
Abstract
Purpose: Humans are increasingly exposed to ionizing radiation (IR). Both low (<100 mGy) and high doses can cause stochastic effects, including cancer; whereas doses above 100 mGy are needed to promote tissue or cell damage. 10–15% of radiotherapy (RT) patients suffer adverse reactions, described as displaying radiosensitivity (RS). Sensitivity to IR’s stochastic effects is termed radiosusceptibility (RSu). To optimize radiation protection we need to understand the range of individual variability and underlying mechanisms. We review the potential mechanisms contributing to RS/RSu focusing on RS following RT, the most tractable RS group.
Conclusions: The IR-induced DNA damage response (DDR) has been well characterized. Patients with mutations in the DDR have been identified and display marked RS but they represent only a small percentage of the RT patients with adverse reactions. We review the impacting mechanisms and additional factors influencing RS/RSu. We discuss whether RS/RSu might be genetically determined. As a recommendation, we propose that a prospective study be established to assess RS following RT. The study should detail tumor site and encompass a well-defined grading system. Predictive assays should be independently validated. Detailed analysis of the inflammatory, stress and immune responses, mitochondrial function and life style factors should be included. Existing cohorts should also be optimally exploited.
1. Introduction
The human population is being increasingly exposed to ionizing radiation (IR), due predominantly to the rising usage of medical radiodiagnostic procedures, in addition to natural background irradiation, including radon in homes built on radon emitting rocks and exposure during flights (McLean et al. Citation2017) (). X-ray exposure during computed tomography (C-T) scanning and interventional radiology are highly beneficial diagnostic tools and their usage has escalated dramatically in the US and in Europe in recent years (Power et al. Citation2016).
Figure 1. Radiation dose received during a range of environmental and medical sources. Background radiation can vary substantially depending on location. Single and repeated exposure from radiodiagnostic procedures can result in exposure to a range of doses and dose rates. For example, during mammography 2 mGy can be delivered in some minutes; for CT examination, 10 to 40 mGy is delivered in tens of minutes; during interventional radiology, 10–200 mGy is received over some hours. Exposure for radiation workers is normally much lower, being limited to 20 mGy per year, but can potentially be delivered (at very low dose rate) during each working day. Exposure during a 1000 km flight represents about 6-7 µSv per hour and chronic exposure to background radiation on Earth ranges between 2 and 70 mSv per year. Importantly, all these exposures can cumulate over a life-time to represent a non-negligible risk (Hall and Brenner Citation2012; Brenner Citation2014). Organ doses are given except for situations marked by asterisks where effective doses are indicated.

Radiation exposure of normal, non-cancer tissue can also arise during radiotherapy (RT). This also represents a significant and increasing aspect of exposure since RT remains a frontline treatment for cancer, with more than fifty percent of cancer patients receiving RT as a single modality, following surgery or in conjunction with chemotherapy (Begg et al. Citation2011). Conventional RT commonly involves daily exposures of about 2 Gy (with total tumor doses of 60–75 Gy being received) although for some cancers (e.g. breast and prostate), lower total doses delivered in fewer, larger daily exposures are being increasingly used (Yarnold Citation2019). Considerable technological advances, such as intensity-modulated RT, image-guided RT and stereotactic RT have enhanced dose conformation, i.e. delivery of the maximal dose to the tumor while sparing healthy tissues. However, some of these new technologies expose a larger volume of healthy tissue to IR than conventional RT (Hall Citation2006; Palm and Johansson Citation2007; Lisbona et al. Citation2010; Tommasino and Durante Citation2015; Ding et al. Citation2018; Durante and Debus Citation2018). A review of imaging practices in five Finnish radiotherapy clinics revealed that the organ absorbed doses depended on the imaging technique and frequency, and could vary widely. Indeed, especially for cone beam computed tomography, the cumulative imaging organ dose could differ as much as tenfold (Siiskonen et al. Citation2017).
Finally, there are now RT procedures that do not use photons as the radiation source but involve proton/particle therapy because of greater tumor targeting potential. These different RT procedures can strongly influence the dose-rate and the total delivered dose, as well as the dose received by the surrounding normal tissue. Nonetheless, currently the dose received by the normal tissue is often greater than 100 mGy.
The health effects of exposure to IR can be divided into two main categories: tissue reactions, formerly referred to as deterministic effects, and stochastic effects (ICRP Citation2007). Deterministic effects generally arise after exposure to higher doses while stochastic effects ensue after lower and higher doses. There is evidence that the incidence of both radiation-induced deterministic and stochastic effects in the human population is individually variable (AGIR Citation2013; Foray et al. Citation2016; Rajaraman et al. Citation2018). Individual variability emerges in epidemiological studies when risk modifying factors such as sex and age at exposure are considered. Radiosensitivity has been defined by the Independent Advisory Group on Ionizing Radiation (AGIR) as a measure of the degree of response of a cell or an organism to radiation with a large response indicating high radiosensitivity (AGIR Citation2013). There has been a recent proposal to define and distinguish radiosensitivity and radiosusceptibility (Foray et al. Citation2016; Foray and Bourguignon Citation2019). Here, we follow this proposal and define radiosensitivity (RS) as any enhanced tissue or cell reaction following exposure to IR compared to that of the majority of individuals, classified as normal responding individuals. We use the term radiosusceptibility (RSu) in relation to stochastic effects, of which IR-induced cancers are an important example. Radiosusceptible individuals, therefore, represent those with an elevated risk of IR-induced cancer. Although these terms distinguish events arising after distinct doses and with different kinetics, and thus likely represent mechanistically distinct processes, there is likely to be overlap since some exposures and outcomes (such as cataracts or heart disease) do not neatly fall into one or other category. Although this terminology clearly has limitations (Wojcik et al. Citation2018), we use it here as a useful, current working terminology.
Although RS and RSu may arise after a range of distinct types of exposures, individual RS is most strikingly evident following RT, where cancer patients treated for a common form of cancer and using the same treatment scheme, can show widely different degrees of tissue reactions (Bentzen and Overgaard Citation1994) (). For stochastic effects the impact of individual variability on risk is less clear (Rajaraman et al. Citation2018), although it is well documented that some rare genetically inherited factors contribute to an enhanced risk of IR-induced cancer (AGIR Citation2013; Foray et al. Citation2016). There is also evidence for combinatorial effects. For example, smokers were suggested to have a significantly higher risk of radon-induced lung cancer compared to nonsmokers (AGIR Citation2013). Finally, although significant differences in the background incidence of certain cancers are observed between human populations, it is not well established how far these differences contribute to the risk of radiation-induced cancers, causing significant uncertainties when radiation-related cancer risks are transferred between populations (AGIR Citation2013).
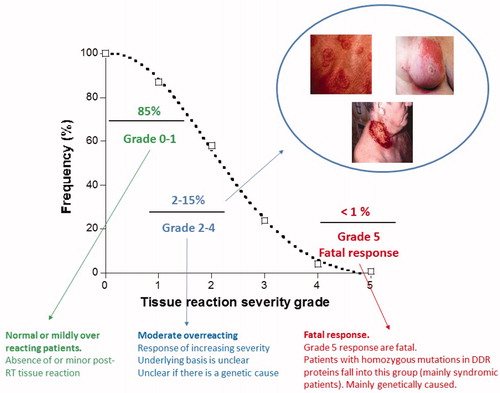
Figure 2. Grade assessment for distribution of responses to Radiotherapy. Radiotherapy doses are chosen to ensure that the majority (at least 85%) of patients display no or minor post-RT tissue reactions. Such patients are defined as showing grade 0-1 reactions. Some patients (up to 15%) show moderate tissue complications that can cause moderate to severe discomfort. Such patients fall into grade 2-3/4 responses). A small subset of patients display a severe response, which for a grade 5 response can be fatal. Patients with mutations in the genes that function in DNA non-homologous end-joining or the ATM-signaling pathway can fall into this category. The precise percentage of patients that are considered to be RS differs slightly between countries and/or departments/hospitals. Particularly critical is the assessment of whether a grade 2 response lies within the normal or RS range. Here, we have placed a grade 2 response as being within the RS category. We give the percentage of RS individuals as ∼15%, which represents an average of estimates presented in the literature.
The goal of radiological protection policy is to protect not only human populations and the environment but also individuals (Cho et al. Citation2018). Inherent to this aim is the need to gain an improved understanding of the degree and mechanisms of individual variability in RS and RSu and, particularly, whether inter-individual differences result in a subset of sensitive individuals being more adversely affected than the average exposed individual for stochastic effects as well as tissue reactions. This requirement is timely and fits with the overall trend of personalizing the health system moving away from a “one size fits all” approach (Jackson and Chester Citation2015). Related to this, is the important question of whether a radiosensitive person can be identified prior to exposure, allowing optimal protection and personalized decision making via individual dose optimization during RT.
Based on the degree of RS, individuals exposed to RT can be classified into graded clinical responses (grade 0–5) (with grade 0–1 corresponding to the absence of or a minor post-RT tissue reaction, grade 2 representing a mild response outside of the normal range and grade 3–5 corresponding to responses of increasing severity. A grade 5 response is a fatal post-RT reaction (). Although there is some variation between countries, most likely due to the categorization of grade 2, generally 10–15% of patients are reported to display a grade 2–4 response (). The above classification is based largely on tissue reactions, which include inflammation and fibrosis. IR-induced heart disease, cognitive decline and cataracts are additional well-reported consequences of IR exposure (Averbeck et al. Citation2018). Although doses >500 mGy were considered to be required to induce these endpoints, there is emerging evidence that they can arise at lower doses (Dauer et al. Citation2017; Hughson et al. Citation2018), making precise dose categorization difficult. Additionally, tissues differ in their RS (and RSu) to IR and, as importantly, the time when tissue damage manifests post IR exposure. Historically, when considering RS, tissues have been divided into early and late responding. Acute or early side effects mainly occur in highly proliferating tissues and develop within 90 days of the onset of radiotherapy. Late effects can be observed several months or even years after radiotherapy, and they occur in both early and late responding tissues (Dorr Citation2015). However, this division is not absolute and, notably due to the diversity of new RT modalities and the different ways of delivering the dose, the precise distinction between early and late effects is poorly defined.
2. Goal of this review
The aim of this paper is to review the mechanisms that could underlie or modulate individual variability in RS and RSu, and to consider future approaches to gain further insight. Understanding the underlying mechanisms will enhance our ability to identify RS and RSu individuals via optimized bioassays and biomarkers. This will not only avoid substantial pain or discomfort arising from an adverse reaction during RT but could allow the use of higher doses for more radioresistant individuals, enhancing the overall efficacy of RT. The understanding of factors conferring RSu and identification of such individuals could help to reduce IR-induced cancer incidence from low dose exposure and from RT. These goals, namely to understand the mechanistic basis underlying RS/RSu and to predict such responses, represents a major challenge, which will necessitate a cross-disciplinary approach involving interface between clinicians, clinical scientists, and basic scientists of distinct disciplines. Additionally, there are several distinct approaches that can provide insight. Thus, we aim to present a co-ordinated set of reviews discussing the available evidence and approaches to evaluate RS/RSu and to promote mechanistic insight into the underlying processes. Review 1 will consider the epidemiological and clinical evidence (Seibold et al. Citation2019). Epidemiological studies are powerful in providing risk estimation and, potentially, when coupled with molecular analysis, insight into mechanisms. Review 3 will consider the evidence for radiation specific biomarkers and the validity of current screening assays (Gomolka et al. Citation2019). Review 4 covers ethical implications (Kalman and Oughton Citation2019). Our focus in this review 2 is to consider the underlying mechanisms conferring RS or RSu, including aspects such as life style factors, that can modify or impact upon RS/RSu. We consider RS and RSu separately since the underlying pathways, although overlapping, are likely to have distinctions. Currently, most insight has been gained from cellular studies aimed at addressing the mechanisms underlying the response to RT, which will form a substantial part of our review. However, other approaches to consider RS/RSu in other contexts are important and will be discussed.
Here, we firstly review and evaluate the main cellular pathways that influence RS/RSu. We also review multiple factors, including life style factors, that can influence RS/RSu by impacting upon these pathways or via distinct mechanisms. Additionally, we discuss non-physiological aspects that are important in evaluating RS/RSu. We include a brief discussion of some potential predictive assays that monitor functionality of the cellular pathways with a focus on the underlying mechanism. Further discussion of the assays as potential biomarkers for RS/RSu is encompassed within review 3. We provide a summary of these findings to aid consideration of the critical questions that need to be addressed and finally propose recommendations for how these can be optimally addressed in a co-ordinated manner.
3. Main cellular pathways determining radiation sensitivity (RS)
3.1. The DNA damage response (DDR)
DNA has long been identified as the major cellular target with DNA double strand breaks (DSBs) being the most biologically significant lesion determining survival following radiation exposure. The response to DSBs encompasses pathways of DNA repair and a signal transduction response, which interface but are mechanistically distinct (Shibata and Jeggo Citation2014). Collectively, this is called the DNA damage response (DDR). Any variation in efficiency of the DDR and its impact on the fidelity of repair is expected to affect cell survival and/or genomic stability. The most significant DNA repair pathway is DNA non-homologous end-joining (NHEJ) (Chang et al. Citation2017). In brief, this pathway involves the binding of the Ku protein to DNA ends, which protects them from degradation. This is followed by recruitment of the DNA dependent protein kinase catalytic subunit (DNA-PKcs) (generating the DNA-PK complex) and finally, ligation by a complex involving DNA ligase IV, XRCC4 and XLF (see (Shibata and Jeggo Citation2014) for a review).
Homologous recombination represents a second DSB repair pathway, which functions uniquely in late S/G2 phase since it necessitates a sister chromatid as a template to repair the DSB (Shibata and Jeggo Citation2014). As a consequence of using an undamaged sister molecule to repair the DSB, HR is often considered an accurate repair process whilst NHEJ is argued to be error-prone. However, although NHEJ certainly has some limitations, it is likely to be relatively accurate in repairing DSBs induced by low doses of low LET radiation. Significantly, although in G2, there is the potential for DSBs to be repaired by HR, the majority of DSBs still undergoes repair by NHEJ; thus, even in G2, NHEJ represents the major DSB repair pathway after low LET radiation (Jeggo et al. Citation2011). However, HR plays a more significant role in G2 in repairing complex DSBs induced by high LET IR (Jeggo et al. Citation2011). Interestingly, recent work has shown that an important subset of DSBs, namely, those within transcriptionally active regions (termed TA-DSBs) appear to preferentially undergo repair by HR (Marnef et al. Citation2017; Yasuhara et al. Citation2018). Factors regulating the choice between HR and NHEJ usage are currently under intense study, of which those determining the use of HR at TA-DSBs, may be particularly important in the context of influencing RS. Since an early step in HR involves DSB end-resection, which serves to preclude the use of NHEJ, factors influencing resection are important in determining pathway choice (Shibata and Jeggo Citation2014). It should be stressed, however, that the major role of HR lies in repairing DSBs that arise following replication fork collapse and in promoting replication fork restart after stalling (Shibata and Jeggo Citation2014).
The ataxia telangiectasia mutated (ATM) kinase lies at the center of the signaling response to DSBs and activates a choreographed assembly of proteins, which collectively orchestrate chromatin changes over large regions around the DSB (Shibata and Jeggo Citation2014; Guleria and Chandna Citation2016). These have been termed irradiation-induced foci (IRIF). ATM has a huge number of substrates and hence impacts upon many modifying pathways, of which cell cycle checkpoint arrest and apoptosis are important in considering RS/RSu. These two responses will be discussed in section 5c below, when discussing predictive assays based on these responses. Although ATM signaling and NHEJ can function independently, ATM has an essential role in a sub-component of NHEJ. Additionally, and of significance here, ATM has a major role in regulating the accuracy of repair via its influence on a range of processes, including the formation of IRIF and cell cycle checkpoint arrest, which will be considered when discussing RSu.
Human and rodent cell lines defective or deficient in NHEJ or ATM signaling proteins display severe RS, reflecting the significance of these pathways in the DSB response. Patients carrying mutations in NHEJ or ATM signaling proteins have also been identified and cells from such patients similarly display pronounced RS (Woodbine et al. Citation2014; Foray et al. Citation2016). In some cases, such patients have received RT and displayed subsequent marked RS (usually with a grade 5 response) (Foray et al. Citation2016). The majority of these patients display syndromic features and are diagnosed at a young age. Efficient diagnosis normally prevents the use of RT based treatments for such patients. However, a small number of patients with weaker impacting mutational changes do not display syndromic features and can escape diagnosis. Patients of this nature have received RT for cancer (they are also prone to carcinogenesis) and have displayed severe RS (see (Riballo et al. Citation1999) for an example). Thus, a level of residual activity of these proteins sufficient to protect against DSBs arising endogenously or from background radiation is insufficient to protect against the higher doses that the normal tissue may receive during RT. Heterozygous carriers of ATM, however, have not generally been identified as over-responding to RT, although there is some evidence that they can show a heightened sensitivity in some cellular-based assays (Royba et al. Citation2017; Aghamohammadi et al. Citation2018). Although single nucleotide polymorphic (SNPs) variations in some DDR proteins have been reported (e.g. in XRCC1 and ATM), weakly impacting polymorphic modifications in the majority of proteins whose loss confers major RS (grade 4–5 responses) are not commonly observed in patients displaying grade 2–3 responses nor in genome-wide association (GWAS) studies (Seibold et al. Citation2015; Andreassen, Rosenstein, et al. Citation2016). An important question is whether deficiency in HR confers RS. Since HR is essential, any mutational changes observed in patients are hypomorphic. Significantly, heterozygous loss of BRCA1 or BRCA2 confers breast and ovarian cancer predisposition and there is evidence that such patients may be RSu (see below). Whether they display RS is less clear (see for syndromes that might confer RS or RSu). We discuss the potential RS caused by mutations in genes that impact upon the DDR, including BRCA2, suggested by the findings from a predictive assay below.
