
Abstract
Background
Adverse outcome pathways (AOPs) describe how a measurable sequence of key events, beginning from a molecular initiator, can lead to an adverse outcome of relevance to risk assessment. An AOP is modular by design, comprised of four main components: (1) a Molecular Initiating Event (MIE), (2) Key Events (KEs), (3) Key Event Relationships (KERs) and (4) an Adverse Outcome (AO).
Purpose
Here, we illustrate the utility of the AOP concept through a case example in the field of ionizing radiation, using the Organisation for Economic Cooperation and Development (OECD) Users' Handbook. This AOP defines a classic targeted response to a radiation insult with an AO of lung cancer that is relevant to radon gas exposure.
Materials and methods
To build this AOP, over 500 papers were reviewed and categorized based on the modified Bradford-Hill Criteria. Data-rich key events from the MIE, to several measurable KEs and KERs related to DNA damage response/repair were identified.
Results
The components for this AOP begin with direct deposition of energy as the MIE. Energy deposited into a cell can lead to multiple ionization events to targets such as DNA. This energy can damage DNA leading to double-strand breaks (DSBs) (KE1), this will initiate repair activation (KE2) in higher eukaryotes through mechanisms that are quick and efficient, but error-prone. If DSBs occur in regions of the DNA transcribing critical genes, then mutations (KE3) generated through faulty repair may alter the function of these genes or may cause chromosomal aberrations (KE4). This can impact cellular pathways such as cell growth, cell cycling and then cellular proliferation (KE5). This will form hyperplasia in lung cells, leading eventually to lung cancer (AO) induction and metastasis. The weight of evidence for the KERs was built using biological plausibility, incidence concordance, dose-response, time-response and essentiality studies. The uncertainties and inconsistencies surrounding this AOP are centered on dose-response relationships associated with dose, dose-rates and radiation quality.
Conclusion
Overall, the AOP framework provided an effective means to organize the scientific knowledge surrounding the KERs and identify those with strong dose-response relationships and those with inconsistencies. This case study is an example of how the AOP methodology can be applied to sources of radiation to help support areas of risk assessment.
Introduction
Decades of research have been conducted to understand the impact of environmental exposures on human health, producing vast amounts of data across many disciplines. Organizing these data in a meaningful way to support risk assessment and decision-making has been a significant challenge. To address this, the OECD formalized the adverse outcome pathway (AOP) framework to consolidate knowledge into measurable key events (KEs) which can be causally linked to disease (OECD Citation2018a). AOPs are hypothetical, modular representations of series of relationships linking chemical interactions with molecules in cells to phenotypic effects (Ankley et al. Citation2010). Thus, AOPs provide a structured approach to utilizing data from various test methods in order to inform how chemicals cause toxic effects. This is of benefit to regulatory decision-making and facilitates the identification of knowledge gaps for further focused research (Ankley et al. Citation2010).
Construction of an AOP involves identifying a molecular initiating event (MIE) and mapping out KEs that lead to the adverse outcome (AO) of interest to risk assessment. The KEs are sequential, measurable and linked through key event relationships (KERs), which define the structural and functional relationships (OECD Citation2018b). Each KE is necessary but not individually sufficient to explain an outcome. KERs are a critical element of the AOP. They are derived from compiling mechanistic understanding (biological knowledge) and empirical evidence that support the notion that the upstream KE is related through quantitative, qualitative or semi-quantitative evidence to the downstream KE. The quality of an AOP is highly dependent on the strength of underlying evidence that supports its KERs. AOPs are not stressor specific because this would otherwise hinder the power the pathway has in risk assessment (OECD Citation2018b). Therefore, the evidence to build an AOP can come from any type of stressor and is not limited to one stressor. The KEs and KERs of an AOP are build in a modular fashion; i.e. they are stand-alone units. This allows simple updating of independent modules as new data/tests are produced, and reuse of modules in other AOPs. A network of related AOPs can emerge that share modules, creating a powerful tool for prediction of AOs that has many potential applications in risk assessment.
