic alterations in the progeny of an irradiated cell (Morgan et al. Citation1996). Many of these changes are similar to those observed a short time after exposure.
It has been proposed that there is an interaction between a reduction in DNA repair, which could facilitate the initiation steps of carcinogenesis, and the loss of cell cycle checkpoint arrest which allows cells with DNA damage to proliferate and ‘fix’ the damage in subsequent daughter cells. This interaction has been postulated to impact the induction of genomic instability (Löbrich and Jeggo Citation2007). It was determined from these studies that there is a threshold dose for the blockage of G2/M at about 400 mGy. The authors concluded: ‘Thus, it seems likely that the G2/M checkpoint does not play any significant role after low radiation doses.’ Further, it was suggested that perhaps the G1/S checkpoint was the master checkpoint regulating genomic stability. Since there was no information on the role of dose rate on these processes in these studies, these results provide little impact on the ability to estimate a DREF for low LET radiation.
Many endpoints have been used as a measure of the induction of genomic instability both in vitro and in vivo. Radiation-induced genomic instability in rodents was dependent on the genetic background of the animals. Two different strains of mice that were either sensitive or resistant to radiation-induced breast cancer were exposed to radiation and both breast cancer frequency and chromosome damage evaluated. Radiation did not induce cancer or genomic instability in the resistant mice, but was demonstrated in the sensitive strain as an increase in late occurring chromosome aberrations and an increase in radiation-induced breast cancer (Ponnaiya et al. Citation1997).
In contrast with the multiple studies that demonstrate radiation induced genomic instability, studies have demonstrated that genomic instability was not induced in stable normal human cells (Dugan and Bedford Citation2003). This failure to demonstrate genomic instability may suggest that genomic instability may play a more limited role in low dose radiation-induced cancer. This point of view was supported by the observation that genomic instability was not detected in lymphocytes of the A-bomb survivors (Hamasaki et al. Citation2009). Thus, there is a continuing discussion and controversy on the role of low dose radiation-induced genomic instability and radiation-induced cancer (Morgan and Bair Citation2013).
A recent review tries to resolve the differences in the induction of genomic instability seen in experimental systems including the failure to demonstrate it in normal human cells and in human populations (Morgan and Sowa Citation2007). Because of the lack of low dose and low dose-rate data it is currently not possible to estimate a DREF for the induction of genomic instability as a key event along the critical pathway to human cancer.
Inflammation and Reactive Oxygen Species (ROS)
High dose rate
It is well established chronic inflammatory disease (Werner and Haller Citation2007) and the level of ROS (Reactive Oxygen Species) in a system can have a marked influence on cancer risk, in this regard, many foods and additives are used to reduce ROS levels. The ROS status of cells and tissues, changes in metabolism and regulation of cytokines represent early changes and are thought to play a role in cancer risk and have been demonstrated to have marked dose and dose-rate dependence. As is the case for many biological endpoints, the ROS status of a cell is a critical variable in the induction and prevention of apoptosis and many other endpoints. It has been demonstrated that very high doses of radiation-induced apoptosis can be modulated by treatment with compounds that inhibit energy metabolism (Hunter et al. Citation2007). This suggests the potential for a link between chronic inflammatory disease (Werner and Haller Citation2007), energy metabolism (Lall et al. Citation2014), low dose-induced changes in cell signaling and gene expression (Berglund et al. Citation2008), the ROS status of cells (Spitz et al. Citation2004) and the induction of apoptosis in the high-dose region, but provides limited information on the responses to low doses. Changing the ROS status of cells can be a protective mechanism. Treatment with vitamin E is thought to inhibit angiogenesis, an essential part of tumor development, by selective induction of apoptosis in proliferating endothelial cells (Dong et al. Citation2007). Such studies show the importance of apoptosis during cancer development and the role that ROS status and radiation-induced changes in ROS status may have on cancer risk.
Pro-inflammatory cytokines are activated following large doses of radiation delivered at a high dose rate and are thought to play an important role in the promotion of cancer. At high doses, these relationships have been well-established. Some studies have demonstrated no changes in cytokines after low dose-rate exposures to radiation (Gridley et al. Citation2001) suggesting that for this endpoint a DREF greater than 1 is indicated but cannot be reliably estimated. Research on the role of low dose and low dose-rate exposures on the immune system need to be expanded (Kaur and Asea Citation2012). It is not possible to directly estimate a DREF from studies on ROS or chronic inflammation but the data suggest that low doses and dose rates may not induce chronic inflammatory disease and have the potential to reduce the ROS in normal tissues.
Low dose rate
Chronic inflammation increases cancer risk. Studies on dogs demonstrated that after exposure to high total doses delivered at a low dose rate, the cancer frequency was very high. In these animals one common observation was the induction of a chronic inflammatory disease of the lung. As the total dose and dose rate were decreased to a level that there was no inflammatory disease in the lung, the cancer frequency decreased to a level that it was not significantly different from that observed in the controls. On the other hand, antioxidant, anti-inflammatory cytokines down-regulate these reactive species and restore homeostasis (Schaue et al. Citation2012). Many anti-inflammatory processes which decrease the ROS have been shown to be activated by low doses and low dose-rate radiation exposures (Amundson et al. Citation2003b; Fan et al. Citation2007). This decrease has been suggested as a mechanism by which exposures activate protective mechanisms. These protective processes involved changes in the mitochondria (Kim et al. Citation2006), the ROS status of the cells (Spitz et al. Citation2004; Gius and Spitz Citation2006), the modification of radioprotective chemicals (Kennedy et al. Citation2007), SH-containing chemicals, MnSOD and SOD2 (Azzam et al. Citation2002; Guo et al. Citation2003; Kim et al. Citation2006).
These protective responses are linked to mitochondrial function. The role of the mitochondria in the total radiation response has been found to be very important. It has been demonstrated that ionizing radiation alters cyclin B1, which is involved in control of cell cycle. This alteration seems to be regulated through NF-κB and through the antioxidant enzyme MnSOD which can modify the oxidative status of cells and act as a protective mechanism against radiation-induced damage (Otsuka et al. Citation2006). The level of MnSOD is increased by exposure to low doses of radiation (Azzam et al. Citation2002). Other research on the relationships that exist between NF-κB and MnSOD, using mouse skin epithelial cells, showed that the interaction between these factors results in a radiation-induced decrease in the ROS status of the cells (Fan et al. Citation2007).