Table 1. All references for these findings are given in (Foray et al. Citation2016).
In summary, patients displaying marked RS (i.e. showing grade 4–5 responses) frequently harbor homozygous or compound heterozygous mutations in NHEJ or ATM signaling genes, such as ATM, LIG4 or NBS1. However, these genetically RS individuals (i.e. those with identified bi-allelic mutations in known DDR proteins) represent a minor subset (less than 1%) of the patients showing adverse tissue reactions following RT although such patients normally display a severe response and hence represent a greater percentage of those displaying a grade 5 reaction. The diagnosis of syndromic patients is reasonably well established because such patients are usually earmarked for RS analysis due to displaying characteristic features. Identifying the very small subset of patients with mildly impacting mutations in DDR proteins (i.e. without syndromic features) is also important and is not carried out routinely due to the lack of the identifying syndromic phenotype. However, it is also important to identify any additional genes that indirectly impact upon the DDR to radiation, particularly if they prove to confer RS.
In summary, deficiency in the DDR proteins clearly confers RS to doses that normal tissues can encounter during RT. However, mutational changes in the encoding genes do not make a major contribution to the group classified as moderate RS individuals (grade 2–4 responses) based on their response to RT. Therefore, in sections 3–5 below we consider additional responses or pathways that may confer RS with a grade 2–4 reaction. It is noteworthy that the proposed factors (or pathways) may modify the DDR or they may have a distinct mode of action, such as causing additive DNA damage or influencing a distinct process, which is additive or synergistic to the DDR.
3.2. The oxidative stress response
3.2.1. Activation of the oxidative stress response and preexisting oxidative stress
Approximately 30% of DNA damage induced by photon irradiation arises directly from interaction of the radiation tracks with DNA. Most indirect DNA damage (i.e. 70% of IR-induced damage) arises from radicals, mainly hydroxyl radicals (OH•) produced by the interaction of IR with water (Nikjoo et al. Citation1999). However, reactive oxygen species (ROS) can also be released from dysfunctional mitochondria directly induced by IR or through the activation of a stress response involving members of the BH3 domain protein family such as Bax or Bak. ROS can furthermore be generated after activation of NADPH oxidases.
The release of ROS by mitochondria and NADPH oxidases during the oxidative stress response elicits the activation of processes to cope with ROS including the deactivation of ROS. In this context, it is noteworthy that ATM influences the stress response by activating ROS scavengers (Bagley et al. Citation2007). However, additional processes can be activated involving a range of transcriptional changes, including processes that can remove oxidized products. Importantly, exposure to IR can activate the stress response. However, additionally, a range of factors, including life style and the presence of a tumor, can activate a stress response and affect preexisting oxidative stress and redox regulation (Zhang et al. Citation2011).
Hydroxyl radicals and other ROS species can exert a range of cellular impacts (see (Spitz et al. Citation2004; Zhou et al. Citation2014) for a review), which includes damage to DNA. However, they can also damage nucleotides within the nucleotide pools, which, unlike DNA, are not protected by chromatin. The damaged nucleotides have the potential to be incorporated into DNA since they can be efficient substrates for DNA polymerases. However, normally only a small component of indirect DNA damage arises via the incorporation of damaged nucleotides since the pools are efficiently sanitized by mechanisms that function in addition to ROS scavenging. 8-oxo-dGTP represents an important damaged nucleotide and Mth1, an 8-oxo-dGTPase, plays a critical role in sanitizing the nucleotide pools by removing 8-oxo-dGTP (Ichikawa et al. Citation2008). This process results in the release of 8-oxo-dG into the urine or serum and precludes its usage during DNA repair or replication. Thus, if Mth1 is impaired in function or becomes saturated, the impact of IR-induced DNA damage arising following incorporation of damaged nucleotides can increase and significantly contribute to the level of IR-induced DNA damage. Support for a model of this nature has been provided by findings showing that the level of 8-oxo-dG increased in the urine of normal responding patients after RT but not in radiosensitive (group 3–4) patients (Haghdoost et al. Citation2001). Further studies involving ex vivo irradiation of blood samples from normal and radiosensitive patients (group 3–4) (Skiold et al. Citation2013; Danielsson et al. Citation2016; Khavari et al. Citation2018) were consistent in revealing distinctions between normal responding and RS patients in their ability to generate 8-oxo-dG. In these studies, the presence of 8-oxo-dG in the urine or serum was argued to arise following Mth1 action on 8-oxo-dGTP in the nucleotide pool, which was consolidated by comparing fibroblasts in which Mth1 was normally expressed or silenced (Haghdoost et al. Citation2006). In follow-up studies, a proteomic approach on a retrospective cohort revealed that RS patients have higher expression of several antioxidant enzymes several years after RT which was not observed in normal responding patients (Skiold et al. Citation2015).
Collectively, these findings suggest that the ability to rapidly remove 8-oxo-dGTP from nucleotide pools is an important response following RT and that the inability to achieve this efficiently may lead to RS (Haghdoost et al. Citation2006). Moreover, RS patients appear to be in a preexisting state of oxidative stress with high levels of 8-oxo-dG in their serum prior to IR exposure, which may preclude the normal response of increased 8-oxo-dG excretion post IR. Thus, it was proposed that RT causes massive dGTP oxidation (8-oxo-dGTP). This could, if not efficiently removed, e.g. due to saturation of the sanitizing response, be incorporated into DNA, causing abasic sites and SSBs following repair processing, potentially enhancing the complexity of IR-induced DNA damage. Aberrant levels of damaged bases in the nucleotide pools may also lead to harmful pool imbalance. Thus, oxidative stress levels prior to RT could be a determinant of RS influencing the response to oxidative stress post RT. Potential biomarkers for analysis have been identified (Haghdoost et al. Citation2001; Danielsson et al. Citation2016). The impact of high endogenous oxidative stress could modulate the response to IR by causing DNA damage that is additive to directly induced damage (as discussed above) or it could impact upon the repair of IR-induced damage, for example by affecting the kinetics and fidelity of repair due to increased damage complexity. The extent to which genetic factors underlie the response to oxidative stress or to which the response is influenced by lifestyle or other non-genetic factors is unclear. It is also possible that the tumor itself influences oxidative stress levels and the radiation response.
3.2.2. Protein oxidation/carbonylation
There is also evidence emerging that oxidative stress can affect protein and membrane functions, which can substantially contribute to RS in living systems. In some extremely radioresistant species, e.g. bacteria such as Deinococcus radiodurans, bdelloid rotifers, tardigrades, nematodes and insect cells, radioresistance can be attributed to specific mechanisms that protect against protein oxidation (Slade and Radman Citation2011; Krisko et al. Citation2012; Chandna et al. Citation2013; Beltran-Pardo et al. Citation2015). Based on these findings, Radman proposed the novel concept that protein damage underlies RS/resistance as well as aging and related diseases (Radman Citation2016). Indeed, the ability to protect against protein carbonylation, a form of oxidative protein damage caused by IR, correlates with resistance to radiation across many species (Daly et al. Citation2004; Aryal and Rao Citation2018). The individual antioxidant defense and capacity to deal with oxidative stress is highly variable (Valko et al. Citation2007; Ruszkiewicz and Albrecht Citation2015; Kurutas Citation2015). Thus, it is possible that the inter-individual response to RT can be influenced by the individual’s antioxidant and antiradical defense capacity, impacting upon protein as well as DNA damage. The oxidation of proteins may, however, affect overall DNA repair capacities. Further, supporting a role for antioxidant protein defenses, there is indirect evidence that such defense processes may play a significant role in the protection against oxidative stress and the induction of premature senescence in human fibroblasts exposed to chronic low dose rate IR (Loseva et al. Citation2014). It is also noteworthy that protein carbonylation by oxidative stress and subsequent diminished DNA repair has been observed after UV irradiation and with melanoma induction (Emanuele et al. Citation2014). Currently, however, protein carbonylation has not been examined in patients receiving RT and any direct link to RS of such patients has not been established.
3.3. Predictive assays for RS based on monitoring the DDR and/or oxidative stress
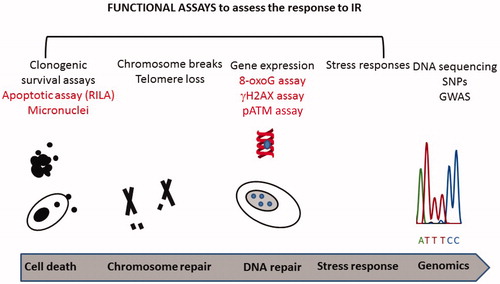
Given the importance of the DDR and the oxidative stress response in determining the response to IR, assays to predict RS have focused on monitoring these responses and several assays have been reported (see for a depiction of some functional assays that can assess the response to IR). Paper 3 discusses the utility of these assays as biomarkers for assessing RS. Here, we discuss two of these assays (namely the RILA assay and an assay involving γH2AX and pATM) from the mechanistic viewpoint and consider how they provide potential insight into the mechanisms underlying RS. The impact of the stress response was discussed above.
Figure 3. Functional assays that can assess the response to IR exposure in cells. A range of assays have been reported that can assess RS in cultured cells. Four of these assays have been reported to correlate with the patient response following RT (highlighted in red). In addition, GWAS studies have been carried out on patients following RT and are included in this Figure. Further assays and biomarkers are discussed in review 3 (Gomolka et al. Citation2019).
3.3.1. The nucleoshuttling of ATM
This functional test uses fibroblasts derived from patients (Bodgi and Foray Citation2016; Granzotto et al. Citation2016). In fibroblasts derived from normal responding patients (designated grade 0 or 1), foci of ATM phosphorylated at S1981 (p-1981 ATM), the autophosphorylation site on ATM required for its DSB-dependent activation, form rapidly in the nucleus after IR exposure. In fibroblasts from RS patients (grade 2–4), nuclear p-1981 ATM foci formation is delayed and does not reach the same yield as in the cells from normal responding patients. At later times, however, the DSBs are recognized and repaired. Cells from extremely radiosensitive patients (designated grade 5) display either very few or no p-1981 ATM foci (e.g. if they are ATM deficient) or they form foci efficiently but display a gross DSB repair defect (e.g. patients mutated in DNA ligase IV, the NHEJ ligase). An explanation for delayed formation of ATM foci in grade 2–4 patients is that the level of nuclear ATM is insufficient to mount an efficient DDR since most ATM in undamaged cells is localized in the cytoplasm as an inactive dimer and after IR converts from the inactive dimer status to an active monomer. Only active monomers are transported to the nucleus. The working model is that the grade 2–4 RS patients specifically express cytoplasmic proteins that form a multiprotein complex with monomeric ATM, sequestering activated ATM in the cytoplasm and precluding its shuttling to the nucleus (hence called delayed ATM-nucleoshuttling). This impedes the rapid formation of MRE11 foci and hence normal ATM and/or MRN function during the DDR. This model has also been used to explain RS of genetic syndromes caused by mutations in cytoplasmic proteins such as Huntington’s disease, neurofibromatosis and Tuberous Sclerosis syndrome (Ferlazzo et al. Citation2014; Ferlazzo et al. Citation2018).
The functional test has predicted the response of 117 patients to RT based on a complete version of the assay (Granzotto et al. Citation2016) and of 30 patients in a faster, modified version, with a high degree of accuracy and independent of cancer site and the early/late nature of the post-RT reaction (Pereira et al. Citation2018; Vogin et al. Citation2018). Of mechanistic importance, the findings strongly suggest that non-core DDR proteins can influence the DDR and that an aberrant DDR contributes to RS. Currently, the findings do not reveal whether the response is genetically determined or due to other factors. Importantly, however, the assay uses patient fibroblasts but is predictive for a range of tissues, suggesting that either there is a genetic determinant or there is a general response arising in multiple tissues or cell types.
Interestingly, the assay has led to the identification of heterozygous mutations in genes that have been proposed to impact on the DDR, such as Neurofibromatosis (NF1), Tuberous Sclerosis (TSC) and the breast cancer susceptibility gene, BRCA2, which functions during HR, in patients showing grade 2–4 responses (Foray et al. Citation2016). Thus, if the findings of the assay are verified, they can provide important insight into factors that by affecting the nucleoshuttling of ATM, can influence the response to IR.
3.3.2. Radiation-induced CD8 T-lymphocyte apoptosis (RILA)
Apoptosis represents an important pathway of programed cell death and can arise as part of the DDR, being activated in a p53-dependent manner. Such apoptosis is attributed to excessive or persisting DNA damage. However, additionally, via a death receptor-mediated process, apoptosis can be induced by generation of sphingolipid ceramide clustering of membrane-bound proteins (receptor proteins) which initiate the activation of a cascade of mitogen-activated protein (MAP) kinases and caspases leading to the fragmentation of nuclear DNA (e.g. (Taha et al. Citation2006)). This latter pathway leading to apoptosis and cell death arises independently of DDR signaling. Thus, cell apoptosis may involve either DNA damage signaling or signaling from damaged membranes. Importantly, apoptosis has been linked to intrinsic RS in peripheral lymphocytes and EBV-transformed lymphoblastoid cell lines (Biechonski et al. Citation2018). Significantly, in the present context, it has been proposed that apoptosis in CD8+ lymphocytes can predict IR-induced late toxicity, from which a test for radiation-induced CD8 T-lymphocyte apoptosis (RILA) was developed (Ozsahin et al. Citation2005; Schnarr et al. Citation2009; Azria et al. Citation2015; Mirjolet et al. Citation2016). In this test, lymphocytes from normal and RS patients are irradiated ex vivo with 2 or 8 Gy and apoptosis is assessed. After exposure to 2 Gy, there is a direct relationship with high apoptosis correlating with the overresponse to RT but at 8 Gy, the apoptotic level is inversely related to response. The validation and predictive capacity of this assay has been undertaken in two studies, and several prospective trials are progressing (West et al. Citation2014; Azria et al. Citation2015; Mirjolet et al. Citation2016). The working model is that radiosensitive patients display aberrant activation of apoptosis, which could be a consequence of changes affecting the DDR or distinct changes that only impact upon apoptosis. However, it should be noted that the RILA assay cannot predict RS alone but needs additional information (e.g. confounding factors such as systemic treatments or tobacco use) to be added to the determinant (Azria et al. Citation2015).
Given the relationship of apoptosis activation to clinical RS and the evidence for a heritable component, studies were undertaken to investigate the relationship between single nucleotide polymorphisms (SNPs) in TRAIL/TNFSF10 with apoptosis in CD8+ lymphocytes and clinical RS endpoints in a series of breast cancer patients (Ozsahin et al. Citation2005; Schmitz et al. Citation2007). Using blood samples from patients a genetic association was found between mRNA levels of the TRAIL/TNFSF10 locus and acute and subacute dermatitis and RS of T4 effector memory lymphocytes suggesting that TRAIL/TNFSF10 genetic variants may be used as markers of individual RS (Baijer et al. Citation2016). These studies reveal a prominent role for the membrane bound protein, TRAIL (mTRAIL) in mediating pro-apoptotic autocrine signaling in T4 lymphocytes (Baijer et al. Citation2016).
3.3.3. Cell cycle checkpoint arrest
Cell cycle checkpoint arrest represents an important component of the DDR, which likely impacts upon both RS and RSu (Jeggo and Lavin Citation2009). Effectively the presence of DNA damage activates a signal transduction response which halts cell cycle progression, precluding entry into cell cycle phases (S and mitosis), where DNA damage can be compounded either following replication or cell division. Following IR exposure, ATM represents the major kinase activating checkpoint arrest, with progression through G1 and G2 phases, being the most significant pathways activated (Jeggo and Lavin Citation2009). Interestingly, failure to implement G1 checkpoint arrest, which occurs in cells lacking p53, does not confer major RS although it may be important in restricting RSu. However, the ability to activate G2/M arrest has been evaluated as a mechanism conferring RS (Roberts et al. Citation1999). Radiosensitivity, in this context, was assessed as chromosomal radiosensitivity, and there was some evidence for heritability of such sensitivity. However, follow up studies have not consolidated this assay as predictive for RS following RT (Hall et al. Citation2017). However, it is possible that the assay can detect genetic conditions conferring predisposition to breast cancer, since breast cancer patients showed evidence of enhanced chromosomal radiosensitivity (Scott Citation2004).
Collectively, the findings of predictive assays of RS can be important not only in providing a biomarker or bioassay for assessing RS but in revealing insight into mechanisms that may confer RS. The findings discussed here raise the possibility that the speed with which the DDR is activated and/or the ability to regulate apoptosis may be factors determining RS.
4. Additional factors influencing RS
Above we have discussed the major pathways (the DDR and the oxidative stress response) which determine RS. However, there are a substantial number of further responses or factors which also influence the response to radiation, which we consider in this section. The separation into a distinct section is more operational than defined and should not undermine their significance, however. These factors or responses, may directly impact upon the DDR or oxidative stress response, but may have distinct effects with a complex interplay. For example, activation of the immune response may result in death of the targeted cell, which is entirely distinct to DDR induced cell death pathways, such as apoptosis. Other factors such as telomere attrition may lead to cell death via activation of DDR signaling.
4.1. The epigenetic status of DNA and influence of chromatin
Epigenetic patterns play a major role in regulating gene expression. DNA methylation and post-translational histone modifications determine the epigenetic status and affect chromatin structure and accessibility of DNA sequences. Unwarranted alterations in epigenetic patterns result in altered cell function and morphology, and hence cellular dysfunction. Epigenetic marks are highly responsive to IR and influence the DDR (Price and D’Andrea Citation2013; Agarwal and Miller Citation2016; Miousse et al. Citation2017) and it has been proposed that “the DNA methylation landscape may influence the tissue response to IR” (Miousse et al. Citation2017). Three types of underlying mechanisms can be defined: i) changes introduced at the DNA damage site and the surrounding chromatin domain in the process of damage recognition, signal transduction and repair, ii) changes related to programed gene expression alterations in the response to IR and iii) changes related to IR-induced disturbances in the availability of enzymes and co-factors (Karabulutoglu et al. Citation2019).