The AOP concept as represented by the OECD framework is relatively new for the radiation community. Globally, more efforts are being put forth to bring awareness to the OECD AOP program. During a recent workshop held in Canada, the role of the AOP concept to support the needs of the radiation community was discussed (Chauhan et al. Citation2019). The workshop highlighted how AOPs can link biological endpoints to AOs to support risk-based decision making. This framework can also help identify knowledge gaps where evidence is lacking. By identifying knowledge gaps, topics of future research can be prioritized and designed to a higher degree of specificity. Due to the AOPs being posted online, they serve as a living document, evolving as knowledge becomes available and readily informing the design of future studies.
To consolidate knowledge surrounding the role of radiation stressors in adverse outcomes, we have developed an AOP using a case example of lung cancer which is relevant to radon gas exposure. Exposure to radon gas has been shown to be associated to lung cancer (Samet and Eradze Citation2000; Samet Citation2006). Following the energy deposition from radiation such as radon, we demonstrate how a sequential chain of key events related to the subsequent DNA damage and (mis)repair can directly lead to lung cancer. There are extensive data to show that the deposited energy from stressors such as radon gas can induce deleterious lesions to the DNA structure by delivering localized energy. This cytogenetic damage is an important biological effector to the downstream development of lung cancer. Using the guidance outlined in the OECD Users' Handbook, we aimed to develop an AOP centered on this widely understood KE, of DNA damage. Here we provide detailed information on adjacent KERs and associated empirical evidence in the form of the biological plausibility, quantitative and essentiality studies that formed the weight of evidence to support the components of this AOP.
Methods
OECD guidance
The AOP was developed using the criteria outlined in the OECD Users' Handbook Supplement and revised guidelines (OECD Citation2018a, Citation2018b), The complete AOP is detailed in the AOP-wiki (www.aopwiki.ca, AOP-272, 2019). This report specifically provides information on the empirical evidence that was used to support the KERs. Any additional information on the KEs and non-adjacent KERs can be found in the AOP-Wiki (www.aopwiki.ca, AOP-272, 2019).
Criteria for building AOP
A literature scan was conducted using various search engines (Google Scholar, PubMed, MEDLINE, and JSTOR) to identify studies investigating the relationship between radiation exposures and their association with the AO (lung cancer) from 1950–2018. The endpoints measured in these studies were used to inform the development of a simplified pathway from the MIE to the AO. The recommended practices featured in the AOP Users Handbook were applied in building this AOP as follows:
The AOP contained only one MIE (starting point) and AO (end-point).
The AOP was mapped in the most simplified and directed means to achieve the AO without the inclusion of biological noise (e.g. detailed metabolic pathway processes).
The evidence used to support the AOP was specific to the MIE and not to the stressor (i.e. data from stressors other than radiation could be used to provide evidence to support the KERs).
All of the proposed KEs were essential (but not necessarily sufficient on their own) to achieve the AO.
Empirical evidence used to support each of the KERs was in the form of dose-responses, incidence-response and temporal-response data.
Empirical evidence was also in the form of semi-quantitative data and qualitative data.
Inconsistencies and uncertainties in the KERs were described.
The domain of applicability for each of the KERs were outlined.
Criteria for identifying studies to support key event relationships
The information and data used to support the KERs were derived from comprehensive and authoritative reviews written by experts in the field and original, high-quality research articles on cell-lines, animals and humans. A summary of the terms and phrases used to conduct the literature search for the KEs (including the MIE and the AO) is shown in . The inclusion of key words such as biological mechanism, dose-response, temporal response, timeframe, knock-in, knock-out, feedback loop, and modulating factors were added to literature searches pertaining to the KERs.
Table 1. Summary of key words used in the literature search for each of the KEs (including the MIE and the AO).
Review articles describing decades of historical research served as a source of biological plausibility, lending confidence to the AOP. For original research, only studies where sufficient information relating to the radiation type used, dose, tissue, time-point, animal model, experimental procedures, and experimental results were considered to assess empirical data for the various components of the AOP. Each study was evaluated to ensure it met the quality control questions related to details on experimental protocols, exposure conditions, sufficient biological replicates, robust statistical approach and conclusions clearly presented. The types of studies that were used included knock-out, knock-in, inhibitor, dose-response, temporal-response and epidemiological. Data to support the KERs were predominantly from stressors that were ionizing radiation-specific; however, in certain cases chemical stressors were also used to support KERs, when evidence from a radiation insult was lacking. The AOP-Wiki was reviewed to identify existing KEs and KERs that could be leveraged to help support the development of this AOP. An AOP on alkylation of DNA leading to heritable mutations (AOP-15, 2019) was identified as containing relevant modules that could be expanded upon for our purposes.