Using proteomic and transcriptomic analyses, it has been determined that mitochondrial dysfunction results in the induction of oxidative stress in cells leading to cell killing through programmed cell death (apoptosis) (Ding et al. Citation2005; Bauer Citation2007; Portess et al. Citation2007). Oxidative stress can also be altered by treatment of cells with Vitamin E analogs that may induce selective apoptosis in proliferating endothelial cells and stop angiogenesis which is critical in cancer growth and spread (Ding et al. Citation2005; Kennedy et al. Citation2007). Such research highlights the role of normal oxidative metabolism in cellular responses and suggests that alterations of this metabolism by any type of stress can have either a protective or detrimental role in the risk for cancer development depending on the radiation dose.
The observations described above demonstrate that the redox status of the cell plays a critical role in signaling in cancer biology (reviewed by Gius and Spitz Citation2006). The role of stress and how it alters gene expression, senescence, redox status of the cells and the risk for cancer has also been reviewed and rather strong links between these factors have been established (Denko and Fornace Citation2005). All this research makes it clear that the ROS status of the cells is a key event during the cell transformation process and important in cancer development. The low dose rate-induced protective responses that alter ROS status are very dependent on the radiation dose and dose rate. Although high doses increase stress and reactive oxygen levels in tissues and cells and results in increased cancer risks, low doses and low dose rates seem to increase the level of MnSOD and decrease the ROS levels. These changes provide a potential mechanism for reducing cancer risks. Such a mechanism would support a large DREF following exposure to high doses delivered at a low dose rate, as is the case in Beagle dogs (Brooks et al. Citation2009). These conclusions further support the need to separate DREF from DDREF. It also suggests the potential need to consider a protective term in risk analysis.
Alteration of metabolic pathways
Recent publications have indicated that low doses of radiation can alter metabolic pathways which can be considered as a key event at the tissue level in the pathway to cancer (Lall et al. Citation2014). It was established that doses as low as 0.1 Gy could result in a large increase in glucose consumption and ECAR while large doses (4.0 Gy) caused either little change or a decrease in these responses ().
Figure 10. This Figure demonstrates that after low doses of ionizing radiation there are marked changes in glucose metabolism not seen after high doses. No data are available on the role of dose rate on these responses so a DREF cannot be determined. The Figure suggests different mechanisms of action in the low dose region which requires additional research.
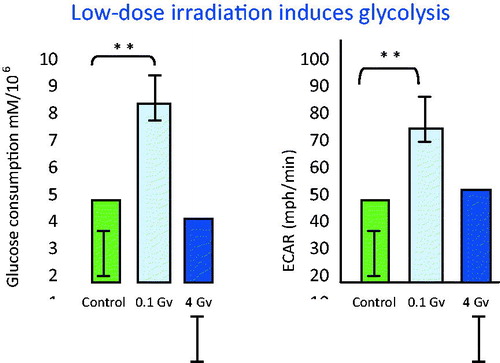
These low-dose-induced changes were related to increased expression of glucose transporters. There is limited evidence that glucose transporters may play an important role in development of cancer (Lall et al. Citation2014). The potential importance of these changes in radiation-induced cancer still remains to be determined and is an important area of future research. Since there are no data on the influence of dose rate on these responses it is not possible at this time to calculate a DREF.
Discussion
The aim of this review article is to evaluate the key events/adverse outcome pathway (AOP) approach for demonstrating how cell and molecular data can potentially enhance the estimation of the DREF and its use in radiation risk assessment. The review does not include information on animal studies and epidemiology studies because these will be addressed in a future review. Data have been published that show the DREF is less than 1, about 1 and greater than 1 (even considerably so). Much of the data that suggest a DREF of 1 or less are derived from DNA damage and repair studies (Rothkamm and Löbrich Citation2003; Rothkamm et al. Citation2007; Grudzenski et al. Citation2010; Hernandez et al. Citation2013). However, other studies using similar DNA endpoints show that at very low doses no DNA damage is detectable following low dose rate exposure but an increased amount of damage is recorded after acute exposures. These two data sets are reviewed in the current review and are in direct conflict. Additional research is required to resolve this conflict. For cellular endpoints the present review illustrates that lower dose rates result in a decreased response for many key events along the cancer adverse outcome pathway. There are data sets where responses for key events change as a function of dose and dose rate but the changes have only been measured under a single exposure condition so it is not possible to determine a slope for high and low dose rate to estimate a DREF. These single values are reported and most support a DREF greater than 1. In addition an increasing amount of data has been published that demonstrates that following low dose or low dose-rate exposure, the response of many key events is even lower than observed in the controls. Such information makes it essential to consider using a negative or protective factor in estimating the DREF. In this manuscript the most useful data sets are those where the total dose is delivered at either a high or low dose rate over a wide range of total doses. For some endpoints the different exposure situations all result in linear dose response relationships so it is possible to make a direct comparison of the slopes of the dose-response relationships. This provides quantitative information to directly calculate a DREF. provides a compilation of data where the key event was measured for single doses for only high or low dose-rate exposure making it impossible to calculate a DREF. We have reviewed these papers and have concluded that the data in the paper can or cannot be used to calculate a DREF or that they support a DREF of either greater or less than 1. Since there is so much information in each manuscript represented in the table it is not possible to summarize in detail the systems used, details of the experimental designs, all the endpoints measured or all the strengths and weaknesses of each paper. provides a summary of the research where dose-response relationships were measured following both high and low dose-rate exposures. Such data are the most useful for considering key events in the critical pathways for estimating a DREF. For these studies, this manuscript directly compared the slopes of dose response relationships following high and low dose-rate exposures and used the ratio to calculate DREF. It is important to stress that key events are empirically observable precursors for an apical endpoint and as such can serve as parameters in some form of a biologically-based dose-response model (Preston Citation2015). Such BBDR models can be used to investigate the development of a reliable value for DREF.