Alterations of epigenetic marks at the damage site or in the chromatin domains marked as IR-induced foci have been well investigated. They have roles in signaling, alteration of chromatin structure to facilitate repair or coordination between different events taking place on DNA (Wilson and Durocher Citation2017). If, after successful repair, these marks are not reverted to their original status, it is likely that long-term, randomly distributed alterations will arise in the genome (O'Hagan Citation2014). The response of cells to IR with activation or repression of specific genes, is accompanied by alterations of epigenetic patterns. If subsequently the original epigenetic patterns are not restored, there may be long-term alterations enriched at IR-responsive genes (Antwih et al. Citation2013). Altered expression or activity of epigenetic enzymes, such as the DNA methyltransferase DNMT1 or changes in metabolism which lead to alterations in the availability of co-factors can also affect epigenetic patterns (Koturbash et al. Citation2016; Miousse et al. Citation2017). This type of long-term epigenetic alteration is expected to be randomly distributed and potentially frequent.
Evidence has accumulated that nutrition and diet can affect the epigenetic pattern, e.g. by providing or limiting co-factors such as S-adenosyl-methionine or acetyl-CoA needed by the histone and DNA modifying enzymes (Etchegaray and Mostoslavsky Citation2016; Feinberg Citation2018). First indications for an effect of physical activity and stress factors on epigenetic patterns have also been provided (Thomas et al. Citation2017; Denhardt Citation2018). Thus, it is conceivable that lifestyle factors may influence the epigenetic pattern present before IR and the resilience to IR-induced long-term alterations. In addition, the epigenetic status present at the damage site can influence the nature of the repair process. For example, DSBs located within highly compacted regions of DNA (heterochromatin) have been reported to be repaired with slow kinetics in an ATM-dependent manner (Goodarzi and Jeggo Citation2012). Finally, by altering epigenetic patterns, lifestyle may modulate any health effects, including tissue effects and IR-induced cancer, caused by IR-induced changes in epigenetic patterns.
The question arises how large are the inter-individual differences in epigenetic patterns related to lifestyle factors. Recent work on the so-called epigenetic clock suggest a major influence of lifestyle on the age-associated accumulation of epigenetic alterations (Quach et al. Citation2017; Declerck and Vanden Berghe Citation2018). Vitamin C is an important modulator of the activity of the methylcytosine dioxygenase TET2, which is involved in DNA regulating methylation status (Agathocleous et al. Citation2017; Cimmino et al. Citation2017). Vitamin C levels in blood serum were observed to vary more than 10-fold in the population (Schleicher et al. Citation2009), and at least part of this variation presumably is due to diet. These and other examples strongly suggest that individual RS and RSu could be modulated by the effects of lifestyle on epigenetic patterns.
4.2. Non-coding RNA
In addition to epigenetic factors influencing histone and DNA modifications, changes in non-coding RNAs can also lead to transcriptomic and proteomic changes (Schofield and Kondratowicz Citation2018). MiRNAs are one such class of small non-coding RNA capable of regulating gene expression post-transcriptionally and influencing the radiation response (Kraemer et al. Citation2011). They influence multiple aspects of cellular responses including DNA damage sensing, signal transduction, DNA repair, cell cycle checkpoint activation and induction of apoptosis (Zhang and Peng Citation2015). MiRNAs, can also confer persistently elevated ROS levels due to an influence on mitochondrial function, causing activation of an oxidative stress response (Kim et al. Citation2006). In addition to endogenous miRNA expression influencing RS, expression of non-coding miRNAs undergoes changes post IR exposure, which can be predictive of the radiation response (Ma et al. Citation2010; Li et al. Citation2018). Indeed, miRNA expression profiles are a hallmark of cancer (Zaheer et al. Citation2019), useful for cancer diagnosis, prognosis and, importantly, in the present context, for influencing and potentially predicting RS (Lacombe and Zenhausern Citation2017). As an example, IR-induced miR-34a expression affects both the response of the tumor and normal tissue to radiotherapy by enhancing the level of DNA damage and the response to it (Kofman et al. Citation2013; Lacombe and Zenhausern Citation2017). Additionally, important components of the DSB repair pathways, NHEJ and HR, are regulated by miRNAs including miR-101 which confers radiosensitivity by targeting DNA-PKcs and miR-107, miR-222 and miR-96, which can target HR (Czochor and Glazer Citation2014). ATM also can be regulated by miRNAs (Kabacik et al. Citation2015). Interestingly, miRNAs can also be regulated by dietary and nutritional compounds, providing a mechanism whereby life style factors can influence RS (Carlos-Reyes et al. Citation2019; Hassan et al. Citation2019; Liu et al. Citation2019).
Another species of non-coding RNAs that may have an impact on cellular and individual RS are the so-called damage-inducible long non-coding RNAs. These are generated from free DNA ends at DSB sites by RNA Polymerase II and serve as precursors of small non-coding DNA damage response RNAs (DDRNAs) (Michelini et al. Citation2017). DDRNAs have been proposed to be necessary for activation of the DDR and as such they may also influence the cellular outcome of radiation-induced DNA damage (Wei et al. Citation2012).
4.3. The response of stem cells
Stem cells (SCs) are effectively the cells that can renew tissues when differentiated cells lose the capacity to replicate. While stem cells are clearly extremely important for consideration of RSu, they are also important in considering RS due to their role in tissue renewal. There is substantial evidence that both the DDR and oxidative stress response in stem cells can differ to that of differentiated cells, especially at low doses (<100 mGy). However, the precise response appears to differ between distinct stem cells. The most commonly studied stem cell systems include the skin, intestinal crypt, hematopoietic stem cells (HSCs) and neural stem cells (Paris et al. Citation2001; Etienne et al. Citation2012; Martin et al. Citation2016; Barazzuol et al. Citation2019; Gault et al. Citation2019). Nonetheless, some general trends have emerged. SCs appear particularly sensitive to ROS, which in some SCs, for example HSCs, can confer hyper-radiosensitivity (Gault et al. Citation2019). Additionally, quiescent stem cells are frequently resistant to IR-induced apoptosis in contrast to progenitor stem cells, which at least in certain tissues can sensitively activate apoptosis. Progenitor stem cells can also undergo proliferation arrest and premature differentiation following low dose IR exposure.
4.4. Telomere attrition
Telomeres (T2AG3), the very end of chromosomes, consist of specialized nucleoprotein structures which protect chromosome ends from being recognized as DSBs, and prevent unwanted activation of the DDR (De Lange Citation2005). Telomere length (TL) varies in humans from 2 to 20 kilobases. TL in somatic proliferative tissues naturally declines with each cell replication cycle at a rate of approximately 50–200 base pairs per population doubling (varying with cell type) (Martens et al. Citation2000) due to the incomplete replication of telomere ends. Telomere maintenance is determined by genetic factors, and both causal and potentiating roles for telomere attrition in human diseases have been described (Blackburn Citation2005). One factor associated with accelerated telomere shortening is oxidative stress (Barnes et al. Citation2019). The impact of oxidative damage and stress on telomere homeostasis raises the possibility that short telomeres are markers for enhanced background oxidative stress.
Telomeres can play a role both in RS (discussed here) and RSu (discussed below). For RS, individuals with short telomeres have higher frequencies of IR-induced micronuclei, a commonly used marker of cell damage, and DSBs, than individuals with longer telomeres (Castella et al. Citation2007). TL indeed correlates well with RS in many in vivo and in vitro studies in telomerase-deficient mouse and human cells (Bouffler et al. Citation2001). These studies have established that there is an inverse correlation between RS and mean TL, implying that telomere shortening enhances RS (Genesca et al. Citation2006). However, it has also been reported that telomere elongation beyond a certain length in tumor cells (>17kb) significantly decreased clonogenic survival after IR (Fairlie and Harrington Citation2015). Consequently, targeting telomerase and telomeres has been proposed to enhance IR effects both in vitro and in vivo (Berardinelli et al. Citation2017).
4.5. Influence of the microenvironment
Tissue microenvironment or stroma represents a dynamic population of cellular and non-cellular constituents maintaining the organ homeostasis. The microenvironment encompasses extracellular matrix components (laminin, fibronectin, collagen and proteoglycans), the immune system, fibroblasts, the vascular network, cytokines, and other secreted factors. All cells are profoundly affected by communication with their microenvironment. Radiation exposure can modify the homeostasis of the microenvironment, which itself can determine the toxicity of a normal tissue, but equally, the response of the tissue can influence the microenvironment. Radiation may induce death or accelerated aging of stroma cells. As an example, acute endothelial apoptosis mediated by the rapid generation of sphingolipid ceramide via the activation of acid sphingomyelinase can enhance intestinal crypt stem cell radiosensitivity leading to intestinal collapse (Paris et al. Citation2001). Long term endothelial senescence through the dysfunction of the respiratory complex II of the mitochondria is observed during intestinal or lung fibrosis (Corre et al. Citation2013; Beach et al. Citation2017; Lafargue et al. Citation2017). Considering all the components of the microenvironment, the ubiquitous immune system has been recently highlighted for its major participation in the normal tissue response to IR exposure (Arnold et al. Citation2018), and this aspect is specifically considered below. Additionally, the inflammatory response secretome released by senescent or activated stroma cells can profoundly influence the microenvironment (Philipp et al. Citation2017), which is discussed further below. Thus, finally the actual tissue response likely arises from the cross talk between the distinct factors influencing the microenvironment. How these factors interplay to influence RS and RSu needs to be better defined.
4.6. Activation of signaling mechanisms including inflammation
Autocrine and paracrine communications between cells directly influence the cellular response to IR. Such communications can include the secretion of factors including cytokines, chemokines, proteases, lipids, DNA and miRNAs which can be present freely in the extracellular milieu or transported via extracellular vesicles such as exosomes. The best studied secreted factors are pro-inflammatory chemokines and cytokines (including TNF-α, IL-1α, IL-1β, IL-6, Il-8, GM-CSF, FAS-L, TGF-β, VEGF), which bind to receptors or membrane proteins to signal to targeted cells which may, in turn, influence their IR response. The inflammatory response can be later down-regulated due to the short half-life of the pool of pro-inflammatory cytokines and the production of anti-inflammatory cytokines, such as IL-4, IL-10 or IL-13 (Koj Citation1998). The impact of secretion is observed for most cell types (including immune cells, vascular cells and fibroblasts). The secretory factors can be either pro- or anti-inflammatory, with the balance being critical in determining a favorable or adverse tissue outcome following IR, namely tissue repair or regeneration, fibrosis and other radiotoxicities (Sun et al. Citation2013). Many different radiobiological parameters can influence the secretome profile, including dose, radiation quality (e.g. protons versus photons) and delivery as fractions (Nielsen et al. Citation2017). The secretome profile changes as a function of time post IR and can be divided into acute and chronic secretory profiles. The two secretome profiles may include different factors causing distinct properties defined by the tissue response. The acute tissue response to IR involves tissue necrosis as well as tissue regeneration, angiogenesis, and immunosuppressive responses. The acute secretory activated phenotype (ASAP) production usually peaks in the first days after IR (Hong et al. Citation1995). However, this secretory profile needs further investigation for improved definition. After high IR doses, it is known to include pro-inflammatory death cytokines such as TNF-α or FAS-L, the sphingolipid ceramide and the related metabolic enzyme, the acid sphingomyelinase, which is involved in apoptosis of endothelial cells after IR (Paris et al. Citation2001), as described above. After doxorubicin treatment, the ASAP phenotype is dependent on the activation of the p38 MAPK molecular cascade. Whether this holds for the ASAP phenotype after other stressors, including IR, requires further investigation. The late and chronic secretion appears several weeks after IR. It is observed in injured tissue through the secretion of pro-inflammatory cytokines such as TGF-β in lung fibrosis (Dadrich et al. Citation2016).
A specific secretome is the one appearing during cellular senescence, a phenotype characterized by permanent cell cycle arrest, the expression of p16 and p21, the overexpression of β galactosidase in the lysosomes and the AMPK and NF-kB-dependent secretion of a pool of pro-inflammatory factors including IL-1α/β, IL-6, IL-8, TGF-β, TNF-α, and matrix metalloproteinases (Michaud et al. Citation2013; Ferrand et al. Citation2015). The initiation and the maintenance of the senescent associated secretory phenotype (SASP) requires the persistence of DNA damage from months to years after IR (Coppe et al. Citation2008; Rodier et al. Citation2009; Strzeszewska et al. Citation2018). IR-induced pro-inflammatory cytokines have a significant effect on the immune system, which can influence tissue RS (Gougerot-Podicalo et al. Citation1996). DNA damage induced breaks may also activate the inflammatory process through the generation of cytoplasmic micronuclei. Cyclic GMP-AMP synthase (cGAS) acts as a cytosolic DNA sensor that is binding to cytosolic DNA and catalyzing the formation of the second messenger molecules, cyclic GMP-AMP (cGAMP) from ATP and GTP. cGAMP binds to the adaptor Stimulator of Interferon Genes (STING), activating a STAT3-dependent signaling pathway, leading to the transcription of pro-inflammatory type I Interferon (Bartsch et al. Citation2017; Harding et al. Citation2017; Mackenzie et al. Citation2017).
These different secretory factors represent interesting markers of acute or late tissue toxicities. However, there are challenges to be overcome. Since inflammation can be encompassed within the umbrella of a trauma response, the timing and the exact profile of the pool of pro-inflammatory factors must be well defined in terms of the IR-induced pathogenesis. Finally, polymorphisms in cytokine signaling may contribute to the differences in the individual response and could contribute to RS (Venkatesh et al. Citation2014).
4.7. The immune response
The immune system can influence RS in multiple ways. Cell and tissue damage induce a so-called sterile inflammatory reaction to eliminate debris and restore cellular homeostasis and tissue function (Chen and Nunez Citation2010). As already mentioned, a wide range of pro-and anti-inflammatory factors are produced after radiation exposure, via for example DNA damage and the presence in the cytosol of micronuclei or dsDNA leaking from damaged mitochondria which activate the cGAS-STING pathway (Bartsch et al. Citation2017; Harding et al. Citation2017; Mackenzie et al. Citation2017) or through senescence induction (Michaud et al. Citation2013; Ferrand et al. Citation2015). In turn, these factors direct the recruitment and activation of immune cells to the site of damage, and therefore modulate and shape the development of these inflammatory reactions. The fate of irradiated cells results from the balance between IR-induced pro-death and pro-survival signaling pathways (Roos et al. Citation2016). The activation of Akt and NF-κB signaling pathways by cytokine/cytokine receptor interactions can alter this balance and promote the survival of damaged cells. Thus, the production of inflammatory factors influences the outcome of IR exposure at several levels and profoundly changes the cellular environment, which also has a major impact on irradiated cell fate, as reviewed in the section above. Impairment of the immune system composition and/or functions will therefore, most likely, result in changes in the response to IR.
Immune functions are, for example, known to decline with time, so that the immune system becomes less efficient in the elderly. This “immunosenescence” results from changes in both the homeostasis and function of innate and adaptive cells and results, among others, in a state of inflammation termed “inflammaging”, with high levels of circulating inflammatory mediators such as IL-6 and TNF-α (Franceschi and Bonafe Citation2003; Olivieri et al. Citation2003; Alvarez-Rodriguez et al. Citation2012; Michaud et al. Citation2013). This preexisting inflammatory milieu can alter the mobilization and function of immune cells in response to radiation exposure. Thus, the age of the immune system at exposure might be a factor influencing the response to IR.
In addition to its direct effects on immune cell homeostasis and function, IR exposure also affects the protective functions of the immune system by accelerating aging and immunosenescence (Richardson Citation2009; Candeias et al. Citation2017). Therefore, IR induces the development of immune responses aimed at counteracting the tissue effects of exposure, but at the same time affects the functionality and efficiency of the immune system. The overall impact on IR-induced health effects, and especially individual RS is currently unknown. RT represents a particular situation characterized by repeated exposure of a defined tissue volume to kill tumor cells or to modify an inflamed microenvironment (Frey et al. Citation2017). However, blood-borne immune cells circulating though the tumor bed are exposed at the same time. In addition, tumor cells shape their micro-environment to favor their growth. They recruit regulatory T cells and myeloid-derived suppressor cells, a category of specialized immune cells that secrete anti-inflammatory, pro-tumor cytokines and prevent the activity of specific anti-tumor T lymphocytes (Candeias and Gaipl Citation2016; Wennerberg et al. Citation2017). Recent work unraveled the very high degree of inter-individual variability in the composition of circulating immune cells (Brodin and Davis Citation2017), but the activation status of these cells may also be a key parameter of the individual response to IR. How these changes are dependent on age at exposure and on the initial inflammatory/activation status of immune cells, and how they subside over time is unknown.
Thus, a key feature of IR effects on the immune system is that localized reactions expand into regional or systemic responses, where complex intercellular communication pathways play a major role. Alterations in the different intercellular communication pathways both within the irradiated microenvironment and at a systemic level can lead to abnormal manifestations of the response. These cell-to-cell communication pathways, mainly via inflammatory factors and extracellular vesicles, participate in the orchestration of the immune system response to IR. Since these are dynamic processes, the outcome is greatly influenced by environmental factors. These can be signals originating from the irradiated microenvironment or signals initiated at a systemic level by exogenous factors (lifestyle, diet, other co-morbidities, etc). These factors can shape the response of a tissue to IR even in the absence of a genetic defect. The importance of extracellular vesicles relies on the fact that they are membrane-bound structures with a complex cargo composed of different nucleic acids (RNAs, small non-coding RNAs), proteins, lipids, lipoproteins, small metabolites through which they are able to transmit a multitude of signals concomitantly and thus are able to induce different biological processes in the recipient cells (Szatmari et al. Citation2019; Zonneveld et al. Citation2019). It has been shown that IR can impact both the quantity and the internal composition of extracellular vesicles (Al-Mayah et al. Citation2012; Xu et al. Citation2015). Several groups have shown that IR (both low and high doses) altered the miRNA composition of extracellular vesicles involved in key cellular pathways related to radiation response and carcinogenesis. Thus, signals delivered to neighboring or distant cells by extracellular vesicles profoundly affect radiation response of recipient cells. For example, delivery of bone marrow-derived extracellular vesicles originating from irradiated mice to non-irradiated naïve animals induced activation of DDR pathways (increased levels of γ-H2AX foci) in the splenocytes of naïve mice (Szatmari et al. Citation2017). A major characteristic of signals transmitted by extracellular vesicles relies in the fact that they transmit information “packages”, which allows much more complex information transfer compared to “traditional” single information units represented by cytokines or chemokines, although chemokines and cytokines rarely act alone. These information packages reflect the complex radiation response of the extracellular vesicles-releasing cells. Their role in mediating radiation effects and response has been recognized relatively recently and it will be important to determine the mechanism by which extracellular vesicles can modulate radiosensitivity and radiosusceptibility.