Criteria to determine overall weight of evidence
The modified Bradford Hill criteria (Fedak et al. Citation2015) were used to assess the overall weight of evidence for this AOP. Briefly, the Bradford Hill criteria are based on nine criterion that includes: (1) strength of association, (2) consistency of response within populations, (3) specificity, (4) temporality (exposure must precede outcome), (5) evidence of dose-response relationship, (6) biological plausibility (epidemiology of an outcome must be related to biology), (7) coherence (biological changes at a molecular level can be observed at higher organizational level), (8) experiment (removal of the stressor declined the outcome) and lastly, analogy (if a strong association is made from one exposure type to a disease, the evidence required for other similar exposure types can be lowered). Among the nine criteria, biological plausibility is weighted the strongest, the remaining criteria can support the relationships and provide a means to identify knowledge gaps. By using these criteria each KER was given an overall assessment on the weight of evidence from strong, moderate to weak.
Results
Summary of the proposed AOP
This AOP focuses on a classic targeted response of energy directly deposited onto the DNA molecule. illustrates the adjacent and non-adjacent KEs and KERs that comprise the AOP. We note that the existing AOP on alkylation of DNA leading to heritable mutations (AOP-15, 2019) had three KEs that could be expanded upon for the purpose of supporting this AOP: DNA damage, inadequate repair, and increases in mutations.
Figure 1. The AOP of direct deposition of energy leading to DNA damage, culminating in the adverse outcome, lung cancer. Labeled in each box are key events (KEs), joined and labeled (in red) by key event relationships (KERs). Nonadjacent relationships are denoted by the dashed lines (not discussed here).
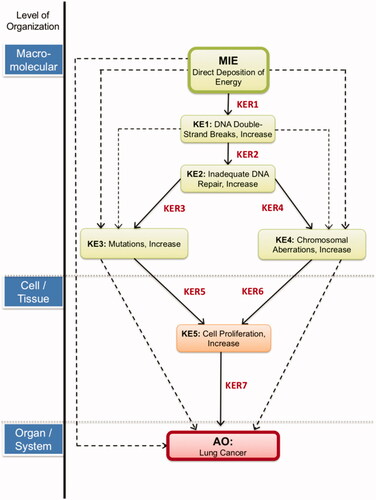
The MIE for this AOP was chosen to be the direct deposition of energy which is relevant to all radiation insults and distinguishable from chemical stressors. Direct energy deposited onto a cell can lead to multiple ionization events to targets such as DNA. The data convincingly show that DNA is a critical target. Radiation-induced energy deposition events interact with DNA strands with the possibility of inducing DNA damage in the form of strand breaks (KE1). There is an extensive array of lesions that can be induced including single strand breaks (SSBs), double strand breaks (DSBs), base damage, mismatches, DNA-DNA cross links and DNA-protein cross-links. DSBs are the most unstable and detrimental to the genome and will be the focus of this AOP. A cell will move to repair the DSBs through the activation of DNA DSB repair machinery. In higher eukaryotes, this can occur through many mechanisms but non-homologous end joining (NHEJ) is the preferred route for DSBs, particularly for noncycling cells in G1 phase of the cell cycle where no homologous chromosomes exist. The process is quick and efficient, but error prone (KE2). If DSBs occur in regions of the DNA transcribing critical genes, then mutations generated through faulty repair may alter the function of these genes (KE3) or may even cause chromosomal aberrations (KE4), resulting in genomic instability and a path toward cancer. This genomic instability will alter many gene products involved in several different cellular pathways such as cell growth, cell cycling, and apoptosis. With these alterations, cell proliferation will prosper (KE5) and form hyperplasia in lung epithelial cells, eventually, toward a path of lung cancer induction and the metastasis of the cancer (AO). Within our AOP-Wiki entry, full descriptions of how each of these KEs work and the methods used to measure them are described, and we direct the reader there for this information (AOP-272, 2019). In the following discussion, we describe the domain of applicability for the AOP and provide a detailed description of the evidence and knowledge supporting each of the KERs. Note that we present information relating to adjacent KERs; non-adjacent KERs are used in the full, aforementioned AOP-Wiki entry as they provide additional support for the overall AOP.