Table 3. (a) Low total dose: Only high or low dose rate data for each key event. No dose-response data needed to estimate a DREF value.
Table 3. (b) Dose response information available following both high and low dose rate. Dose response data make it possible to estimate DREF.
Summary
A consideration of the uncertainty associated with radiation cancer risk estimates clearly shows that one of the most significant uncertainties is the selection of a value for DREF. A major reason for this is that, in general, human data are not available for low dose-rate exposures and it is unclear which animal and cellular data sets are the most appropriate for use in the calculation of DREF. To address this issue, it has been proposed that the most informative animal and cellular data are those that are the key events along the adverse outcome pathway (AOP) leading to a cancer or non-cancer outcome. We present such an approach in the present review. Thus, as a key component, we provide a comprehensive discussion of how the level of low dose-rate effects can vary with variations in cell cycle times and how these interactions influence many of the measurements made at low dose rates. A review is also provided of the radiation-induced differences in the molecular, cellular and tissue responses for exposures and comparisons where possible at high or low dose rate. Two ) were constructed that make it possible to quickly review the data presented in this review for evaluating the responses to key events at molecular and cellular levels of biological organization. This allows the reader to further research and follow-up on any points or estimates that are of particular interest. These tables show that for key events along the pathway to cancer, exposures to ionizing radiation at low dose rates result in a decrease in the biological responses when compared to the responses after exposure to the same dose delivered at a high dose rate. Such data support the need for a dose-rate effectiveness factor that is greater than 1. In addition, these data provide a unique combination of molecular, cellular and tissue outcomes that have not, in the past, been used for estimating DREF. In future publication it may be possible to combine such biological information with animal and human cancer data to support a mechanism-based dose-rate effectiveness factor to be used in cancer risk estimates. This will in turn, reduce the overall uncertainty in the setting of dose limits based on these risk values. In summary, it seems prudent to conclude that the majority of the data support a DREF greater than 1.
Acknowledgements
The authors would like to acknowledge the editorial help of Careina C. Brooks and Lezlie A. Couch and the scientific input from Dr Donald Cool and Phung Than. This work was supported by the Electric Power Research Institute (EPRI Agreement No. 10000432 Charge number 1-003171-02-01).
Disclosure statement
The authors report no conflicts of interest. The authors alone are response for the content and writing of the paper.
References
- Adeleye Y, Andersen M, Clewell R, Davies M, Dent M, Edwards S, Fowler P, Malcomber S, Nicol B, Scott A, Scott S, Sun B, Westmoreland C, White A, Zhang O, Charmichael PL. 2015. Implementing toxicity testing in the 21st century (TT21C): making safety decisions using toxicity pathways and progress in a prototype risk assessment. Toxicology. 332:102–111.
- Amundson SA, Bittner M, Fornace AJ. 2003a. Functional genomics as a window on radiation stress signaling. Oncogene. 22:5828–5833.
- Amundson SA, Lee RA, Koch-Paiz CA. 2003b. Differential responses of stress genes to low dose-rate γ irradiation. Mol Cancer Res. 1:445–452.
- Asaithamby A, Chen D. 2009. Cellular responses to DNA double-strand breaks after low dose gamma-irradiation. Nucleic Acids Res. 37:3012–3923.
- Azzam EI, de Toledo SM, Raaphorst GP, Mitchel REG. 1996. Low-dose ionizing radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in C3H 10T1/2 cells. Radiat Res. 146:369–373.
- Azzam EI, de Toledo SM, Spitz DR, Little JB. 2002. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res. 62:5436–5442.
- Bao S, Harwood PW, Chrisler WB, Groch KM, Brooks AL. 1997. Comparative clastogenic sensitivity of respiratory tract cells to gamma rays. Radiat Res. 148:90–97.
- Bauer G. 2007. Low dose radiation and intercellular induction of apoptosis: potential implications for the control of oncogenesis. Int J Radiat Biol. 83:873–888.
- Bedford JS, Mitchell JB. 1973. Dose rate effects in synchronous mammalian cells in culture. Radiat Res. 54:316–327.
- Bender MA, Awa AA, Brooks AL, Evans HJ, Groer PG, Littlefield LG, Perira C, Preston RJ, Wachholz BW. 1988. Current status of cytogenetic procedures to detect and quantify previous exposures to radiation. Mutat Res. 196:103–159.
- Berglund SR, Rocke DM, Dai J, Schwietert CW, Santana A, Stern RL, Lehmann J, Hartmann Siantar CL, Goldburg Z. 2008. Transient genome-wide transcriptional response to low-dose ionizing radiation in vivo in humans. Int J Radiat Oncology Biol Phys. 70:229–234.
- Blakely EA. 2012. Lauriston S. Taylor Lecture on radiation protection and measurements: what makes particle radiation so effective? Health Phys. 103:508–528.
- Bogdandi EN, Balogh A, Felgyinszki N, Szatmari G, Luminszky K. 2010. Effects of low-dose radiation on the immune system of mice after total body irradiation. Radiat Res. 174:480–489.
- Boss MK, Bristow R, Dewhirst MW. 2014. Linking the history of radiation biology to the hallmarks of cancer. Radiat Res. 181:561–577.
- Brenner DJ, Elliston CD. 2004. Estimated radiation risks potentially associated with full-body CT screening. Radiology. 232:735–738.
- Brenner DJ, Hall EJ. 2007. Computed tomography – an increasing source of radiation exposure. New Engl J Med. 357:2277–2284.
- Brooks AL. 1975. Chromosome damage in liver cells from low dose rate alpha, beta and gamma irradiation: derivation of RBE. Science. 190:1090–1092.
- Brooks AL. 1980. Low dose and low dose-rate effects on cytogenetics. In: Meyn RE, Withers HR, editors. Radiation biology in cancer research. New York: Raven Press. pp. 263–276.
- Brooks AL. 1986. Whole animal and in vivo cell systems for detecting mutations. In: Lloyd WE, editor. Safety evaluation of drugs and chemicals (ch. 12). Washington: Hemisphere Publishing Corp. pp. 167–183.