4.8. Potential influence of life-style factors on cellular pathways
The impact of life-style factors such as smoking, alcohol consumption, diet, degree of physical activity, physical injury, hygiene and medication as factors modulating the degree of RS has received increasing interest. While clinicians are well aware of the response-modifying effects of at least some of these factors, systematic overviews are still scarce. Some of these factors may, in fact, impact upon the response via a cellular mechanism, as considered above for how life-style may influence the epigenetic status of a cell. However, we have included these factors in this additional category, since the impact may be highly complex and multifactorial. Within the scope of this review, we concentrate on diet and smoking, since these lifestyle factors have been investigated more deeply than the others.
Due to the pivotal role of reactive species in the radiation response, both for immediate damage induction and for later-occurring effects including inflammatory processes, the influence of spontaneous oxidative stress levels and the status of detoxifying systems has been investigated in detail. Oxidative stress markers exhibit wide inter-individual variability and can be affected by nutritional factors (Alajbeg et al. Citation2017; Bjorklund and Chirumbolo Citation2017; Saha et al. Citation2017). Since chronic oxidative stress is a major issue in normal tissue injury after radiotherapy, in addition to radical scavenging medication, a diet rich in anti-oxidants may help to reduce or delay side effects caused by oxidative stress (Citrin and Mitchell Citation2017). A recent systematic review on the influence of various natural and derivative compounds and diets in vitro and in vivo on radiation effects concluded that antioxidants such as ascorbic acid and N-acetyl-cysteine, but also polyphenols, lactoferrin and others can act as radioprotectors and reduce induction of DNA damage (Smith et al. Citation2017). Overall, in the studies analyzed little attention was given to the second wave of (cellular) ROS production, although some of the studies involved prolonged administration of the radioprotective compound. The authors concluded that diet modification alone could provide radioprotective effects given that the radioprotective agents are often phytonutrients frequent in a plant-based diet (Smith et al. Citation2017). Amongst these phytonutrients, polyphenols have attracted interest as potentially mitigating factors in acute radiation syndrome and radiation-induced bone loss (Schreurs et al. Citation2016; Singh et al. Citation2017). Diet may also affect pathways involved in late, inflammation-associated effects. For example, lower levels of pro-inflammatory cytokines were detected in head and neck cancer patients consuming whole foods and diet rich in vegetables and fruits (Arthur et al. Citation2014). Concerning gastrointestinal radiation toxicity, the preventive activity of plant products appears to rely on their fiber content, in addition to antioxidant and anti-inflammatory activities (Wedlake et al. Citation2017; Pathak et al. Citation2019). Since diet, and especially fiber composition, affect the intestinal microbiome, it is conceivable that the microbiome participates in the pathogenesis of radiation-induced bowel injury (Kumagai et al. Citation2018). There is also evidence that caloric restriction or a ketogenic diet can reduce radiation-induced side effects, possibly by altering metabolic and inflammatory pathways (O'Flanagan et al. Citation2017), although the tolerance and acceptance of these interventions may be limited (Zahra et al. Citation2017). However, nutritional deficiencies were found associated with higher risk of hemorrhagic cystitis after pelvic RT (Platzer et al. Citation2018) and of adverse events in head and neck cancer patients treated with RT (Kono et al. Citation2017).
Another life-style factor frequently implicated in affecting adverse effects of RT is smoking habit. By causing additional DNA damage and oxidative stress, smoking may affect the ability of cells to handle radiation damage. A small study investigating induction, processing and fixation of DNA damage after ex vivo irradiation of lymphocytes in pairs of monozygotic twins discordant for their smoking habit did not find that smoking habits influence cellular radiosensitivity (Marcon et al. Citation2013). In contrast, in a recent report of the US Surgeon General about 90% of all studies including results for the association between cigarette smoking and increased toxicity of cancer treatment showed a positive association (National Center for Chronic Disease Prevention Citation2014). An exception to this may be radiation-induced lung injury including fibrosis, for which smokers may carry a lower risk (Kong and Wang Citation2015; Menoux et al. Citation2018).
4.9. Other personal factors
While sex and age at exposure are routinely considered as risk modifying factors in epidemiological studies on radiation-associated carcinogenesis (i.e. RSu), their role in RS, while certainly acknowledged by clinicians, has received less vigorous assessment. Increased age has consistently been associated with higher RS including endpoints such as cardiac toxicity and endocrine deficiency (AGIR Citation2013; Bouffler Citation2016; Marliere et al. Citation2016; Rose et al. Citation2016). However, the contribution of age to radiation-induced lung toxicity is less clear (Kong and Wang Citation2015). For several, but not all, organs increased risk of physiological abnormalities is seen following exposure of children (e.g. brain) compared to adults. In addition, children can develop radiation-induced impairment of growth and maturation (UNSCEAR Citation2013). In general, sex is not regarded as a major factor in RS and if differences are observed, they may by caused by lifestyle factor variability (AGIR Citation2013; Kong and Wang Citation2015; Bouffler Citation2016). However, in animal models differential biological responses of both sexes have been observed, such as differential gene expression patterns and epigenetic alterations (Narendran et al. Citation2019).
4.10. Diseases potentially conferring RS
In the discussion above, we have highlighted some genetic syndromes that are associated with RS, such as ataxia telangiectasia and LIG4 Syndrome, due to mutations in known DDR components. Such proteins also function during the development of the immune response and during neuronal development. Thus, in some instances, immunodeficiency or microcephaly can be associated with radiosensitivity. Many disease states can cause activation of inflammatory responses or metabolic changes, which, as discussed above, can also impact on RS. Diabetes is an important example, with chronic diabetes being associated with microvascular occlusion, atherosclerosis and tissue hypoxia. Multiple groups have observed a hyper-response of diabetic patients following RT (AGIR Citation2013). One of the more recent studies found that diabetes could be a contributing factor for RS when Standardized Total Average Toxicity scores were assessed for late radiotoxicity in breast cancer patients (Barnett et al. Citation2012). It is unclear, however, if such a correlation could be due to the predisposition to breast cancer and diabetes (especially type 2 diabetes) associated with deficiency in DDR proteins, such as ATM or whether any RS is a direct consequence of diabetes via, for example, the activation of inflammatory responses (Ditch and Paull Citation2012). ATM affects insulin signaling, glucose metabolism and the response to oxidative stress (Ditch and Paull Citation2012). Thus, it is possible that changes, genetic or through a disease state, that affect these pathways, feedback and impact upon RS. A striking more recent report revealed that diabetic individuals with prostate cancer had lower survival rates and worse adverse effects from RT compared with non-diabetic patients, independently of whether they took insulin or other anti-diabetic medicine (Zaorsky et al. Citation2017). Collectively, these findings suggest that diabetes does influence RS, although the underlying mechanism is unclear.
Collagen vascular disease (CVD), which encompasses rheumatoid arthritis, systemic lupus erythematosus, polymyositis and systemic schloerosis, represents another disease state characterized by immune dysregulation and inflammation. For these diseases, there also appears to be heightened late toxicity reactions following RT (AGIR Citation2013).
5. Non-physiological factors that can modulate RS
The aspects above relate to individual differences in patient responses. However, consideration should additionally be given to non-patient derived factors that can influence the apparent response of individuals.
5.1. Definition and delineation of RS
A system for grading adverse effects of any treatment has been defined and is used in the assessment of the response to RT (see https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. for details). However, the precise delineation of grades lacks rigor and can differ between hospitals, professionals as well as countries. Grade 2 responses are considered to be on the edge of the normal response in some countries but a well-defined over-response in others. One important compounding aspect is the time at which responses arise; the response can be observed at early times after RT (categorised as early effects) but can arise multiple years later (late effects). Although specific tissues can be classified as early or late responding, a feature determined predominantly by their rate of proliferation, some tissues such as skin can be both early and late responding. It is currently unclear whether the early or late responses really represent distinct classes of responses or a spectrum, not least because there is no rigorously defined time scale shut off for these two types of responses. Another important consideration is the impact of tumor type and site on the timing of the response. Frequently, patients are classified as showing a grade 1–5 early or late response without any consideration of the tumor site or tissue. Thus, the specificity of IR responses of different organs should also be considered. To attempt to evaluate the underlying cause for RS, these factors need to be defined and monitored. Currently, it is unclear whether early or late responses are caused by similar or distinct genetic or non-genetic factors. Finally, the diversity of new RT modalities and the manner of dose delivery have contributed to the complexity of defining early/late reactions, notably by accelerating or decelerating tissue reactions and the responses need to be evaluated within this context.
5.2. Treatment planning and dosimetry
Significant differences exist between hospitals and RT professionals in contouring the primary tumor volume PTV and estimation of organs at risk. Such differences could potentially modulate the response to RT for certain patients. Another consideration is variation in dosimetry estimations and distinctions between treatment planning systems.
5.3. Radiation quality
Although X-rays (low linear energy transfer (LET) are used for most RT, increasingly proton and carbon ion therapy (high LET) are being used. Thus, it is important to evaluate the influence of the radiation quality (Relative Biological Effectiveness) when considering individual RS. The local distribution of radiation insults and damage differs with low compared to high LET irradiation. Sparsely ionizing, low LET radiations such as photons induce multiple DNA lesions that include a high fraction of single-strand breaks and base damage, and a lower level of DSBs. Densely ionizing, high LET radiations (such as heavy ions and alpha rays) induce more localized and complex lesions, giving rise to complex clustered lesions and complex exchange aberrations (Sabatier et al. Citation1992; Mladenov et al. Citation2018). Such lesions are more difficult to repair accurately (Stewart et al. Citation2011; Sage and Shikazono Citation2017). Hence, the impact on cells, tissues and organs, including cell death, genomic instability and cancer induction, differs between low and high LET radiation and is in general considered to be more severe for high LET radiation (Sabatier et al. Citation1987). Interestingly, there is both in vitro and in vivo data suggesting that tumors exposed to charged particles are less invasive than tumors exposed to low LET radiation, a notion supported by recent “omics” analysis suggesting that the signaling pathways associated with angiogenesis, vasculogenesis, migration and invasion are downregulated when exposed to charged particles (Story and Durante Citation2018). The mitochondrial respiratory chain appears to be significantly upregulated after heavy ion exposure (Fan et al. Citation2019). A further important advantage of high LET radiation such as carbon ions, is that most of the energy is delivered with defined localization, termed the Bragg Peak, resulting in better sparing of normal tissue which is relevant when considering the response of the normal tissue during RT. Additionally, the assay monitoring nucleo-shuttling of ATM (described above) predicted the radiation response in human quiescent cells irrespective of LET, and provided evidence that high-LET particles provided protection against the cytoplasmic sequestering of ATM (Maalouf et al. Citation2019). Collectively these findings reveal that the quality of the radiation needs to be considered when assessing RS.
5.4. The shape of the dose-response curve for tissue effects and the influence of chance
Classically, the phenotype of an organism is defined as the interaction of the genotype and the environment. However, a third contributing component is chance, that is the outcome of stochastic intracellular processes influencing the expression of genes (Burga and Lehner Citation2012). When dealing with radiation effects, a fourth component is the stochastic nature of radiation damage, leading to an unpredicted fate of an irradiated cell. Above, we have considered the impact of genetics and the environment. The influence of genetics on individual RS is clear, as demonstrated by the high RS of people with genetic diseases such as ataxia telangectasia or Nijmegen breakage syndrome (AGIR Citation2013). The impact of the environment is equally demonstrated by the observations that surgery increases the radiotoxicity of RT (AGIR Citation2013) and that mouth hygiene modulates the risk of mucositis in patients treated by RT for head and neck cancers (Sroussi et al. Citation2017). The impact of chance is difficult to prove. However, it is reasonable to assume that the sigmoid dose-response curves for tumor control after IR arise due to the stochastic nature of IR-induced cell killing of tumor cells (Munro and Gilbert Citation1961). The dose response curves for normal tissue complications are also sigmoidal, and it is likely that severe normal tissue complications are a stochastic accumulation of radiation-induced DNA lesions leading to cell death. That is, some patients will lie at the extreme edge of the normal distribution simply by chance. This explanation should be seriously considered given the inability to find a validated genetic fingerprint of RS in RT treated patients having normal tissue complications (AGIR Citation2013). Indeed, as mentioned above, the very extreme RS patients (groups 4–5) are those with defined mutations in the known DDR genes, but a proportion of the less severely over-responding patients (groups 2–3) could, thus, represent those that arise simply by chance. The important question is what is the level of this chance contribution versus the impact of genetic or life style factors. Whilst it is imperative that an assessment of the underlying causes of RS is undertaken, the possible existence of the “chance” component should not be neglected.
6. A consideration of radiosusceptibility (RSu); cellular pathways conferring RSu
As discussed above, RSu represents the sensitivity to IR-induced stochastic endpoints, of which cancer is the most significant. Although IR is not a strong carcinogen, given our increased exposure to low doses, it is vital to assess cancer risks from background and diagnostic radiation exposure, and to identify whether there are RSu individuals. This is more challenging than gaining insight into RS, partly because, although there is evidence emerging for a signature for IR-induced cancers, currently such cancers cannot be distinguished from those arising from a distinct origin (Behjati et al. Citation2016; Wilke et al. Citation2018). They may also represent a small subset of cancers compared to those arising via other exposures/causes. It is also possible that RSu individuals will be susceptible to carcinogenesis from other exposures. Indeed, patients with increased cancer predisposition due to genetic alterations have been described (e.g. Li Fraumeni, Retinoblastoma patients and BRCA1/2 heterozygous carriers) and some may display enhanced RSu. Whether IR exposure contributes to the cancer susceptibility of such patients remains unclear albeit with some evidence supporting this possibility (Berrington de Gonzalez et al. Citation2009; Obdeijn et al. Citation2016; Bernstein et al. Citation2017). There may also be individuals without marked overall cancer susceptibility but with RSu. Such limitations, however, should not undermine the importance of gaining insight into RSu.
Multiple lines of evidence have demonstrated that low level IR exposure can enhance cancer levels, raising the possibility that there could be RSu individuals. In addition to the well reported data derived from the A-bomb survivors, epidemiological studies have provided strong evidence for an increased risk of tumors correlating with exposure to diagnostic X-rays. A-T and BRCA1/2 carriers have increased cancer risk, and their encoded proteins function in the response to IR-induced damage, raising the possibility that they display RSu, with indeed some supporting evidence (Berrington de Gonzalez et al. Citation2009; Obdeijn et al. Citation2016).
All the pathways and factors influencing RS are likely to also affect RSu. Below we include a discussion of mechanisms or aspects that may be of specific relevance for considering RSu compared to RS.
6.1. The fidelity of DSB repair
RS strongly correlates with unrepaired DSBs (which may be induced directly via IR exposure or arise as a consequence of misrepair). Such DSBs can trigger responses such as senescence, apoptosis, permanent checkpoint arrest or mitotic catastrophe. In contrast, a cancer cell must be viable to be able to develop into a tumor. For IR-induced carcinogenesis, it is therefore likely that DNA misrepair is an underlying causal event. For a DSB, misrepair can involve small deletions at the DSB site or translocation events, which effectively represent the rejoining of the incorrect DNA ends. Translocations often cause interstitial deletions (which may cause cell death) but balanced or reciprocal translocations compatible with cell survival can also arise. Significantly, a recent study searching for an IR-induced DNA signature in cancers that were potentially IR-induced found evidence for elevated small deletions and balanced translocations (Behjati et al. Citation2016). In considering factors conferring RSu, therefore, we can consider mechanisms that promote inaccurate DSB repair. Although loss of NHEJ proteins causes a reduced level of DSB repair, indirect evidence suggests that there can be increased misrepair leading to increased aberrations even though unrepaired DSBs and lethality increases (Kemp and Jeggo Citation1986). The ATM signal transduction pathway has a major role in ensuring both the accuracy of DSB repair as well as the activation of pathways (such as checkpoint arrest) that remove damaged cells (Jeggo and Lavin Citation2009; Clouaire et al. Citation2017). Indeed, cells lacking ATM have substantially enhanced IR-induced translocations (Cornforth and Bedford Citation1987). There is also evidence for a role of BRCA1 and homologous recombination (HR) in the repair of transcriptionally active DSBs, which may be prone to translocation formation if misrepaired (Marnef et al. Citation2017; Yasuhara et al. Citation2018). Additionally, HR plays a major role in regulating the appropriate response to replication fork stalling or collapse, and diminished HR function can promote carcinogenesis due to translocations arising following replication. Importantly, in contrast to RS, there is strong evidence that heterozygosity for HR genes confers an elevated risk of carcinogenesis (e.g. as observed in cancer predisposed BRCA1/2 carriers). It is possible that such heterozygous individuals display elevated RSu although it is unclear whether IR exposure provides a significant contribution to the enhanced tumor levels in such patients.