Domain of applicability
This AOP is relevant to all mammals and is independent of sex (Eymin and Gazzeri Citation2010; Barron et al. Citation2014; Kurgan et al. Citation2017). The pathway leading to the development of lung cancer often occurs during adulthood but may be applicable at earlier life stages (Liu et al. Citation2015) and is independent of sex. In humans, however, genetic abnormalities/mutations suggestive of lung cancer risk seem to be influenced by confounders such as smoking history, age, sex, ethnicity and genotype (Lim et al. Citation2009; Sanders and Albitar Citation2010; Kim et al. Citation2012; Paik et al. Citation2012; Lloyd et al. Citation2013; Cortot et al. Citation2014; Minina et al. Citation2017).
Molecular initiating event
The MIE represents the first critical physical interaction within the cell through the direct deposition of energy onto the DNA molecule. The word ‘direct’ is used to distinguish the MIE from instances of ‘indirect’ damage to the DNA from reactive species generation through early dispersion of radiolytic products. It was hypothesized that the distributions and/or type of DSBs may be different from a chemical insult and it was therefore important to differentiate this effect by including a MIE that was specific to radiation. As research surrounding the link between the deposition of energy and the induction of DSBs advances, more critical KEs between the MIE and DSBs could emerge, and it would become important to capture these in the AOP. Direct deposition of energy refers to events where subatomic particles or electromagnetic waves of sufficient energy cause direct ionization to genetic material through which they transverse (Kaczmarska et al. Citation2017). The energy of these subatomic particles or electromagnetic waves is dependent on the source and type of radiation. When this energy is deposited, it may cause the ejection of electrons from atoms and molecules, thereby breaking different types of chemical bonds and ionizing the atoms and molecules. Not all electromagnetic radiation is ionizing; the incident radiation must have sufficient energy to free electrons from the atom or the molecule’s electron orbitals. Many of the downstream biochemical effects of energy deposition are due to the initial geometry of the physical energy deposition event, known as the track structure (Desouky et al. Citation2015). Thus, the ionization produced is usually localized on a track. Energy deposition can affect cells at the molecular, tissue and organ level.
KER1: Direct deposition of energy (Mie) leading to DNA double strand breaks (KE1)
It is well established that ionizing radiation can cause damage to the DNA in the form of strand breaks through direct (focus of this AOP) and indirect mechanisms (reviewed in Lomax et al. Citation2013). A significant fraction of strand breaks can also be generated by the splitting of water molecules near DNA (indirect). This can result in a significant quantity of reactive oxygen species in the form of hydroxyl free radicals. These short-lived but highly reactive molecules can attack nearby DNA, producing single-strand DNA breaks; if several single-strand breaks are in close proximity to each other, they may spontaneously convert into a DSB (Ward Citation1988; Yamaguchi et al. Citation2005). DSBs are considered the most lethal and deleterious type of DNA damage. If DSBs are left unrepaired, they can drive the cell toward apoptosis or senescence. If they are misrepaired, the DSBs can lead to clastogenic lesions, resulting in genomic instability and potentially tumorigenesis through the activation of oncogenes or inactivation of tumor-suppressor genes (Raynard et al. Citation2017).
The most direct path to generate DSBs entails a collision between a high-energy particle or photon (e.g. X-rays and gamma-rays) and a strand of DNA. The high-energy subatomic particles interact with the orbital electrons of the DNA; this results in ionization and excitation (Joiner and Van Der Kogel Citation2009) ultimately breaking the phosphodiester backbone. Additionally, excitation of secondary electrons in the DNA allows for a cascade of ionization events to occur, which can lead to the formation of multiple damage sites (Joiner and Van Der Kogel Citation2009). High linear energy transfer (LET) radiation, such as alpha particle radiation, can deposit larger quantities of energy within a single track than low LET radiation, such as gamma-ray radiation. High LET radiation has a propensity to produce clustered damage, meaning that as the radiation transverses the media, it induces complex DNA damage comprised of closely spaced DNA lesions (Hada and Georgakilas Citation2008). The complexity and yield of clustered DNA damage increases with ionizing density (Ward Citation1988; Goodhead Citation2006).