- Brooks AL. 1999. Biomarkers of exposure, sensitivity and disease. Int J Radiat Biol. 75:1481–1503.
- Brooks AL, Bao S, Harwood PW, Wood BH, Chrisler WB, Gies RA, Cross FT. 1997. Induction of micronuclei in respiratory tract following radon inhalation. Int J Radiat Biol. 72:485–495.
- Brooks AL, Dauer LT. 2014. Advances in radiation biology: effect on nuclear medicine. Semin Nucl Med. 44:179–186.
- Brooks AL, Eberlein PE, Couch LA, Boecker BB. 2009. The role of dose-rate on risk from internally-deposited radionuclides and the potential need to separate dose-rate effectiveness factor (DREF) from the dose and dose-rate effectiveness factor (DDREF). Health Phys. 97:458–469.
- Brooks AL, Mead DK, Peters RF. 1971a. Effect of chronic exposure to 60Co on the frequency of metaphase chromosome aberrations in the liver cells of the Chinese hamster (in vivo). Int J Radiat Biol. 20:599–604.
- Brooks AL, McClellan RO, Benjamin SA. 1972. The effects of 144Ce-144Pr on the metaphase chromosomes of the Chinese hamster liver cells in vivo. Radiat Res. 52:481–498.
- Brooks AL, McClellan RO. 1968. Cytogenetic effects of Strontium-90 on the bone marrow of the Chinese Hamster. Nature. 219:761–763.
- Brooks AL, McClellan RO. 1969. Chromosome aberrations and other effects produced by 90Sr-90Y in Chinese hamsters. Int J Radiat Biol. 16:545–561.
- Brooks AL, Miick R, Buschbom RL, Murphy MK, Khan MA. 1995. The role of dose rate in the induction of micronuclei in deep-lung fibroblasts in vivo after exposure to 60Co gamma rays. Radiat Res. 144:114–118.
- Brooks AL, Lei XC, Rithidech K. 2003. Changes in biomarkers from space radiation may reflect dose not risk. Adv Space Res. 31:1505–1512.
- Brooks AL, Peters RF, Rollag MD. 1971b. Metaphase chromosome aberrations in Chinese hamster liver cells in vivo after single acute 60Co exposure. Radiat Res. 45:191–201.
- Coleman MA, Wyrobek AJ. 2006. Differential transcript modulation of genes after low vs. high doses of ionizing radiation. In: Wolbarst AB, Samenhof RG, Hendee WH, editors. Advances in medical physics. Madison, WI: Medical Physics Publishing. pp. 273–276.
- Dauer LT, Brooks AL, Hoel DG, Morgan WF, Stram D, Tran P. 2010. Review and evaluation of updated research on the health effects associated with low-dose ionising radiation. Radiat Prot Dos. 140:103–136.
- Day TK, Zeng G, Hooker AM, Bhat M, Scott BR, Turner DR, Sykes PJ. 2007. Adaptive response for chromosomal inversions in pKZ1 mouse prostate induced by low doses of X radiation delivered after a high dose. Radiat Res. 167:682–692.
- De Toledo SM, Asaad N, Venkatachalam P, Li L, Howell RW, Spitz DR, Azzam EI. 2006. Adaptive responses to low-dose/low-dose-rate gamma rays in normal human fibroblasts: The role of growth architecture and oxidative metabolism. Radiat Res. 166:849–857.
- Denko NC, Fornace AJ Jr. 2005. Editorial comment: Mutation Research Special Issue on stress response. Mutat Res. 569:1–2.
- Ding LH, Shingyoji M, Chen FQ, Hwang JJ, Burma S, Lee C, Cheng JF, Chen DJ. 2005. Gene expression profiles of normal human fibroblasts after exposure to ionizing radiation: a comparative study of low and high doses. Radiat Res. 164:17–26.
- Dong L-F, Swettenham E, Eliasson J, Wang X-F, Gold M, Medunic Y, Stantic M, Low P, Prochazka L, Witting PK. 2007. Vitamin E analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: the role of oxidative stress. Cancer Res. 67:11906–11913.
- Dugan LC, Bedford JS. 2003. Are chromosomal instabilities induced by exposure of cultured normal human cells to low- or high-LET radiation? Radiat Res. 159:301–311.
- Edwards AA, Lindholm C, Darroudi F, Stephan G, Romm II, Barquinero J, Barrios L, Caballin MR, Roy L, Whitehouse CA, Tawn EJ, Moquet J, Lloyd DC, Voisin P. 2005. Review of translocations detected by FISH for retrospective biological dosimetry applications. Radiat Prot Dos. 133:396–402.
- Edwards SW, Tan Y-M, Villeneuve DL, Meek ME, McQueen CA. 2016. Adverse outcome pathways – organizing toxicological information to improve decision making. J Pharmacol Exp Ther. 356:170–181.
- Eidemuller M, Holmberg E, Jacob P, Lundell M, Karlsson P. 2011. Breast cancer risk after radiation treatment at infancy: potential consequences of radiation-induced genomic instability. Radiat Prot Dosim. 143:375–379.
- Elmore E, Lao XY, Kapadia R, Redpath JL. 2006. The effect of dose rate on radiation-induced neoplastic transformation in vitro by low doses of Low-LET radiation. Radiat Res. 166:832–838.
- EPA (U.S. Environmental Protection Agency). 2005. Guidelines for carcinogen risk assessment. In: EPA/630/P 03/001F Washington, DC: US EPA, 1–166.
- Erixon K, Cedervall B. 1995. Linear induction of DNA double-strand breakage with X-ray dose, as determined from DNA fragment size distribution. Radiat Res. 142:153–162.
- Fachin AL, Mello SS, Sandrin-Garcia P, Junta CM, Ghilardi-Netto T, Danadi EA, Passos Gad S, Sakamoto-Hojo ET. 2009. Gene expression profiles in radiation workers occupationally exposed to ionizing radiation. J Radiat Res. 50:61–71.
- Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. 2007. Nuclear factor-k[kappa]B and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 67:3220–3228.
- Feinendegen LE, Brooks AL, Morgan WF. 2011. Biological consequences and health risks of low-level exposure to ionizing radiation: commentary on the workshop. Health Phys. 100:247–259.