6.2. Diminished cell death or checkpoint arrest pathways
Apoptosis is a significant cell death pathway, which is down-regulated in many cancers. Patients, such as Li Fraumeni Syndrome patients, with heterozygous mutations in genes controlling such cell death pathways display elevated cancer predisposition. As for heterozygosity for HR genes, it is likely that such patients would display elevated RSu, but the relative contribution of radiation exposure to their cancer predisposition is unclear. Indeed, reduced expression of any tumor suppressor gene could enhance IR-induced carcinogenesis, although the specific contribution of IR exposure remains unclear. Importantly, apoptosis is subject to intricate regulatory mechanisms. Thus, many of the factors discussed above for RS, could also influence RSu. As one example, many factors involved in apoptosis can be regulated by miRNA-dependent post-transcriptional changes with the degree of changes being highly complex (Czochor and Glazer Citation2014; Heider et al. Citation2017).
Cell cycle checkpoint arrest can also represent a cell death pathway if the arrest is permanent, and thus represents an important pathway to remove damaged cells, which have the potential to initiate carcinogenesis. Additionally, by restricting the traversal of damaged cells through replication and mitosis, the activation of checkpoint arrest has the potential to enhance the accuracy of DSB repair. Thus, efficient activation of cell cycle checkpoint arrest is likely to be significant in maintaining genomic stability in the presence of DNA damage and thus in restricting Rsu. G1/S arrest is p53-dependent and most likely contributes to the tumor suppressor function of p53 (Wahl et al. Citation1997). Failure to activate G2/M arrest and chromosomal radiosensitivity has also been reported to be elevated in breast cancer patients, as well as colorectal, head and neck and some childhood cancers. However, whether this confers RSu specifically or broader cancer predisposition remains unconfirmed (Scott Citation2004).
6.3. Telomere attrition
Above, we have considered the role of telomere length in determining RS. Here, we consider its impact on RSu. For this endpoint, it is important to appreciate that carcinogenesis is a complex, multi-step process. Telomere-driven instability can aid the acquisition of certain hallmarks of cancer, as it generates mutations, chromosomal rearrangements, and other types of genetic alterations which, upon accumulation in oncogenes or tumor suppressor genes, can fuel tumor progression (Beerenwinkel et al. Citation2007; Hanahan and Weinberg Citation2011). Exposure to IR induces multiple recessive mutations via long-term IR-induced genomic instability that remain silent until unmasked when they can potentially accelerate the process of carcinogenesis (Morgan Citation2003). End-to-end dicentric chromosomes cause cell cycle arrest in normal cells with intact cell cycle checkpoints. However, cells lacking normal checkpoints continue to divide, and unstable dicentric chromosomes can break during cell division, leading to the propagation of chromosomal instability via breakage-fusion-bridge (B/F/B) cycles (Murnane and Sabatier Citation2004). Loss of whole chromosomes or partial chromosomal arms may arise due to chromosomal instability caused by telomere loss, leading to massive loss of heterozygosity (LOH), a phenomenon that has been coined “TELOLOH” (Pommier et al. Citation1995). Thus, in a single step via telomere-induced chromosomal imbalances, recessive mutations of hundreds of genes that have accumulated in the genome can become unmasked. These mutant cells may gain a proliferative advantage, leading to cell immortalization and eventually, carcinogenesis (Ducray et al. Citation1999; Ayouaz et al. Citation2008).
There are inter-individual and intra-individual differences in TL; that is, TL varies among healthy individuals (Gilson and Londono-Vallejo Citation2007), and it also varies for each chromosome arm in a cell. Indeed, the particular shortest telomeres can vary between individuals (Pommier and Sabatier Citation2002). Moreover, this inherent cellular heterogeneity of TL is conserved throughout the lifespan of the individual (Graakjaer et al. Citation2004). Therefore, the inherent TL and TL heterogeneity both per individual and within the individual could contribute to individual RSu (as well as RS) resulting from cells with chromosomal instability gaining a proliferative advantage (Shim et al. Citation2014).
6.4. Immune response
As stated for RS, the immune response can have multiple impacts on RSu. The changes in transformed cells can trigger immune responses aimed at their elimination, which effectively can function as a significant cell death pathway (Dunn et al. Citation2004). However, tumor cells use several strategies to escape immune surveillance, from the recruitment of immunosuppressive myeloid derived suppressor cells to the tumor bed (Candeias and Gaipl Citation2016) to the expression of immune checkpoint proteins such as the Programmed cell Death Ligand (PD-L1) which inhibits the activity of tumor-specific cytotoxic T lymphocytes by engaging their inhibitory Programmed Death (PD)-1 receptor (Bardhan et al. Citation2016). There is increasing evidence that DNA damage and IR exposure, in particular, can further activate PDL-1 expression on tumor cells, effectively diminishing the capacity of the immune response to remove them (Sato et al. Citation2017; Sato et al. Citation2019). The significance of these effects are being increasingly recognized and inhibitors of immune checkpoint responses, such as PD-L1 inhibitors, have the potential to be used in conjunction with RT (Boustani et al. Citation2019; Portella and Scala Citation2019). However, PD-L1 up-regulation after genotoxic stress seems to be limited to “established” cancer cells (Hagiwara et al. Citation2018), and the importance of this DNA-damage induced mechanism in RSu and the “de novo” development of IR-induced cancer is, therefore, currently unknown. Notwithstanding, the expression of PD-L1 can be induced in normal cells by stimulation with cytokines such as IFNγ (Hagiwara et al. Citation2018), suggesting that an IR-induced inflammatory environment might be able to promote PD-L1-mediated escape of neo-transformed cells from immunosurveillance. A chronic low-grade inflammation is, indeed, thought to be one of the key factors in ageing and in the development of age-related chronic diseases, including cancers (Fougere et al. Citation2017).
6.5. Responses of stem cells
Stem and primitive progenitor cells are considered to be the cell of origin for most cancers, partly because they are sufficiently long-lived cells that can accumulate the required mutational changes for carcinogenesis. For considering RSu, exposure to low dose IR may be particularly important, given that survival of the cell is required. IR exposure can particularly enhance the risk of leukemia, making the response of HSCs of importance. Significantly, differences in the response of hematopoietic stem and progenitor cells to low dose radiation compared to more differentiated cells have been reported and could contribute to leukemia risk following IR exposure (Gault et al. Citation2019). Several studies have focused on the specific response of stem cells to low-dose IR (Squillaro et al. Citation2018). An important response in considering RSu is the sensitivity of many primitive progenitor cells to cell death via apoptosis, although this is not observed in all stem cell compartments. (Revenco et al. Citation2017; Barazzuol et al. Citation2019). Interestingly, bulge epidermal stem cells do not commit to death via apoptosis after low dose IR, due to a hypoxia-inducible Factor 1a response, and low dose IR conferred elevated carcinogenesis specifically in the epidermis in mice lacking the tumor suppressor Ptch1, supporting the notion that the apoptotic response in stem cells may be important in protecting against carcinogenesis. A further interesting concept is the ability of specific mutational changes, such as mutations in p53, that arise after low dose IR to confer a growth advantage within a stem cell compartment in a manner related to oxidative stress (Fernandez-Antoran et al. Citation2019).
6.6. Influence of personal and lifestyle factors
Epidemiological assessments of cancer risks after radiation exposure use risk models that, in addition to dose, dose rate and radiation quality, also include a number of intrinsic factors such as effect modifiers, among which age-at-exposure and sex have major importance (Wakeford Citation2012). Since modification of radiation risks by these factors is well-characterized, they are explicitly considered in risk calculations for NASA astronauts, which can be viewed as a step toward personalized risk assessing (Locke and Weil Citation2016). Analysis of the Life-Span Study (LSS) of atomic bomb survivors and various other studies have indicated that, for many cancer types, younger age at the time of exposure confers a greater cancer risk from a given radiation dose than higher age. However, the mechanistic basis of this difference has not entirely been clarified (UNSCEAR Citation2013; Ozasa Citation2016).
Concerning the influence of sex, excess relative rates (ERR) per dose for many cancer types are higher for females than males (Grant et al. Citation2017), which, at least in part, reflects lower baseline levels in females for many tumor types (Ozasa Citation2016); consideration of excess absolute rates (EAR) tends to reduce the observed sex difference. In transferring risk estimates from one population to another, relative risk coefficients are considered important, since it is assumed that radiation may interact mechanistically with other risk factors in a way that both absolute risks are supra-additive, i.e. the excess risk from both factors is greater than the sum of the risk of either factor alone (Wakeford Citation2012). Thus, one could assume that a variety of cancer risk factors (smoking, BMI, UV exposure etc.) affect the radiation-associated risk by increasing the mutational load and modulating other carcinogenic processes. However, smoking, which is the best-investigated of these potential factors, has been shown to interact with radiation in a complex manner with different effects for low-moderate vs. heavy smokers on lung cancer risk after radiation exposure (Cahoon et al. Citation2017).
7. State of the art: Summary of what we know
7.1. Are RS and RSu genetically determined
Worldwide, a subset of patients (usually around 10–15%) receiving RT display pronounced reactions assessed via a grading system of post-IR responses (). Such patients have been termed RS. Although factors associated with the delivery of RT and the precise response assessment differs between hospitals and countries, the relative uniformity in identifying such individuals is striking. Only a minor fraction (<10% of RS patients) can be attributed to mutations in known DDR genes (). Such patients, however, tend to be those with dramatic RS (grade 4–5) reactions and, despite being a minor subset, are important to identify. For these patients, RS can be attributed to genetic factors. For the large majority of less marked RS individuals (those with grade 2–4 reactions), it is currently unclear if the cause is predominantly genetic or a consequence of non-genetic factors. Significantly, several candidate gene studies and genome wide association studies (GWAS) have suggested evidence for genetic signatures of RS. However, the number of investigated patients was usually quite small resulting in insufficient statistical power (Andreassen, Schack, et al. Citation2016). Thus, a more profound exploration of the emerging field of radiogenomics is important to take advantage of the full potential of genomics technologies and their application to personalized RT medicine.
Several lines of evidence raise the possibility that RS is intrinsic to the patient and linked to defined cellular responses. Firstly, as discussed above, functional tests appear able, at least to some degree, to predict or correlate with RS (). Additional predictive assays have also been described (Vandevoorde et al. Citation2016). These tests utilize defined cell types (e.g. lymphocytes or fibroblasts), which may not be equivalent to the precise tissue showing RS, suggesting a fundamental sensitivity to IR. It is important to note, however, that these findings do not necessitate a genetic cause; e.g. an activated stress response could be a consequence of non-genetic or genetic factors. Additionally, it is unclear if these functional tests have a common underlying basis and identify the same patients or whether they identify patients with distinct mechanistic explanations for their RS. Establishing the validity of these assays and the fraction of RS patients they can detect will be helpful in assessing the contribution of life-style factors and whether the basis for RS is genetically based.
Finally, biomarkers which correlate with RS have been described (Pernot et al. Citation2012; Hall et al. Citation2017). Although such biomarkers may be a consequence of RS rather than defining the underlying basis, their existence provides evidence that RS can be intrinsic to the patient. Importantly, as discussed above, classification of RS remains poorly defined and the link with life style factors has not been rigorously examined. Thus, for the majority of patients who suffer painful and occasionally fateful responses to RT, there is no insight into the underlying cause and whether there is a genetic etiology or whether there are causal non-genetic factors.
7.2. Combinatorial impacts underlying RS
Our discussion here provides clear evidence that for some patients there is a genetic basis underlying their RS, and potentially RSu. Such patients have marked RS, however. We have also highlighted how multiple additional factors can influence the DDR and oxidative stress pathways. They may also lead to more endogenous DNA damage that can be compounded by IR exposure. Additionally, however, factors such as the degree of inflammation or the status of the immune response, can exert impacts on the response, either via an influence on the DDR and oxidative stress pathways or via distinct mechanisms. Thus, as discussed above, there are multiple factors that act in a combinatorial manner to determine the individual response to IR exposure, both underlying RS and RSu. The fact that most GWAS studies have not identified major causal genetic defects, could, arguably, be evidence for such an explanation. Thus, the future challenge may well be to gain insight into this complex interplay of factors underlying RS/RSu and a consideration of the potential factors and mechanism as considered here will be key to this.
8. Questions that need to be addressed
In the discussion below, we will predominantly focus on the majority of RS patients who do not have mutational changes in known DDR genes (but will make it clear when we specifically discuss this small subset of patients). The important questions to be addressed include:
What are the pathways causing RS following RT?
Is there a single major pathway or can multiple factors contribute?
Is the underlying cause genetically determined or due to other host factors?
If, as is likely, there are multiple factors, what is their interplay?
Can we identify RS individuals with functional tests or via the use of specific biomarkers?
Do the described functional tests identify the same or distinct RS patients?
Do they identify patients with tumors of specific tissues or are they broader?
Do they identify patients displaying early and late enhanced responses?
What is the mechanistic basis underlying these functional tests?
What is the role of the immune system in RS/RSu?
Finally, to address the questions above, we need to define and delineate the description of RS individuals?
9. Recommendations
As evident above, RT is an important situation where RS exerts a profound impact. Given the significant number of patients who receive RT and the fact that treatment plans are designed such that 10–15% of RT patients display some level of RS, the strongest pressing need is to establish the underlying cause. To achieve this, patient cohorts and interactions, between and within scientists and clinicians, need to be established. Our recommendations discussed below focus particularly on RS following RT. RSu, i.e. sensitivity to carcinogenesis following IR exposure, represents a more difficult analysis since the identification of patient cohorts is challenging. Nonetheless, we also make some recommendations to help pursue analysis of RSu individuals. In assembling these proposed recommendations, we consider responses and analysis that can be conveniently undertaken using patient derived material.
9.1. A newly established prospective study to assess RS following RT
We recommend that a prospective study is undertaken to allow a coordinated analysis of the multiple endpoints (discussed above) that might impact upon RS after RT on a defined cohort of cancer patients receiving RT. This analysis should include all RT patients irrespective of their response to provide a suitable control cohort. The cohort should be subdivided for specific tumor sites, however, since this is a critical variable. Parameters important to include for analysis are:
Good quality data that includes the time-frame of RS (early or late response) and assessment of grading, using a rigorously defined system to enable comparison between hospitals and radiation oncologists. Details should include the site and hypoxic environment of the tumor.
Follow up information should be obtained at 3, 6 months and then annually post RT for at least 10 years.
The functional tests discussed above (plus any other tests that appear reproducible and suitable following review) should be undertaken to allow the coordinated analysis on the same patients. This will involve establishing fibroblast cultures from patients as well as blood, urine and/or plasma samples.
Detailed analysis of the inflammatory response and other paracrine factors.
Detailed mechanistic analysis of the dynamics of the immune response before and after RT, taking into account the preexisting status. This should include inflammatory responses, cell-cell communication (including the role of extracellular vesicles), and adaptive and innate immune responses.
Mitochondrial function, including an assessment of oxygen and glucose metabolism.
The age of the patient and any signs of premature ageing should be recorded.
Extensive details of life style factors should be recorded, including smoking, alcohol consumption, diet, exercise, hygiene and relevant environmental exposure. Questionnaire-derived data could be complemented by metabolite analysis using blood or urine samples.
It would be beneficial to include a high throughput analysis of epigenetic markers, including DNA methylation status, histone modifications by ChIP-Seq analysis
The goal is to evaluate whether there are multiple single factors and/or the interplay between factors influences the response to RT. To achieve this goal, integrative analysis of the multi-modal data will be required. We also note that this type of approach requires close interface between clinicians, research scientists and data analysts, and we place a priority on achieving this.
9.2. Use of existing cohorts
We recognize that establishing a new cohort involving all RT patients will take time, be costly and require rigid co-ordination. We, therefore, consider that existing patient cohorts can also be exploited to address some of the questions delineated above. Data and material established from the REQUITE program should be exploited where possible.
9.3. Functional test consolidation
As a priority, we believe that the functional tests discussed above should be consolidated by independent analysis and, preferably on the same group of patients to establish if they identify same or distinct patients.
9.4. Analysis of a genetic contribution
We recognize that the recommendations above will not establish whether there is a genetic contribution to RS after RT. Once a predictive functional test (or tests) becomes validated and/or mechanistic insight is gained, it will be essential to consider this important question. To achieve this, advancing sequencing technology could be exploited to help identify polymorphisms or mutations that may link to RS, i.e. whether there is a genetic component and the candidate genes. We have not included such sequencing analysis in the list above due to cost constraints but this will become a priority once functional tests or mechanistic insight are gained.
9.5. Establishment of tissue specific tests for analysis of RS individuals
We note that currently functional tests use cell types that can be readily established from patients, either lymphocytes or fibroblasts. However, it is highly possible that an individual RS could be tissue specific. For example, a patient may have an over response (due to genetic or other factors), which only affects a specific type of tissue. We, therefore, recommend that studies are pursued to enable the analysis of specific tissues in RS patients. This can be achieved via the analysis of organoid cultures although it is time consuming to carry out on a large patient cohort. Thus, we recommend that such analysis be progressed on a subset of control and RS patients.
9.6. Assessment of RSu
We need to identify and/or consolidate an IR-induced signature to help establish the origin of the cancer.
For RSu, there is not a defined cohort of patients due to the difficulty of identification. Thus, for this sensitivity, more epidemiological studies are required and more assessment of the underlying mechanisms.
10. Summary
In summary, there is strong evidence that individuals differ in their response to radiation exposure, and it is likely that this will hold for RS and RSu. However, the underlying basis is likely to be multifactorial given that a range of processes, which may function additively or even synergistically, can influence RS and RSu (). Understanding the response of the individual to radiation is a current goal, which will certainly enhance our opportunity to benefit from medical applications exploiting radiation and protect individuals from radiation encountered during daily life. To achieve this goal requires a broad approach, including the assessment of biomarkers and the use of epidemiological studies. However, understanding the mechanisms underlying RS and RSu is essential, not least to help identify biomarkers and to direct epidemiological studies. Our understanding of the DDR is now at a sophisticated level but we need to learn more about the breadth of interfacing processes. In this article, we have provided an in depth discussion of mechanisms that could influence RS and RSu, and presented a framework for a future strategy to achieve this important goal.