- Feinendegen LE, Graessle D. 2002. Energy deposition in tissue during chronic irradiation and the biological consequences. In: Chronic irradiation: tolerance and failure in complex biological systems. British Institute of Radiology. Br J Radiol (Suppl.)26:6–14.
- Feinendegen LE, Polycove M, Neumann RD. 2007. Whole-body responses to low-level radiation exposure: new concepts in mammalian radiobiology. Exper Hematol. 35:37–46.
- Fornace AJ, Amundson SA, Bittner M, Myers TG, Meltzer P, Weinsten, JN, Trent J. 1999. The complexity of radiation stress responses: analysis by informatics and functional genomic approaches. Gene Expr. 7:387–400.
- Ghandhi SA, Smilenov LB, Elliston CD, Chowdhury M, Amundson SA. 2015. Radiation dose-rate effects on gene expression for human biodosimetry. BMC Med Genom. 8:22–32.
- Gius D, Spitz DR. 2006. Redox signaling in cancer biology. Antioxid Redox Signal 9(7&8):1249–1252.
- Gridley DS, Pecaut MJ, Miller GM, Moyers MF, Nelson GA. 2001. Dose and dose rate effects of whole-body γ-irradiation: II. Hematological variables and cytokines. In vivo. 15:209–216.
- Grudzenski S, Raths A, Conrad S, Rube CE, Löbrich M. 2010. Inducible response required for repair of low-dose radiation damage in human fibroblasts. PNAS. 12:14205–14210.
- Guerrero-Carbajal C, Edwards AA, Lloyd DC. 2003. Induction of chromosome aberrations in human lymphocytes and its dependence on x ray energy. Radiat Prot Dos. 106:131–135.
- Guo G, Yan-Sanders Y. Lyn-Cook BD, Wang T, Tamae D, Ogi J, Khaletskiy A, Li Z, Weydert C, Longmate JA, Huang TT, Spitz DR, Oberley LW, Li JJ. 2003. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol. 23:2362–2378.
- Hall E. 2000. Radiobiology for the radiologist. Philadelphia, PA: Lippincott Williams and Wilkins.
- Hamasaki K, Kusunoki Y, Nakashima E. 2009. Clonally expanded T lymphocytes from atomic bomb survivors in vitro show no evidence of cytogenetic instability. Radiat Res. 172:234–243.
- Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell. 100:57–70.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674.
- Heddle JA, Cimino MC, Hayashi M, Romagna F, Shelby MD, Tucker JD, Vanparys P, McGregor JT. 1991. Micronuclei as an index of cytogenetic damage: past, present and future. Environ Mol Mutagen. 18:277–291.
- Heidenreich WF, Rosemann M. 2012. Genetic background and 227-Thorium as risk factors in biologically based models for induction of bone cancer in mice. Radiat Environ Biophys. 51:179–185.
- Heidenreich WF, Saran A, Atkinson M, Pazzaglia S. 2013. A mechanistic model for medulloblastoma induction in mice. Radiat Res. 179:610–614.
- Hernandez L, Twerradas M, Feijoo P, Soler D, Tusell L, Genesca A. 2013. Increased mammogram-induced DNA damage in mammary epithelial cells aged in vitro. PLos One. 2013;8:e63052.
- Hoel DG, 2015. Comments on the DDREF estimate of the BEIR VII committee. Health Phys. 108:351–356.
- Hooker AM, Morley AA, Tilley WD, Sykes PJ. 2004. Cancer-associated genes can affect somatic intrachromosomal recombination early in carcinogenesis. Mutat Res-Fund Mole Mech Mutag. 550:1–10.
- Hsie AW, O’Neill JP, Couch DB, San Sebastian JR, Brimer PA, Mackanoff P, Fuscoe JC, Li AP, Forbes NL, Hsie MH. 1978. Quantitative analyses of radiation- and chemical-induced lethality and mutagenesis in Chinese hamster ovary cells. Radiat Res. 76:471–492.
- Hsieh WA, Deng W, Chang WP, Galvan N, Owens CL, Morrison DP, Gale KL, Lucas JN. 1999. Alpha coefficient of dose-response for chromosome translocations measured by FISH in human lymphocytes exposed to chronic 60Co gamma rays at body temperature. Int J Radiat Biol. 75:435–439.
- Hsu W-L, Preston DL, Soda M, Sugiyama H, Funomoto S, Kodama K, Kimura A, Kamada N, Dohy H, Tomonaga M, Iwanaga M, Miyazaki Y, Cullings HM, Suyama A, Ozasa K, Shore RE, Mabuchi K. 2013. The incidence of leukemia lymphoma and multiple myeloma among atomic bomb survivors. Radiat Res. 179:361–382.
- Hunter AJ, Hendrikse AS, Renan MJ. 2007. Can radiation-induced apoptosis be modulated by inhibitors of energy metabolism? Int J Radiat Biol. 83:105–114.
- International Agency for Research on Cancer (IARC). 2012. IARC Monographs on the evaluation of carcinogenic risks to humans, Vol. 100F: chemical agents and related occupations. Lyon, France: WHO Press.
- International Commission on Radiological Protection (ICRP). 1991. Recommendations of the International Commission on Radiological Protection. ICRP Publication 60, Ann. ICRP 21(1–3).
- International Commission on Radiological Protection (ICRP). 2007. International Commission on Radiological Protection. 2007. The 2007 recommendations of the International Commission on Radiological Protection. New York: Elsevier; ICRP Publication 103; Ann ICRP 37(2–4).
- Ishizaki K, Hayashi Y, Nakamura H, Yasui Y, Komatsu K, Tachibana A. 2004. No indication of p53 phosphorylation and few focus formation of phosphorylated H2AX suggest efficient repair of DNA damage during chronic low-dose-rate irradiation in human cells. J Radiat Res (Tokyo). 45:521–525.
- Joiner MC, Maples B, Lambin P, Short SC. 2001. Low dose hypersensitivity: current status and possible mechanisms. Int J Radiat Oncol Biol Phys. 49:379–384.