Figure 4. Potential factors or responses that could enhance RS and their interplay. The figure displays the responses that could potentially impact upon RS as discussed in the text. These responses could be regulated genetically or they could be influenced by environmental or life style factors. They could interplay to impact upon the DDR (left hand side of the figure) or could act independently but enhance DNA damage or an adverse tissue reaction (right hand side).
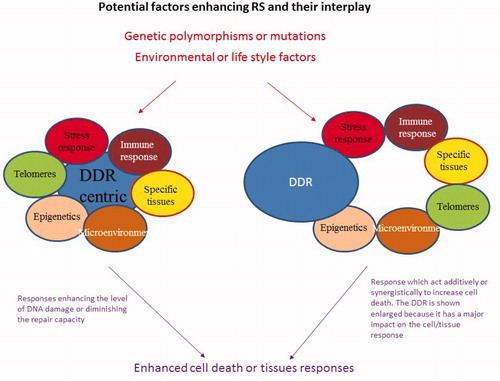
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes on contributors
Dietrich Averbeck
Dietrich Averbeck, Ph.D. is Director of Research Emeritus at CNRS (France) from the Curie Institute, Orsay (France) (1974–2008) involved in low dose radiation research with the MELODI association (2009–2019).
Serge Candéias
Serge Candéias, PhD, is a scientist in the Laboratory of Chemistry and Biology of Metals, a mixed research unit affiliated with the CEA, the CNRS and the UGA in the Interdisciplinary Research Institute of Grenoble (IRIG) in Grenoble, France.
Sudhir Chandna
Sudhir Chandna, PhD, is a Chief Scientist heading the Division of Radiation Biosciences in the Institute of Nuclear Medicine and Allied Sciences (Defense R&D Organisation), Delhi, India. He works on mechanisms of high dose radioresistance in model cell systems.
Nicolas Foray
Nicolas Foray, PhD is radiobiologist and Researcher at the French National Health Institute (INSERM). He is the author of 100 publications about individual radiosensitivity and DSB repair. Since 2019, he is the Director of the Research UA8 Unit “Radiations: Defense, Health and Environment” based in Lyon (France).
Anna A. Friedl
Anna A. Friedl is Adjunct Professor for Human Genetics and head of the Molecular Radiation Biology group in the Department of Radiation Oncology at the hospital of the Ludwig Maximilians University Munich.
Siamak Haghdoost
Siamak Haghdoost, PhD, is Professor of Radiobiology at University of Caen Normandie, Cimap-Laria, Advanced Resource Center for HADrontherapy in Europe (ARCHADE,), Caen, France and Center for Radiation Protection Research, Dept. of Molecular Biosciences, the Wenner-Gren Institute, Stockholm University, Sweden.
Penelope A. Jeggo
Penny Jeggo is Emeritus Professor at the Genome Damage and Stability Center, School of Life Sciences at the University of Sussex.
Katalin Lumniczky
Katalin Lumniczky, MD, PhD, is the head of the Department of Radiation Medicine, Division of Radiobiology and Radiohygiene, National Public Health Center Budapest, Hungary
Francois Paris
François Paris is Research Director at French National Health InstituTe (Inserm). He leads the team “Plasticity of the ecosystem of the tumour after Radiotherapy and Immunotherapy” in the Cancer research Center at the University of Nantes.
Roel Quintens
Roel Quintens, PhD is a senior scientist at the Radiobiology Unit of the Belgian Nuclear Research Center (SCK·CEN), Mol, Belgium.
Laure Sabatier
Laure Sabatier is the scientific advisor of the cytogenetics laboratory in CEA. She was the PI and head of the lab for more than 20 years before moving to the coordination of biology and health programs and infrastructures.
Related Research Data
References
- Agarwal P, Miller KM. 2016. The nucleosome: orchestrating DNA damage signaling and repair within chromatin. Biochem Cell Biol. 94(5):381–395.
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. 2017. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 549(7673):476–481.
- Aghamohammadi A, Akrami SM, Yaghmaie M, Rezaei N, Azizi G, Yaseri M, Nosrati H, Zaki-Dizaji M. 2018. Individual radiosensitivity assessment of the families of ataxia-telangiectasia patients by G2-checkpoint abrogation. Sultan Qaboos Univ Med J. 18(4):e440–e446.
- AGIR. 2013. Human radiosensitivity. Report of the Independetn Advisory Group on Ionising Radiation. Chilton, Doc HPA, RCE-21, 1–152.
- Al-Mayah AH, Irons SL, Pink RC, Carter DR, Kadhim MA. 2012. Possible role of exosomes containing RNA in mediating nontargeted effect of ionizing radiation. Rad Res. 177(5):539–545.
- Alajbeg I Z, Lapić I, Rogić D, Vuletić L, Andabak Rogulj A, Illeš D, Knezović Zlatarić D, Badel T, Vrbanović E, Alajbeg I. 2017. Within-subject reliability and between-subject variability of oxidative stress markers in saliva of healthy subjects: a longitudinal pilot study. Dis Markers. 2017:1–11.
- Alvarez-Rodriguez L, Lopez-Hoyos M, Munoz-Cacho P, Martinez-Taboada VM. 2012. Aging is associated with circulating cytokine dysregulation. Cell Immunol. 273:124–132.
- Andreassen C N, Rosenstein B S, Kerns S L, Ostrer H, De Ruysscher D, Cesaretti J A, Barnett G C, Dunning A M, Dorling L, West C M.L, et al. 2016. Individual patient data meta-analysis shows a significant association between the ATM rs1801516 SNP and toxicity after radiotherapy in 5456 breast and prostate cancer patients. Radiother Oncol. 121(3):431–439.
- Andreassen CN, Schack LM, Laursen LV, Alsner J. 2016. Radiogenomics – current status, challenges and future directions. Cancer Lett. 382(1):127–136.
- Antwih DA, Gabbara KM, Lancaster WD, Ruden DM, Zielske SP. 2013. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics. 8(8):839–848.
- Arnold KM, Flynn NJ, Raben A, Romak L, Yu Y, Dicker AP, Mourtada F, Sims-Mourtada J. 2018. The impact of radiation on the tumor microenvironment: effect of dose and fractionation schedules. Cancer Growth Metastasis. 11:1179064418761639.
- Arthur AE, Peterson KE, Shen J, Djuric Z, Taylor JM, Hebert JR, Duffy SA, Peterson LA, Bellile EL, Whitfield JR, et al. 2014. Diet and proinflammatory cytokine levels in head and neck squamous cell carcinoma. Cancer. 120(17):2704–2712.
- Aryal B, Rao VA. 2018. Specific protein carbonylation in human breast cancer tissue compared to adjacent healthy epithelial tissue. PLoS One. 13(3):e0194164.
- Averbeck D, Salomaa S, Bouffler S, Ottolenghi A, Smyth V, Sabatier L. 2018. Progress in low dose health risk research: novel effects and new concepts in low dose radiobiology. Mutat Res. 776:46–69.
- Ayouaz A, Raynaud C, Heride C, Revaud D, Sabatier L. 2008. Telomeres: hallmarks of radiosensitivity. Biochimie. 90(1):60–72.
- Azria D, Riou O, Castan F, Nguyen TD, Peignaux K, Lemanski C, Lagrange JL, Kirova Y, Lartigau E, Belkacemi Y, et al. 2015. Radiation-induced CD8 T-lymphocyte apoptosis as a predictor of breast fibrosis after radiotherapy: results of the prospective multicenter French trial. EBioMedicine. 2(12):1965–1973.
- Bagley J, Singh G, Iacomini J. 2007. Regulation of oxidative stress responses by ataxia-telangiectasia mutated is required for T cell proliferation. J Immunol. 178(8):4757–4763.
- Baijer J, Dechamps N, Perdry H, Morales P, Kerns S, Vasilescu A, Baulande S, Azria D, Romeo PH, Schmitz A. 2016. TNFSF10/TRAIL regulates human T4 effector memory lymphocyte radiosensitivity and predicts radiation-induced acute and subacute dermatitis. Oncotarget. 7(16):21416–21427.
- Barazzuol L, Hopkins SR, Ju L, Jeggo PA. 2019. Distinct response of adult neural stem cells to low versus high dose ionising radiation. DNA Repair. 76:70–75.
- Bardhan K, Anagnostou T, Boussiotis VA. 2016. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 7:550
- Barnes RP, Fouquerel E, Opresko PL. 2019. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev. 177:37–45.
- Barnett GC, West CM, Coles CE, Pharoah PD, Talbot CJ, Elliott RM, Tanteles GA, Symonds RP, Wilkinson JS, Dunning AM, et al. 2012. Standardized total average toxicity score: a scale- and grade-independent measure of late radiotherapy toxicity to facilitate pooling of data from different studies. Int J Radiat Oncol Biol Phys. 82(3):1065–1074.
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A, et al. 2017. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet. 26(20):3960–3972.
- Beach TA, Johnston CJ, Groves AM, Williams JP, Finkelstein JN. 2017. Radiation induced pulmonary fibrosis as a model of progressive fibrosis: contributions of DNA damage, inflammatory response and cellular senescence genes. Exp Lung Res. 43(3):134–149.
- Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, Nowak MA. 2007. Genetic progression and the waiting time to cancer. PLoS Comput Biol. 3(11):e225.
- Begg AC, Stewart FA, Vens C. 2011. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 11(4):239–253.
- Behjati S, Gundem G, Wedge DC, Roberts ND, Tarpey PS, Cooke SL, Van Loo P, Alexandrov LB, Ramakrishna M, Davies H, et al. 2016. Mutational signatures of ionizing radiation in second malignancies. Nat Commun. 7(1):12605..
- Beltran-Pardo E, Jonsson KI, Harms-Ringdahl M, Haghdoost S, Wojcik A. 2015. Tolerance to gamma radiation in the tardigrade hypsibius dujardini from embryo to adult correlate inversely with cellular proliferation. PLoS One. 10:e0133658.
- Bentzen SM, Overgaard J. 1994. Patient-to-patient variability in the expression of radiation-induced normal tissue injury. Semin Radiat Oncol. 4(2):68–80.
- Berardinelli F, Coluzzi E, Sgura A, Antoccia A. 2017. Targeting telomerase and telomeres to enhance ionizing radiation effects in in vitro and in vivo cancer models. Mutat Res. 773:204–219.
- Bernstein JL, Group WSC, Concannon P. 2017. ATM, radiation, and the risk of second primary breast cancer. Int J Radiat Biol. 93(10):1121–1127.
- Berrington de Gonzalez A, Berg CD, Visvanathan K, Robson M. 2009. Estimated risk of radiation-induced breast cancer from mammographic screening for young BRCA mutation carriers. J Natl Cancer Inst. 101(3):205–209.
- Biechonski S, Olender L, Zipin-Roitman A, Yassin M, Aqaqe N, Marcu-Malina V, Rall-Scharpf M, Trottier M, Meyn MS, Wiesmuller L, et al. 2018. Attenuated DNA damage responses and increased apoptosis characterize human hematopoietic stem cells exposed to irradiation. Sci Rep. 8(1):6071.
- Bjorklund G, Chirumbolo S. 2017. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition. 33:311–321.
- Blackburn EH. 2005. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 579(4):859–862.
- Bodgi L, Foray N. 2016. The nucleo-shuttling of the ATM protein as a basis for a novel theory of radiation response: resolution of the linear-quadratic model. Int J Radiat Biol. 92(3):117–131.
- Bouffler SD. 2016. Evidence for variation in human radiosensitivity and its potential impact on radiological protection. Ann ICRP. 45(1_suppl):280–289.
- Bouffler SD, Blasco MA, Cox R, Smith PJ. 2001. Telomeric sequences, radiation sensitivity and genomic instability. Int J Radiat Biol. 77(10):995–1005.
- Boustani J, Grapin M, Laurent PA, Apetoh L, Mirjolet C. 2019. The 6th R of radiobiology: reactivation of anti-tumor immune response. Cancers (Basel). 11(6): E860.
- Brenner DJ. 2014. What we know and what we don't know about cancer risks associated with radiation doses from radiological imaging. Br J Radiol. 87(1035):20130629.
- Brodin P, Davis MM. 2017. Human immune system variation. Nat Rev Immunol. 17(1):21–29.
- Burga A, Lehner B. 2012. Beyond genotype to phenotype: why the phenotype of an individual cannot always be predicted from their genome sequence and the environment that they experience. Febs J. 279(20):3765–3775.
- Cahoon EK, Preston DL, Pierce DA, Grant E, Brenner AV, Mabuchi K, Utada M, Ozasa K. 2017. Lung, laryngeal and other respiratory cancer incidence among Japanese atomic bomb survivors: an updated analysis from 1958 through 2009. Radiat Res. 187(5):538–548.
- Candeias SM, Gaipl US. 2016. The immune system in cancer prevention, development and therapy. Anticancer Agents Med Chem. 16(1):101–107.
- Candeias SM, Mika J, Finnon P, Verbiest T, Finnon R, Brown N, Bouffler S, Polanska J, Badie C. 2017. Low-dose radiation accelerates aging of the T-cell receptor repertoire in CBA/Ca mice. Cell Mol Life Sci. 74:4339–4351.
- Carlos-Reyes A, Lopez-Gonzalez JS, Meneses-Flores M, Gallardo-Rincon D, Ruiz-Garcia E, Marchat LA, Astudillo-de la Vega H, Hernandez de la Cruz ON, Lopez-Camarillo C. 2019. Dietary compounds as epigenetic modulating agents in cancer. Front Genet. 10:79.
- Castella M, Puerto S, Creus A, Marcos R, Surralles J. 2007. Telomere length modulates human radiation sensitivity in vitro. Toxicol Lett. 172(1–2):29–36.
- Chandna S, Suman S, Chandna M, Pandey A, Singh V, Kumar A, Dwarakanath BS, Seth RK. 2013. Radioresistant Sf9 insect cells undergo an atypical form of Bax-dependent apoptosis at very high doses of gamma-radiation. Int J Radiat Biol. 89(12):1017–1027.
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 18(8):495–506.
- Chen GY, Nunez G. 2010. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 10(12):826–837.
- Cho KW, Cantone MC, Kurihara-Saio C, Le Guen B, Martinez N, Oughton D, Schneider T, Toohey R, ZoLzer F. 2018. ICRP publication 138: ethical foundations of the system of radiological protection. Ann ICRP. 47(1):1–65.
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. 2017. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 170(6):1079–1095 e1020.
- Citrin DE, Mitchell JB. 2017. Mechanisms of normal tissue injury from irradiation. Semin Radiat Oncol. 27(4):316–324.
- Clouaire T, Marnef A, Legube G. 2017. Taming tricky DSBs: ATM on duty. DNA Repair. 56:84–91.
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6:2853–2868.
- Cornforth MN, Bedford JS. 1987. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat Res. 111(3):385–405.
- Corre I, Guillonneau M, Paris F. 2013. Membrane signaling induced by high doses of ionizing radiation in the endothelial compartment. Relevance in radiation toxicity. Int J Mol Sci. 14(11):22678–22696.
- Czochor JR, Glazer PM. 2014. microRNAs in cancer cell response to ionizing radiation. Antioxid Redox Signal. 21(2):293–312.
- Dadrich M, Nicolay NH, Flechsig P, Bickelhaupt S, Hoeltgen L, Roeder F, Hauser K, Tietz A, Jenne J, Lopez R, et al. 2016. Combined inhibition of TGFbeta and PDGF signaling attenuates radiation-induced pulmonary fibrosis. Oncoimmunology. 5(5):e1123366.
- Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, et al. 2004. Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science. 306(5698):1025–1028.
- Danielsson D, Brehwens K, Halle M, Marczyk M, Sollazzo A, Polanska J, Munck-Wikland E, Wojcik A, Haghdoost S. 2016. Influence of genetic background and oxidative stress response on risk of mandibular osteoradionecrosis after radiotherapy of head and neck cancer. Head Neck. 38(3):387–393.
- Dauer LT, Ainsbury EA, Dynlacht J, Hoel D, Klein BEK, Mayer D, Prescott CR, Thornton RH, Vano E, Woloschak GE, et al. 2017. Guidance on radiation dose limits for the lens of the eye: overview of the recommendations in NCRP Commentary No. 26. Int J Radiat Biol. 93(10):1015–1023.
- De Lange T. 2005. Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol. 70(0):197–204.
- Declerck K, Vanden Berghe W. 2018. Back to the future: epigenetic clock plasticity towards healthy aging. Mech Ageing Dev. 174:18–29.
- Denhardt DT. 2018. Effect of stress on human biology: epigenetics, adaptation, inheritance, and social significance. J Cell Physiol. 233(3):1975–1984.
- Ding GX, Alaei P, Curran B, Flynn R, Gossman M, Mackie TR, Miften M, Morin R, Xu XG, Zhu TC. 2018. Image guidance doses delivered during radiotherapy: quantification, management, and reduction: report of the AAPM Therapy Physics Committee Task Group 180. Med Phys. 45(5):e84–e99.
- Ditch S, Paull TT. 2012. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 37(1):15–22.
- Dorr W. 2015. Radiobiology of tissue reactions. Ann ICRP. 44(1 Suppl):58–68.
- Ducray C, Pommier JP, Martins L, Boussin FD, Sabatier L. 1999. Telomere dynamics, end-to-end fusions and telomerase acivation during the human fibroblast immortalization process. Oncogene. 18(29):4211–4223.
- Dunn GP, Old LJ, Schreiber RD. 2004. The three Es of cancer immunoediting. Annu Rev Immunol. 22(1):329–360.
- Durante M, Debus J. 2018. Heavy charged particles: does improved precision and higher biological effectiveness translate to better outcome in patients? Semin Radiat Oncol. 28(2):160–167.
- Emanuele E, Spencer JM, Braun M. 2014. From DNA repair to proteome protection: new molecular insights for preventing non-melanoma skin cancers and skin aging. J Drugs Dermatol. 13(3):274–281.
- Etchegaray JP, Mostoslavsky R. 2016. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell. 62(5):695–711.
- Etienne O, Roque T, Haton C, Boussin FD. 2012. Variation of radiation-sensitivity of neural stem and progenitor cell populations within the developing mouse brain. Int J Radiat Biol. 88(10):694–702.
- Fairlie J, Harrington L. 2015. Enforced telomere elongation increases the sensitivity of human tumour cells to ionizing radiation. DNA Repair. 25:54–59.
- Fan PC, Zhang Y, Wang Y, Wei W, Zhou YX, Xie Y, Wang X, Qi YZ, Chang L, Jia ZP, et al. 2019. Quantitative proteomics reveals mitochondrial respiratory chain as a dominant target for carbon ion radiation: Delayed reactive oxygen species generation caused DNA damage. Free Radic Biol Med. 130:436–445.
- Feinberg AP. 2018. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 378(14):1323–1334.
- Ferlazzo ML, Bach-Tobdji MKE, Djerad A, Sonzogni L, Devic C, Granzotto A, Bodgi L, Bachelet JT, Djefal-Kerrar A, Hennequin C, et al. 2018. Radiobiological characterization of tuberous sclerosis: a delay in the nucleo-shuttling of ATM may be responsible for radiosensitivity. Mol Neurobiol. 55(6):4973–4983.
- Ferlazzo ML, Sonzogni L, Granzotto A, Bodgi L, Lartin O, Devic C, Vogin G, Pereira S, Foray N. 2014. Mutations of the Huntington's disease protein impact on the ATM-dependent signaling and repair pathways of the radiation-induced DNA double-strand breaks: corrective effect of statins and bisphosphonates. Mol Neurobiol. 49(3):1200–1211.
- Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, Jones PH. 2019. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell. 25(3):329–341 e326.
- Ferrand M, Kirsh O, Griveau A, Vindrieux D, Martin N, Defossez PA, Bernard D. 2015. Screening of a kinase library reveals novel pro-senescence kinases and their common NF-kappaB-dependent transcriptional program. Aging (Aging). 7(11):986–1003.
- Foray N, Bourguignon M. 2019. Comment on 'considerations on the use of the terms radiosensitivity and radiosusceptibility' by Wojcik. J Radiol Prot. 39(1):309–313.
- Foray N, Bourguignon M, Hamada N. 2016. Individual response to ionizing radiation. Mutat Res. 770:369–386.
- Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. 2017. Chronic Inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. 72:1218–1225.
- Franceschi C, Bonafe M. 2003. Centenarians as a model for healthy aging. Biochem Soc Trans. 31(2):457–461.
- Frey B, Ruckert M, Deloch L, Ruhle PF, Derer A, Fietkau R, Gaipl US. 2017. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol Rev. 280(1):231–248.
- Gault N, Verbiest T, Badie C, Romeo PH, Bouffler S. 2019. Hematopoietic stem and progenitor cell responses to low radiation doses - implications for leukemia risk. Int J Radiat Biol. 95(7):892–899.
- Genesca A, Martin M, Latre L, Soler D, Pampalona J, Tusell L. 2006. Telomere dysfunction: a new player in radiation sensitivity. Bioessays. 28:1172–1180.
- Gilson E, Londono-Vallejo A. 2007. Telomere length profiles in humans: all ends are not equal. Cell Cycle. 6(20):2486–2494.
- Gomolka M, Blyth B, Bourguignon M, Badie C, Schmitz A, Talbot C, Hoeschen C, Salomaa S. 2019. Potential screening assays for individual radiation sensitivity and susceptibility and their current validation state. Int J Radiat Biol. 1–17. doi:10.1080/09553002.2019.1642544.
- Goodarzi AA, Jeggo PA. 2012. The heterochromatic barrier to DNA double strand break repair: how to get the entry visa. Int J Mol Sci. 13(12):11844–11860.
- Gougerot-Podicalo MA, Elbim C, Chollet-Martin S. 1996. Modulation of the oxidative burst of human neutrophils by pro- and anti-inflammatory cytokines. Pathol Biol. 44(1):36–41.
- Graakjaer J, Pascoe L, Der-Sarkissian H, Thomas G, Kolvraa S, Christensen K, Londono-Vallejo JA. 2004. The relative lengths of individual telomeres are defined in the zygote and strictly maintained during life. Aging Cell. 3(3):97–102.
- Grant EJ, Brenner A, Sugiyama H, Sakata R, Sadakane A, Utada M, Cahoon EK, Milder CM, Soda M, Cullings HM, et al. 2017. Solid cancer incidence among the life span study of atomic bomb survivors: 1958–2009. Radiat Res. 187(5):513–537.
- Granzotto A, Benadjaoud MA, Vogin G, Devic C, Ferlazzo ML, Bodgi L, Pereira S, Sonzogni L, Forcheron F, Viau M, et al. 2016. Influence of nucleoshuttling of the ATM protein in the healthy tissues response to radiation therapy: toward a molecular classification of human radiosensitivity. Int J Radiat Oncol Biol Phys. 94(3):450–460.
- Guleria A, Chandna S. 2016. ATM kinase: much more than a DNA damage responsive protein. DNA Repair. 39:1–20.
- Haghdoost S, Sjolander L, Czene S, Harms-Ringdahl M. 2006. The nucleotide pool is a significant target for oxidative stress. Free Radic Biol Med. 41(4):620–626.
- Haghdoost S, Svoboda P, Naslund I, Harms-Ringdahl M, Tilikides A, Skog S. 2001. Can 8-oxo-dG be used as a predictor for individual radiosensitivity? Int J Radiat Oncol Biol Phys. 50(2):405–410.
- Hagiwara Y, Sato H, Permata TBM, Niimi A, Yamauchi M, Oike T, Nakano T, Shibata A. 2018. Analysis of programmed death-ligand 1 expression in primary normal human dermal fibroblasts after DNA damage. Hum Immunol. 79(8):627–631.
- Hall EJ. 2006. Intensity-modulated radiation therapy, protons, and the risk of second cancers. Int J Radiat Oncol Biol Phys. 65(1):1–7.
- Hall EJ, Brenner DJ. 2012. Cancer risks from diagnostic radiology: the impact of new epidemiological data. Br J Radiol. 85(1020):e1316–e1317.
- Hall J, Jeggo PA, West C, Gomolka M, Quintens R, Badie C, Laurent O, Aerts A, Anastasov N, Azimzadeh O, et al. 2017. Ionizing radiation biomarkers in epidemiological studies – an update. Mutat Res. 771:59–84.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell. 144(5):646–674.
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 548(7668):466–470.
- Hassan FU, Rehman MS, Khan MS, Ali MA, Javed A, Nawaz A, Yang C. 2019. Curcumin as an alternative epigenetic modulator: mechanism of action and potential effects. Front Genet. 10:514
- Heider T, Mutschelknaus L, Radulovic V, Winkler K, Kimmel J, Anastasov N, Atkinson MJ, Moertl S. 2017. Radiation induced transcriptional and post-transcriptional regulation of the hsa-miR-23a∼27a∼24-2 cluster suppresses apoptosis by stabilizing XIAP. Biochim Biophys Acta Gene Regul Mech. 1860(11):1127–1137.
- Hong JH, Chiang CS, Campbell IL, Sun JR, Withers HR, McBride WH. 1995. Induction of acute phase gene expression by brain irradiation. Int J Radiat Oncol Biol Phys. 33(3):619–626.
- Hughson RL, Helm A, Durante M. 2018. Heart in space: effect of the extraterrestrial environment on the cardiovascular system. Nat Rev Cardiol. 15(3):167–180.
- Ichikawa J, Tsuchimoto D, Oka S, Ohno M, Furuichi M, Sakumi K, Nakabeppu Y. 2008. Oxidation of mitochondrial deoxynucleotide pools by exposure to sodium nitroprusside induces cell death. DNA Repair (Amst). 7(3):418–430.
- ICRP. 2007. ICRP Publication 103: The Recommendations of the International Committee on Radiological Protection.
- Jackson SE, Chester JD. 2015. Personalised cancer medicine. Int J Cancer. 137(2):262–266.
- Jeggo PA, Geuting V, Lobrich M. 2011. The role of homologous recombination in radiation-induced double-strand break repair. Radiother Oncol. 101(1):7–12.
- Jeggo P, Lavin MF. 2009. Cellular radiosensitivity: how much better do we understand it?. Int J Radiat Biol. 85(12):1061–1081.
- Kabacik S, Manning G, Raffy C, Bouffler S, Badie C. 2015. Time, dose and ataxia telangiectasia mutated (ATM) status dependency of coding and noncoding RNA expression after ionizing radiation exposure. Radiat Res. 183(3):325–337.
- Kalman C, Oughton D. 2019. Ethical considerations related to radiosensitivity and radiosusceptibility. Int J Radiat Biol. 1–4. doi:10.1080/09553002.2019.1665210.
- Karabulutoglu M, Finnon R, Imaoka T, Friedl AA, Badie C. 2019. Influence of diet and metabolism on hematopoietic stem cells and leukemia development following ionizing radiation exposure. Int J Radiat Biol. 95(4):452–479.
- Kemp LM, Jeggo PA. 1986. Radiation-induced chromosome damage in X-ray-sensitive mutants (xrs) of the Chinese hamster ovary cell line. Mutat Res. 166:255–263.
- Khavari A-P, Liu Y, He E, Skog S, Haghoost S. 2018. Serum 8-Oxo-dG as a predictor of sensitivity and outcome of radiotherapy and chemotherapy of upper gastrointestinal tumours. Oxid Med Cell Longev. 2018:4153574.
- Kim GJ, Chandrasekaran K, Morgan WF. 2006. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 21:361–367.
- Kofman AV, Kim J, Park SY, Dupart E, Letson C, Bao Y, Ding K, Chen Q, Schiff D, Larner J, et al. 2013. microRNA-34a promotes DNA damage and mitotic catastrophe. Cell Cycle. 12(22):3500–3511.
- Koj A. 1998. Termination of acute-phase response: role of some cytokines and anti-inflammatory drugs. Gen Pharmacol. 31(1):9–18.
- Kong FM, Wang S. 2015. Nondosimetric risk factors for radiation-induced lung toxicity. Semin Radiat Oncol. 25(2):100–109.
- Kono T, Sakamoto K, Shinden S, Ogawa K. 2017. Pre-therapeutic nutritional assessment for predicting severe adverse events in patients with head and neck cancer treated by radiotherapy. Clin Nutr. 36(6):1681–1685.
- Koturbash I, Miousse IR, Sridharan V, Nzabarushimana E, Skinner CM, Melnyk SB, Pavliv O, Hauer-Jensen M, Nelson GA, Boerma M. 2016. Radiation-induced changes in DNA methylation of repetitive elements in the mouse heart. Mutat Res. 787:43–53.
- Kraemer A, Anastasov N, Angermeier M, Winkler K, Atkinson MJ, Moertl S. 2011. MicroRNA-mediated processes are essential for the cellular radiation response. Radiat Res. 176(5):575–586.
- Krisko A, Leroy M, Radman M, Meselson M. 2012. Extreme anti-oxidant protection against ionizing radiation in bdelloid rotifers. Proc Natl Acad Sci USA. 109(7):2354–2357.
- Kumagai T, Rahman F, Smith AM. 2018. The microbiome and radiation induced-bowel injury: evidence for potential mechanistic role in disease pathogenesis. Nutrients. 10(10):1405.
- Kurutas EB. 2015. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 15(1):71.
- Lacombe J, Zenhausern F. 2017. Emergence of miR-34a in radiation therapy. Crit Rev Oncol Hematol. 109:69–78.
- Lafargue A, Degorre C, Corre I, Alves-Guerra MC, Gaugler MH, Vallette F, Pecqueur C, Paris F. 2017. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic Biol Med. 108:750–759.
- Li AL, Chung TS, Chan YN, Chen CL, Lin SC, Chiang YR, Lin CH, Chen CC, Ma N. 2018. microRNA expression pattern as an ancillary prognostic signature for radiotherapy. J Transl Med. 16(1):341.
- Lisbona A, Averbeck D, Supiot S, Delpon G, Ali D, Vinas F, Diana C, Murariu C, Lagrange JL. 2010. IMRT combined to IGRT: increase of the irradiated volume. Consequences? Cancer Radiother. 14(6–7):563–570.
- Liu Y, Sun H, Makabel B, Cui Q, Li J, Su C, Ashby CR, Jr., Chen Z, Zhang J. 2019. The targeting of noncoding RNAs by curcumin: facts and hopes for cancer therapy (Review. Oncol Rep. 42:20–34.
- Locke PA, Weil MM. 2016. Personalized cancer risk assessments for space radiation exposures. Front Oncol. 22:38.
- Loseva O, Shubbar E, Haghdoost S, Evers B, Helleday T, Harms-Ringdahl M. 2014. Chronic low dose rate ionizing radiation exposure induces premature senescence in human fibroblasts that correlates with up regulation of proteins involved in protection against oxidative stress. Proteomes. 2(3):341–362.
- Ma S, Liu X, Jiao B, Yang Y, Liu X. 2010. Low-dose radiation-induced responses: focusing on epigenetic regulation. Int J Radiat Biol. 86(7):517–528.
- Maalouf M, Granzotto A, Devic C, Bodgi L, Ferlazzo M, Peaucelle C, Bajard M, Giraud JY, Balosso J, Herault J, et al. 2019. Influence of linear energy transfer on the nucleo-shuttling of the ATM Protein: a novel biological interpretation relevant for particles and radiation. Int J Radiat Oncol Biol Phys. 103(3):709–718.
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 548(7668):461–465.
- Marcon F, Carotti D, Andreoli C, Siniscalchi E, Leopardi P, Caiola S, Biffoni M, Zijno A, Medda E, Nistico L, et al. 2013. DNA damage response in monozygotic twins discordant for smoking habits. Mutagenesis. 28(2):135–144.
- Marliere S, Vautrin E, Saunier C, Chaikh A, Gabelle-Flandin I. 2016. Radiation-related heart toxicity: Update in women]. Ann Cardiol Angeiol (Paris). 65:411–419.
- Marnef A, Cohen S, Legube G. 2017. Transcription-coupled DNA double-strand break repair: active genes need special care. J Mol Biol. 429(9):1277–1288.
- Martens UM, Chavez EA, Poon SS, Schmoor C, Lansdorp PM. 2000. Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp Cell Res. 256(1):291–299.
- Martin MT, Vulin A, Hendry JH. 2016. Human epidermal stem cells: role in adverse skin reactions and carcinogenesis from radiation. Mutat Res. 770:349–368.
- McLean AR, Adlen EK, Cardis E, Elliott A, Goodhead DT, Harms-Ringdahl M, Hendry JH, Hoskin P, Jeggo PA, Mackay DJC, et al. 2017. A restatement of the natural science evidence base concerning the health effects of low-level ionizing radiation. Proc Biol Sci. 284(1862):20171070.
- Menoux I, Le Fevre C, Noel G, Antoni D. 2018. Radiation-induced lung toxicity predictors after stereotactic radiation therapy for non-small cell lung carcinoma stage I. Cancer Radiother. 22(8):826–838.
- Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, Cesari M, Nourhashemi F. 2013. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 14(12):877–882.
- Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I, Iannelli F, et al. 2017. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat Cell Biol. 19(12):1400–1411.
- Miousse IR, Kutanzi KR, Koturbash I. 2017. Effects of ionizing radiation on DNA methylation: from experimental biology to clinical applications. Int J Radiat Biol. 93(5):457–469.
- Mirjolet C, Merlin JL, Dalban C, Maingon P, Azria D. 2016. Correlation between radio-induced lymphocyte apoptosis measurements obtained from two French centres. Cancer Radiother. 20(5):391–394.
- Mladenov E, Saha J, Iliakis G. 2018. Processing-challenges generated by clusters of DNA double-strand breaks underpin increased effectiveness of high-LET radiation and chromothripsis. Adv Exp Med Biol. 1044:149–168.
- Morgan WF. 2003. Non-targeted and delayed effects of exposure to ionizing radiation: I. radiation-induced genomic instability and bystander effects in vitro. Radiat Res. 178:AV223–236.
- Munro TR, Gilbert CW. 1961. The relation between tumour lethal doses and the radiosensitivity of tumour cells. Br J Radiol. 34(400):246–251.
- Murnane JP, Sabatier L. 2004. Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. Bioessays. 26(11):1164–1174.
- Narendran N, Luzhna L, Kovalchuk O. 2019. Sex difference of radiation response in occupational and accidental exposure. Front Genet. 10:260
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. 2014. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention
- Nielsen S, Bassler N, Grzanka L, Swakon J, Olko P, Andreassen CN, Overgaard J, Alsner J, Sorensen BS. 2017. Differential gene expression in primary fibroblasts induced by proton and cobalt-60 beam irradiation. Acta Oncol. 56(11):1406–1412.
- Nikjoo H, O'Neill P, Terrissol M, Goodhead DT. 1999. Quantitative modelling of DNA damage using Monte Carlo track structure method. Radiat Environ Biophys. 38(1):31–38.
- O'Flanagan CH, Smith LA, McDonell SB, Hursting SD. 2017. When less may be more: calorie restriction and response to cancer therapy. BMC Med. 15:106.
- O'Hagan HM. 2014. Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environ Mol Mutagen. 55:278–291.
- Obdeijn IM, Heijnsdijk EA, Hunink MG, Tilanus-Linthorst MM, de Koning HJ. 2016. Mammographic screening in BRCA1 mutation carriers postponed until age 40: Evaluation of benefits, costs and radiation risks using models. Eur J Cancer. 63:135–142.
- Olivieri F, Bonafe M, Giovagnetti S, Stecconi R, Cardelli M, Cavallone L, Spazzafumo L, Marchegiani F, Carrieri G, Mugianesi E, et al. 2003. In vitro IL-6 production by EBV-immortalized B lymphocytes from young and elderly people genotyped for -174 C/G polymorphism in IL-6 gene: a model to study the genetic basis of inflamm-aging. Mech Ageing Dev. 124(4):549–553.