- Jostes RF, Fleck EW, Morgan TL, Stiegler GL, Cross FT. 1994. Southern blot and polymerase chain-reaction exon analyses of HPRT(-) mutation induced by radon and radon progeny. Radiat Res. 137:371–379.
- Julien E. Bobbis AR, Olin SS. 2009. The ILSI research foundation threshold working group. The key events dose-response framework: a cross disciplinary Mode-of-Action based approach to examining dose-response and thresholds. Crit Rev Food Sci Nutrit. 49:682–689.
- Kaur P, Asea A. 2012. Radiation-induced effects and the immune system in cancer. Front Oncol. 2:1–10.
- Kennedy AR, Guan J, Ware JH. 2007. Countermeasures against space radiation induced oxidative stress in mice. Radiat Environ Biophys. 46:201–203.
- Khan MA, Cross FT, Bushbom RL, Brooks AL. 1995. Inhaled radon-induced genotoxicity in Wistar rat, Syrian hamster and Chinese hamster deep-lung fibroblasts in vivo. Mutat Res. 334:131–137.
- Kim GJ, Fiskum GM, Morgan WF. 2006. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 66:10377–10383.
- Ko M, Lao X-Y, Kapadia E, Elmore E, Redpath JL. 2006. Neoplastic transformation in vitro by low doses of ionizing radiation: role of adaptive response and bystander effects. Mutat Res. 597:11–17.
- Krewski D, Zielinski JM, Hazelton WD, Garner MJ, Moolgavkar SH. 2003. The use of biologically based cancer risk models in radiation epidemiology. Rad Protect Dosim. 104:367–376.
- Lall R, Ganapathy S, Yang M, Xiao S, Xu T, Su H, Shadfan M, Asara JM, Ben-Sahra I, Manning BD, Little JB, Yuan Z-M. 2014. Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ. 4:1–9.
- Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. 2005. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 569:123–132.
- Limoli CL, Corcoran JJ, Milligan JR, Ward JF, Morgan WF. 1999. Critical target and dose and dose-rate responses for the induction of chromosomal instability by ionizing radiation. Radiat Res. 151:677–685.
- Little MP, Haylock RGE, Muirhead CR. 2002. Modelling lung tumour risk in radon-exposed uranium miners use generalizations of the two-mutation model of Moolgavkar, Venzon and Knudson. Int J Radiat Biol. 78:49–68.
- Little MP, Li G. 2007. Stochastic modelling of colon cancer: is there a role for genomic instability? Carcinogenesis. 28:479–487.
- Little MP, Tawn EJ, Tzoulaki I, Wakeford R, Hildebrandt G, Paris F, Tapio S, Elliott P. 2008a. A systematic review of epidemiological associations between low and moderate doses of ionizing radiation and late cardiovascular effects, and their possible mechanisms. Radiat Res. 169:99–109.
- Little MP, Heidenreich WF, Moolgavkar SH, Schollnberger H, Thomas DC. 2008b. Systems biological and mechanistic modelling of radiation-induced cancer. Radiat Environ Biophys. 47:39–47.
- Löbrich M, Rief N, Kuhne M, Heckmann M, Fleckenstein J, Rube C, Uder M. 2005. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc Natl Acad Sci USA. 102:8984–8989.
- Löbrich M, Jeggo PA. 2007. The impact of negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev/Cancer. 7:861–869.
- Lucas BD, Eberle R, Bailey SM, Cornforth MN. 2004. Influence of dose rate on the induction of simple and complex chromosome exchanges by gamma rays. Radiat Res. 162:339–349.
- Lucas JN, Hill F, Fester T, Straume T. 1999. Dose-response curve for chromosome translocations measured in human lymphocytes exposed to 60Co gamma rays. Health Phys. 68:761–765.
- Manning G, Kabacik S, Finnon P, Bouffler S, Badie C. 2013. High and low dose responses of transcriptional biomarkers in ex vivo X-irradiated human blood. Int J Radiat Biol. 89:512–522.
- Marples B, Collis SJ. 2008. Low-dose hyper-radiosensitivity: past, present, and future. Int J Radiat Onco Biol Phys. 70:1310–1318.
- Marples B, Wouters BG, Joiner MC. 2003. An association between the radiation-induced arrest of G2-phase cells and low-dose hyper-radiosensitivity: a plausible underlying mechanism? Radiat Res. 160:38–45.
- Mendonca MS, Howard KL, Farrington DL, Desmond LA, Temples TM, Mayhugh BM, Pink JJ, Boothman DA. 1999. Delayed apoptotic responses associated with radiation-induced neoplastic transformation of human hybrid cells. Cancer Res. 59:3972–3979.
- Mettler FA Jr, Thomadsen BR, Bhargavan M, Gilley DB, Gray JE, Lipoti JA, McCrohan J, Yoshizumi TT, Mahesh M. 2008. Medical radiation exposure in the U.S. in 2006: preliminary results. Health Phys. 95:502–507.
- Meyn RE, Milas L, Ang KK. 2009. The role of apoptosis in radiation oncology. Int J Radiat Biol. 85:107–115.
- Mill AJ, Well SC, Butler A. 1996. Micronuclei induction in human lymphocytes: comparative effects of x rays, alpha particles, beta particles and neutrons and implications for biological dosimetry. Radiat Res. 145:575–585.
- Moolgavkar SH, Knudson AG Jr. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66:1037–1052.
- Morandi E, Severini C, Quercioli D, Perdichizzi S, Mascolo MG, Horn W, Vaccari M, Nucci MC, Lodi V, Violante FS, Bolognesi C, Grilli S, Silingardi P, Colacci A. 2009. Gene expression changes in medical workers exposed to radiation. Radiat Res. 172:500–508.
- Morgan WF, Bair WJ. 2013. Issues in low doe radiation biology: the controversy continues. Perspective. Radiat Res. 179:501–510.
- Morgan WF, Day JP, Kaplan MI, McGhee EM, Limoli CL. 1996. Genomic instability induced by ionizing radiation. Radiat Res. 146:247–258.
- Morgan WF, Sowa MB. 2007. Non-targeted bystander effects induced by ionizing radiation. Mutat Res-Fund Mole Mech. 616:159–164.