- Ozasa K. 2016. Epidemiological research on radiation-induced cancer in atomic bomb survivors. J Radiat Res. 57(S1):i112–i117.
- Ozsahin M, Crompton NE, Gourgou S, Kramar A, Li L, Shi Y, Sozzi WJ, Zouhair A, Mirimanoff RO, Azria D. 2005. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clin Cancer Res. 11(20):7426–7433.
- Palm A, Johansson KA. 2007. A review of the impact of photon and proton external beam radiotherapy treatment modalities on the dose distribution in field and out-of-field; implications for the long-term morbidity of cancer survivors. Acta Oncol. 46(4):462–473.
- Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R. 2001. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 293(5528):293–297.
- Pathak R, Shah SK, Hauer-Jensen M. 2019. Therapeutic potential of natural plant products and their metabolites in preventing radiation enteropathy resulting from abdominal or pelvic irradiation. Int J Radiat Biol. 95(4):493–505.
- Pereira S, Bodgi L, Duclos M, Canet A, Ferlazzo ML, Devic C, Granzotto A, Deneuve S, Vogin G, Foray N. 2018. Fast and binary assay for predicting radiosensitivity based on the theory of ATM nucleo-shuttling: development, validation, and performance. Int J Radiat Oncol Biol Phys. 100(2):353–360.
- Pernot E, Hall J, Baatout S, Benotmane MA, Blanchardon E, Bouffler S, El Saghire H, Gomolka M, Guertler A, Harms-Ringdahl M, et al. 2012. Ionizing radiation biomarkers for potential use in epidemiological studies. Mutat Res. 751(2):258–286.
- Philipp J, Azimzadeh O, Subramanian V, Merl-Pham J, Lowe D, Hladik D, Erbeldinger N, Ktitareva S, Fournier C, Atkinson MJ, et al. 2017. Radiation-induced endothelial inflammation is transferred via the secretome to recipient cells in a STAT-mediated process. J Proteome Res. 16(10):3903–3916.
- Platzer V, Perez G, Galinier A, Genestal M, Riu-Poulenc B, Gonzalez L, Huyghe E, Caspar-Bauguil S. 2018. Protein and micronutrient deficiencies in patients with radiation cystitis and outcome after hyperbaric oxygen therapy. Clin Nutr ESPEN. 23:141–147.
- Pommier JP, Lebeau J, Ducray C, Sabatier L. 1995. Chromosomal instability and alteration of telomere repeat sequences. Biochimie. 77(10):817–825.
- Pommier JP, Sabatier L. 2002. Telomere length distribution. Digital image processing and statistical analysis. Methods Mol Biol. 191:33–63.
- Portella L, Scala S. 2019. Ionizing radiation effects on the tumor microenvironment. Semin Oncol. 46(3):254–260. doi:10.1053/j.seminoncol.2019.07.003.
- Power S P, Moloney F, Twomey M, James K, O’Connor O J, Maher M M. 2016. Computed tomography and patient risk: facts, perceptions and uncertainties. World J Radiol. 8(12):902–915.
- Price B D, D’Andrea A D. 2013. Chromatin remodeling at DNA double-strand breaks. Cell. 152(6):1344–1354.
- Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, Ritz B, Bandinelli S, Neuhouser ML, Beasley JM. 2017. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Aging). 9:419–446.
- Radman M. 2016. Protein damage, radiation sensitivity and aging. DNA Repair. 44:186–192.
- Rajaraman P, Hauptmann M, Bouffler S, Wojcik A. 2018. Human individual radiation sensitivity and prospects for prediction. Ann ICRP. 47(3–4):126–141.
- Revenco T, Lapouge G, Moers V, Brohee S, Sotiropoulou PA. 2017. Low dose radiation causes skin cancer in mice and has a differential effect on distinct epidermal stem cells. Stem Cells. 35(5):1355–1364.
- Riballo E, Critchlow S.E, Teo S-H, Doherty A.J, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett C.F, et al. 1999. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 9(13):699–702.
- Richardson RB. 2009. Ionizing radiation and aging: rejuvenating an old idea. Aging (Aging). 1(11):887–902.
- Roberts SA, Spreadborough AR, Bulman B, Barber JB, Evans DG, Scott D. 1999. Heritability of cellular radiosensitivity: a marker of low-penetrance predisposition genes in breast cancer? Am J Hum Genet. 65(3):784–794.
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. 2009. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 11(8):973–979.
- Roos WP, Thomas AD, Kaina B. 2016. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 16(1):20–33.
- Rose SR, Horne VE, Howell J, Lawson SA, Rutter MM, Trotman GE, Corathers SD. 2016. Late endocrine effects of childhood cancer. Nat Rev Endocrinol. 12(6):319–336.
- Royba E, Miyamoto T, Natsuko Akutsu S, Hosoba K, Tauchi H, Kudo Y, Tashiro S, Yamamoto T, Matsuura S. 2017. Evaluation of ATM heterozygous mutations underlying individual differences in radiosensitivity using genome editing in human cultured cells. Sci Rep. 7(1):5996.
- Ruszkiewicz J, Albrecht J. 2015. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem Int. 88:66–72.
- Sabatier L, Al Achkar W, Hoffschir F, Luccioni C, Dutrillaux B. 1987. Qualitative study of chromosomal lesions induced by neutrons and neon ions in human lymphocytes at G0 phase. Mutat Res. 178(1):91–97.
- Sabatier L, Dutrillaux B, Martin MB. 1992. Chromosomal instability. Nature. 357(6379):548.
- Sage E, Shikazono N. 2017. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic Biol Med. 107:125–135.
- Saha SK, Lee SB, Won J, Choi HY, Kim K, Yang GM, Dayem AA, Cho SG. 2017. Correlation between oxidative stress, nutrition, and cancer initiation. Int J Mol Sci. 18(7):E1544.
- Sato H, Jeggo PA, Shibata A. 2019. Regulation of PD-L1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci. 110(11):3415–3423.
- Sato H, Niimi A, Yasuhara T, Permata T B M, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. 2017. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 8(1):1751.
- Schleicher RL, Carroll MD, Ford ES, Lacher DA. 2009. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 90(5):1252–1263.
- Schmitz A, Bayer J, Dechamps N, Goldin L, Thomas G. 2007. Heritability of susceptibility to ionizing radiation-induced apoptosis of human lymphocyte subpopulations. Int J Radiat Oncol Biol Phys. 68(4):1169–1177.
- Schnarr K, Boreham D, Sathya J, Julian J, Dayes IS. 2009. Radiation-induced lymphocyte apoptosis to predict radiation therapy late toxicity in prostate cancer patients. Int J Radiat Oncol Biol Phys. 74(5):1424–1430.
- Schofield PN, Kondratowicz M. 2018. Evolving paradigms for the biological response to low dose ionizing radiation; the role of epigenetics. Int J Radiat Biol. 94(8):769–781.
- Schreurs AS, Shirazi-Fard Y, Shahnazari M, Alwood JS, Truong TA, Tahimic CG, Limoli CL, Turner ND, Halloran B, Globus RK. 2016. Dried plum diet protects from bone loss caused by ionizing radiation. Sci Rep. 6(1):21343.
- Scott D. 2004. Chromosomal radiosensitivity and low penetrance predisposition to cancer. Cytogenet Genome Res. 104(1–4):365–370.
- Seibold P, Auvinen A, Averbeck D, Bourguignon M, Hartikainen JM, Hoeschen C, Laurent O, Noël G, Sabatier L, Salomaa S, et al. 2019. Clinical and epidemiological observations on individual radiation sensitivity and susceptibility. Int J Radiat Biol. 1–16. doi:10.1080/09553002.2019.1665209.
- Seibold P, Behrens S, Schmezer P, Helmbold I, Barnett G, Coles C, Yarnold J, Talbot CJ, Imai T, Azria D, et al. 2015. XRCC1 polymorphism associated with late toxicity after radiation therapy in breast cancer patients. Int J Radiat Oncol Biol Phys. 92(5):1084–1092.
- Shibata A, Jeggo PA. 2014. DNA double-strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 26(5):243–249.
- Shim G, Ricoul M, Hempel WM, Azzam EI, Sabatier L. 2014. Crosstalk between telomere maintenance and radiation effects: a key player in the process of radiation-induced carcinogenesis. Mutat Res Rev Mutat Res. 760:1–17.
- Siiskonen T, Kaijaluoto S, Florea T. 2017. Imaging practices and radiation doses from imaging in radiotherapy. Phys Med. 42:247–252.
- Singh VK, Hanlon BK, Santiago PT, Seed TM. 2017. A review of radiation countermeasures focusing on injury-specific medicinals and regulatory approval status: part III. Countermeasures under early stages of development along with 'standard of care' medicinal and procedures not requiring regulatory approval for use. Int J Radiat Biol. 93:885–906.
- Skiold S, Azimzadeh O, Merl-Pham J, Naslund I, Wersall P, Lidbrink E, Tapio S, Harms-Ringdahl M, Haghdoost S. 2015. Unique proteomic signature for radiation sensitive patients; a comparative study between normo-sensitive and radiation sensitive breast cancer patients. Mutat Res. 776:128–135.
- Skiold S, Naslund I, Brehwens K, Andersson A, Wersall P, Lidbrink E, Harms-Ringdahl M, Wojcik A, Haghdoost S. 2013. Radiation-induced stress response in peripheral blood of breast cancer patients differs between patients with severe acute skin reactions and patients with no side effects to radiotherapy. Mutat Res. 756:152–157.
- Slade D, Radman M. 2011. Oxidative stress resistance in Deinococcus radiodurans. Microbiol Mol Biol Rev. 75(1):133–191.
- Smith TA, Kirkpatrick DR, Smith S, Smith TK, Pearson T, Kailasam A, Herrmann KZ, Schubert J, Agrawal DK. 2017. Radioprotective agents to prevent cellular damage due to ionizing radiation. J Transl Med. 15(1):232.
- Spitz DR, Azzam EI, Li JJ, Gius D. 2004. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 23(3/4):311–322.
- Squillaro T, Galano G, De Rosa R, Peluso G, Galderisi U. 2018. Concise review: the effect of low-dose ionizing radiation on stem cell biology: a contribution to radiation risk. Stem Cells. 36(8):1146–1153.
- Sroussi HY, Epstein JB, Bensadoun RJ, Saunders DP, Lalla RV, Migliorati CA, Heaivilin N, Zumsteg ZS. 2017. Common oral complications of head and neck cancer radiation therapy: mucositis, infections, saliva change, fibrosis, sensory dysfunctions, dental caries, periodontal disease, and osteoradionecrosis. Cancer Med. 6(12):2918–2931.
- Stewart RD, Yu VK, Georgakilas AG, Koumenis C, Park JH, Carlson DJ. 2011. Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat Res. 176(5):587–602.
- Story MD, Durante M. 2018. Radiogenomics. Med Phys. 45(11):e1111–e1122.
- Strzeszewska A, Alster O, Mosieniak G, Ciolko A, Sikora E. 2018. Insight into the role of PIKK family members and NF-кB in DNAdamage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis. 9(2):44
- Sun Y, Cheng MK, Griffiths TR, Mellon JK, Kai B, Kriajevska M, Manson MM. 2013. Inhibition of STAT signalling in bladder cancer by diindolylmethane: relevance to cell adhesion, migration and proliferation. Curr Cancer Drug Targets. 13(1):57–68.
- Szatmari T, Kis D, Bogdándi E N, Benedek A, Bright S, Bowler D, Persa E, Kis E, Balogh A, Naszályi L N, et al. 2017. Extracellular vesicles mediate radiation-induced systemic bystander signals in the bone marrow and spleen. Front Immunol. 8:347
- Szatmari T, Persa E, Kis E, Benedek A, Hargitai R, Safrany G, Lumniczky K. 2019. Extracellular vesicles mediate low dose ionizing radiation-induced immune and inflammatory responses in the blood. Int J Radiat Biol. 95:12–22.
- Taha TA, Mullen TD, Obeid LM. 2006. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim Biophys Acta. 1758(12):2027–2036.
- Thomas RJ, Kenfield SA, Jimenez A. 2017. Exercise-induced biochemical changes and their potential influence on cancer: a scientific review. Br J Sports Med. 51(8):640–644.
- Tommasino F, Durante M. 2015. Proton radiobiology. Cancers (Basel). 7(1):353–381.
- UNSCEAR. 2013. Annex B: Effects of radiation exposure of children. New York United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR), United Nations. Sources, Effects and Risks of Ionizing Radiation, Vol II.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. 2007. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 39(1):44–84.
- Vandevoorde C, Depuydt J, Veldeman L, De Neve W, Sebastia N, Wieme G, Baert A, De Langhe S, Philippe J, Thierens H, et al. 2016. In vitro cellular radiosensitivity in relationship to late normal tissue reactions in breast cancer patients: a multi-endpoint case-control study. Int J Radiat Biol. 92(12):823–836.
- Venkatesh GH, Manjunath VB, Mumbrekar KD, Negi H, Fernandes DJ, Sharan K, Banerjee S, Bola Sadashiva SR. 2014. Polymorphisms in radio-responsive genes and its association with acute toxicity among head and neck cancer patients. PLoS One. 9(3):e89079.
- Vogin G, Bastogne T, Bodgi L, Gillet-Daubin J, Canet A, Pereira S, Foray N. 2018. The phosphorylated ATM immunofluorescence assay: a high-performance radiosensitivity assay to predict postradiation therapy overreactions. Int J Radiat Oncol Biol Phys. 101(3):690–693.
- Wahl GM, Linke SP, Paulson TG, Huang LC. 1997. Maintaining genetic stability through TP53 mediated checkpoint control. Cancer Surv. 29:183–219.
- Wakeford R. 2012. Radiation effects: modulating factors and risk assessment – an overview. Ann ICRP. 41(3–4):98–107.
- Wedlake L, Shaw C, McNair H, Lalji A, Mohammed K, Klopper T, Allan L, Tait D, Hawkins M, Somaiah N, et al. 2017. Randomized controlled trial of dietary fiber for the prevention of radiation-induced gastrointestinal toxicity during pelvic radiotherapy. Am J Clin Nutr. 106(3):849–857.
- Wei W, Ba Z, Gao M, We Y, Amiard S, Whiste CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell. 2012. 149:101–112.
- Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones KA, Garcia-Martinez E, Rudqvist NP, Formenti SC, Demaria S. 2017. Barriers to radiation-induced in situ tumor vaccination. Front Immunol. 8:229
- West C, Azria D, Chang-Claude J, Davidson S, Lambin P, Rosenstein B, De Ruysscher D, Talbot C, Thierens H, Valdagni R, et al. 2014. The REQUITE project: validating predictive models and biomarkers of radiotherapy toxicity to reduce side-effects and improve quality of life in cancer survivors. Clin Oncol (R Coll Radiol). 26(12):739–742.
- Wilke CM, Braselmann H, Hess J, Klymenko SV, Chumak VV, Zakhartseva LM, Bakhanova EV, Walch AK, Selmansberger M, Samaga D, et al. 2018. A genomic copy number signature predicts radiation exposure in post-Chernobyl breast cancer. Int J Cancer. 143(6):1505–1515.
- Wilson MD, Durocher D. 2017. Reading chromatin signatures after DNA double-strand breaks. Philos Trans R Soc Lond B Biol Sci. 372:20160280.
- Wojcik A, Bouffler S, Hauptmann M, Rajaraman P. 2018. Considerations on the use of the terms radiosensitivity and radiosusceptibility. J Radiol Prot. 38(3):N25–N29.
- Woodbine L, Gennery AR, Jeggo PA. 2014. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair. 16:84–96.
- Xu S, Wang J, Ding N, Hu W, Zhang X, Wang B, Hua J, Wei W, Zhu Q. 2015. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 12(12):1355–1363.
- Yarnold J. 2019. Changes in radiotherapy fractionation-breast cancer. Br J Radiol. 92(1093):20170849. doi:10.1259/bjr.20170849.
- Yasuhara T, Kato R, Hagiwara Y, Shiotani B, Yamauchi M, Nakada S, Shibata A, Miyagawa K. 2018. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell. 175(2):558–570 e511.
- Zaheer U, Faheem M, Qadri I, Begum N, Yassine HM, Al Thani AA, Mathew S. 2019. Expression profile of MicroRNA: an emerging hallmark of cancer. Curr Pharm Des. 25(6):642–653.
- Zahra A, Fath M A, Opat E, Mapuskar K A, Bhatia S K, Ma D C, Iii S N. R, Snyders T P, Chenard C A, Eichenberger-Gilmore J M, et al. 2017. Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: the University of Iowa experience of two phase 1 clinical trials. Radiat Res. 187(6):743–754.
- Zaorsky NG, Shaikh T, Ruth K, Sharda P, Hayes SB, Sobczak ML, Hallman MA, Smaldone MC, Chen DY, Horwitz EM. 2017. Prostate cancer patients with unmanaged diabetes or receiving insulin experience inferior outcomes and toxicities after treatment with radiation therapy. Clin Genitourin Cancer. 15(2):326–335 e323.
- Zhang Y, Du Y, Le W, Wang K, Kieffer N, Zhang J. 2011. Redox control of the survival of healthy and diseased cells. Antioxid Redox Signal. 15(11):2867–2908.
- Zhang C, Peng G. 2015. Non-coding RNAs: an emerging player in DNA damage response. Mutat Res Rev Mutat Res. 763:202–211.
- Zhou D, Shao L, Spitz DR. 2014. Reactive oxygen species in normal and tumor stem cells. Adv Cancer Res. 122:1–67.
- Zonneveld MI, Keulers TGH, Rouschop K. 2019. Extracellular vesicles as transmitters of hypoxia tolerance in solid cancers. Cancers (Basel). 11(2):154.