- National Council for Radiation Protection and Measurements (NCRP). 1980. Influence of dose and its distribution in time on dose-response relationships for low LET radiation. Bethesda, MD: NCRP Report No. 64.
- National Council for Radiation Protection and Measurements (NCRP). 1987. Genetic effects of from internally deposited radionuclides. Bethesda, MD: NCRP Report No. 89.
- National Council for Radiation Protection and Measurements (NCRP). 1993. Limitation of exposure to ionizing radiation. Bethesda, MD: NCRP Report No. 116.
- National Council for Radiation Protection and Measurements (NCRP). 2001. Liver cancer risks from internally-deposited radionuclides. Bethesda, MD: NCRP Report No. 135.
- National Council for Radiation Protection and Measurements (NCRP). 2009. Ionizing radiation exposure of the population of the United States. Bethesda, MD: NCRP Report No. 160.
- National Council on Radiation Protection and Measurements (NCRP). 2012. Uncertainties in the Estimation of Radiation Risks and Probability of Disease Causation. Bethesda, MD: NCRP Report No. 171.
- National Council on Radiation Protection and Measurements (NCRP). 2015. Where are the radiation professionals (WARP)? Synopsis of NCRP Statement No. 12. Bethesda, MD: NCRP.
- National Research Council/National Academy of Sciences (NRC/NAS). 2006. Health risks from exposure to low levels of ionizing radiation (BEIR VII Phase 2). Committee to assess health risks from exposure to low levels of ionizing radiation. Washington, DC: National Academy Press.
- Neel JV. 1998. Genetic studies at the Atomic Bomb Casualty Commission-Radiation Effects Foundation: 1946–1997. Proc Natl Acad Sci USA. 95:5432–5436.
- Neumaier T, Swenson J, Pham C, Plyzos A, Lo AT, Yang P, Dyball J, Asithamby A, Chen DJ, Bissell MJ, Thalhammer S, Cotes SV. 2012. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells Proc Natl Acad Sci USA. 109:443–448.
- Organization for Economic Co-operation and Development (OECD). 2015. Report of the workshop on a framework for the development and use of integrated approaches to testing and assessment. In: Environment Directorate Joint Meeting of the Chemicals Committee and the Working Party on Chemicals. Paris: OECD.
- Ojima M, Furutani A, Ban N, Kai M. 2011. Persistence of DNA double-strand breaks in normal human cells induced by radiation-induced bystander effects. Radiat Res. 175:90–96.
- Olipitz W, Wiktor-Brown D, Shuga J, Pang B, McFaline J, Lonkar P, Thomas A, Mutamba JT, Greenberger JS, Sampson LD, Dedon PC, Yanch JC, Engelward BP. 2012. Integrated molecular analysis indicates undetectable DNA damage in mice after continuous irradiation at ∼400-fold natural background radiation. Environ Health Perspect. 120:1130–1135.
- Otsuka K, Koana T, Tauchi H. 2006. Activation of antioxidative enzymes induced by low dose-rate whole body gamma irradiation: adaptive response in terms of initial DNA damage. Radiat Res. 166:474–478.
- Ozasa K, Shimizu Y, Suyama A, Kasagi F, Soda M, Grant EJ, Sakata R, Sugiyama H, Kodama K. 2012. Studies of the mortality of atomic bomb survivors, Report 14, 1950–2003: an overview of cancer and non-cancer diseases. Radiat Res. 177:27–39.
- Paul S, Amundson SA. 2008. Development of gene expression signatures for practical radiation biodosimetry. Int J Radiat Oncol Biol Phys. 73:1236–1244.
- Paul S, Smilenov LB, Elliston CD, Amundson SA. 2015. Radiation dose-rate effects on gene expression in mouse biodosimetry model. Radiat Res. 184:24–32.
- Ponnaiya B, Cornforth MN, Ullrich RL. 1997. Radiation-induced chromosomal instability in BALB/c and C57BL/6 mice: the difference is as clear as black and white. Radiat Res. 147:121–125.
- Portess DL, Bauer G, Hill MA, O’Neill P. 2007. Low-dose irradiation of nontransformed cells stimulates the selective removal of precancerous cells via intercellular induction of apoptosis. Cancer Res. 67:1246–1253.
- Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, Mabuchi K, Komada K. 2007. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res. 168:1–64.
- Preston RJ. 2015. Integrating basic radiobiological science and epidemiological studies: why and how. Health Phys. 108:125–130.
- Purrott RJ, Reeder E. 1976. The effect of changes in dose rate on the yield of chromosome aberrations in human lymphocytes exposed to gamma rays. Mutat Res. 35:437–444.
- Redpath JL, Liang D, Taylor TH, Christie C, Elmore E. 2001. The shape of the dose-response curve for radiation-induced neoplastic transformation in vitro: evidence for an adaptive response against neoplastic transformation at low doses of low-LET radiation. Radiat Res. 156:700–707.
- Redpath JL. 2007. In vitro radiation-induced neoplastic transformation: suppressive effects at low doses. Radiat Res. 167:345–346.
- Redpath JL. 2006. Health risks of low photon energy imaging. Radiat Prot Dos. 122:528–533.
- Redpath JL, Lu Q, Lao X, Molloi S, Elmore E. 2003. Low doses of diagnostic energy X-rays protect against neoplastic transformation in vitro. Int J Radiat Biol. 79:235–240.
- Rogers BE, Baker RJ. 2000. Frequencies of micronuclei in bank voles from zones of high radiation at Chernobyl, Ukraine. Environ Tox Chem. 6:1644–1648.
- Rogers BE, Chesser RK, Wickliffe JK, Phillips CJ, Baker RJ. 2001. Subchronic exposure of BALB/C and C57BL/6 strains of Mus musculus to the radioactive environment of the Chernobyl, Ukraine exclusion zone. Environ Tox Chem. 20:2830–2835.
- Rothkamm K, Löbrich M. 2003. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray dose. Proc Natl Acad Sci USA. 100:5057–5062.
- Rothkamm K, Balrrop S, Shekhdar J, Fernie P, Goh V. 2007. Leukocyte DNA damage after multi-detector row CT: a quantitative biomarker of low-level radiation exposure. Radiology. 242:244–251.
- Rowley JD. 1980. The Philadelphia chromosome translocation. A paradigm for understanding leukemia. Cancer. 65:2178–2184.
- Ruhm W, Woloschak GE, Shore RE, Azizova TV, Grosche B, Niwa O, Akiba S, Ono T, Suzuki K, Iwasaki T, Ban N, Kai M, Clement CH, Bouffler S, Toma H, Hamada N. 2015. Dose and dose-rate effects on ionizing radiation: a discussion in the light of radiological protection. Radiat Environ Biophys. (DOI 10.1007/s00411-015-0613-6).
- Russell WL, Matter BE. 1980. Whole animal mutagenesis tests: evaluation of five methods. Mutat Res. 75:279–302.
- Russell WL, Russell LB, Cupp MB. 1959. Dependence of mutation frequency on radiation dose-rate in female mice. Proc Natl Acad Sci USA. 45:18–23.
- Russell WL, Russell LB, Kelly EM. 1958. Radiation dose rate and mutation frequency. Science. 128:1546–1550.
- Russell WL. 1968. Recent studies on genetic effects of radiation in mice. Pediatrics. 41:223–230.
- Russell LB, Hunsicker PR. 2012. The effect of dose rate on the frequency of specific-locus mutations in mouse spermatogonia is restricted to larger lesions: a retrospective analysis of historical data. Radiat Res. 177:555–564.
- Schaue D, Kachikwu EL, McBride WH. 2012. Cytokines in radiobiological responses: a review. Radiat Res. 178:505–523.
- Schulte-Hermann R, Grasl-Kraupp B, Bursch W. 2000. Dose-response and threshold effects in cytotoxicity and apoptosis. Mutat Res. 464:13–18.
- Scott BR. 2004. A biological-based model that links genomic instability, bystander effects, and adaptive response. Mutat Res. 568:129–143.
- Scott BR. 2007. Low-dose radiation induced protective process and implications for risk assessment, cancer prevention, and cancer therapy. Dose Res Int J Hormes Soc. 5:131–149.
- Shimizu Y, Kodama K, Nishi N, Kasagi F, Sutyama A, Soda M, Grant EJ, Sugiyama H, Sakata R, Moriwaki H, Hayashi M, Konda M, Shore RE. 2010. Radiation exposure and circulatory disease risk. BMJ. 340:b5349.
- Shuryak I, Sachs RK, Brenner DJ. 2010. Cancer risks after radiation exposure in middle age. J Natl Cancer Inst. 102:1628–1636.
- Simon T, Simons SS Jr, Preston RJ, Boobis AR, Cohen SM, Doerrer NG, Fenner-Crisp PA, McMullin T, McQueen CA, Rowlands JC. 2014. The use of mode of action information in risk assessment: quantitative key events/dose-response framework (Q-KEDRF) for modeling the dose-response for key events. Crit Rev Toxicol. 44(Suppl. 3):17–43.
- Spitz DR, Azzam EI, Li JJ, Gius D. 2004. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 23:311–322.
- Strahlenschutzkommission (SSK). 2014. Dose and dose-rate effectiveness factor (DDREF) Recommendationas by German Commission on Radiological Protection with scientific grounds: Chair: Beckow J. Members: Atkinson M, Dietze G, Kiefer J, Ruhm W. Adopted at the 268th meeting of the German Commission on Radiological Protection; 13–14. February 2014.
- Sturbaum B, Brooks AL, McClellan RO. 1970. Tissue distribution and dosimetry of 144Ce in Chinese hamsters. Radiat Res. 44:359–367.
- Sykes PJ, Zeng G, Day TK, Hooker AM, Blyth BJ, Bhat M, Tilley WD. 2006. Non-linear chromosomal inversion response in prostate after low dose X-radiation exposure. Mutat Res. 602:65–73.
- Tomasetti C, Vogelstein G. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 347:78–100.
- Tubiana M, Aurengo A, Averbeck D, Bonnin A, Le Guen B, Masse R, Monier R, Valleron AJ, de Vathaire F. 2005. Dose-effect relationships and estimation of the carcinogenic effects of low doses of ionization radiation. Paris: Academie National de Medicine, Institute de France-Academie des Sciences [English translation].
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). 1993. United Nations Scientific Committee on the Effects of Atomic Radiation. Annex F: Influence of dose and dose-rate on stochastic effects of radiation. New York: United Nations. pp. 620–728.
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) 2008. UNSCEAR 2006 Report. Annex B. Epidemiological evaluation of cardiovascular disease and other non-cancer disease following radiation exposure. New York: United Nations. pp. 325–383.
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) 2012. Biological mechanisms of radiation actions at low doses. A white paper to guide the Scientific Committee’s future programme of work. New York: United Nations. pp. 1–35.
- Ward JF. 1994. The complexity of DNA damage: relevance to biological consequences. Int J Radiat Biol. 66:427–432.
- Werner T, Haller D. 2007. Intestinal epithelial cell signaling and chronic inflammation: from the proteome to specific molecular mechanisms. Mutat Res. 622:42–57.
- Wickliffe JK, Rogers BE, Chesser RK, Phillips CJ, Gaschak SP, Baker RJ. 2002. Mitochondrial DNA heteroplasmy in laboratory mice experimentally enclosed in the radioactive Chernobyl environment. Radiat Res. 159:458–464.
- Wolff S. 1995. Adaptation of human and other mammalian cells to low doses of radiation. Radiat Res. 141:115–117.
- Yin E, Nelson DO, Coleman MA, Peterson LE, Wyrobek AJ. 2003. Gene expression changes in mouse brain after exposure to low-dose ionizing radiation. Int J Radiat Biol. 79:759–775.
- Zanzonico P, Stabin MG. 2014. Qunatitative benefit-risk analysis of medical radiation exposures. Semin Nucl Med. pp. 210–214.
- Zeng G, Day TK, Hooker AM, Blyth BJ, Bhat M, Tilley WD, Sykes PJ. 2006. Non-linear chromosomal inversion response in prostrate after low dose X-radiation exposure. Mutat Res. 602:65–73.