The mechanisms of this KER (biological plausibility) are widely understood and there are many reviews available on this topic. Results from in vitro (Sutherland et al. Citation2000; Rogakou et al. Citation1999; de Lara et al. Citation2001; Rothkamm and Lobrich Citation2003; Kuhne et al. Citation2005; Sudprasert et al. Citation2006; Beels et al. Citation2009; Grudzenski et al. Citation2010; Antonelli et al. Citation2015; Shelke and Das Citation2015; Fernandez-Antoran et al. Citation2019), in vivo (Sutherland et al. Citation2000; Rube et al. Citation2008; Beels et al. Citation2009; Grudzenski et al. Citation2010), ex vivo (Rube et al. Citation2008; Flegal et al. Citation2015) and simulation studies that used biological experimental data (Charlton et al. Citation1989) demonstrate that there is a dose-dependent increase in DSBs with increasing deposition of energy across a wide range of ionizing radiation (iron ions, X-rays, ultrasoft X-rays, gamma-rays, photons, and alpha particles) and doses (1 mGy–100 Gy). There is also considerable evidence suggesting a time concordance. A number of different models have documented DSBs at 10–30 minutes after exposure to ionizing radiation (Rogakou et al. Citation1999; Rube et al. Citation2008; Kuefner et al. Citation2009; Beels et al. Citation2009, Grudzenski et al. Citation2010; Antonelli et al. Citation2015), with evidence of DSBs occurring as early as 3–5 minutes postirradiation (Rogakou et al. Citation1999; Rothkamm and Lobrich Citation2003; Rube et al. Citation2008; Grudzenski et al. Citation2010). In the majority of cases, the number of DSBs decline to near baseline levels by 24 hours after irradiation (Rothkamm and Lobrich Citation2003; Rube et al. Citation2008; Grudzenski et al. Citation2010; Antonelli et al. Citation2015), with the sharpest decrease documented within the first 5 hours (Rogakou et al. Citation1999; Rube et al. Citation2008; Kuefner et al. Citation2009; Grudzenski et al. Citation2010; Shelke and Das Citation2015). DSBs were found to be more persistent when they were induced by higher LET radiation (Antonelli et al. Citation2015). summarizes the quantitative evidence of the aforementioned studies for this KER. Reviews do not appear in the table.
Table 2. Summary of articles forming the empirical evidence (EE) for KER1 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
KER2: double-strand breaks (KE1) leading to inadequate repair (KE2)
The maintenance of DNA integrity is essential for genomic stability. As such, when DNA DSBs occur, the cell reacts by initiating repair machinery. To fix this potentially lethal damage, eukaryotic cells have evolved a variety of repair pathways that rely on homologous and illegitimate recombination, also called nonhomologous DNA end joining (NHEJ), which may induce small scale mutations and chromosomal aberrations (Pfeiffer et al. Citation2000). Homologous recombination (HR) is used by cells when there is a homologous piece of DNA. In mitotic cells NHEJ occurs throughout all phases of the cell cycle, whereas HR is largely restricted to the S and G2 phases when the sister chromatid is available to mediate the repair process (Raynard et al. Citation2017). The type of DSB repair pathway that activates can vary widely among species, between different cell types of a single species, and during different cell cycle phases (Shrivastav et al. Citation2008). NHEJ is initiated in G1 and early S phases of the cell cycle (Lieber et al. Citation2003) and is preferentially used to repair DSB damage (Godwin et al. Citation1994), as it is more rapid than HR (Iliakis Citation1991; Jeggo Citation1998; Mao et al. Citation2008). In higher-order eukaryotes such as humans, NHEJ is relied upon when homology of DNA is scarce or unavailable, as NHEJ is highly flexible and can deploy multiple enzymes to process the various types of DNA ends (Pannunzio et al. Citation2018). NHEJ can occur through one of two subtypes: canonical NHEJ (C-NHEJ) or alternative non-homologous end joining (alt-NHEJ). C-NHEJ, as the name suggests, simply ligates the broken ends back together. In contrast, alt-NHEJ occurs when one strand of the DNA on either side of the break is resected to repair the lesion (Betermier et al. Citation2014). Both DSB repair mechanisms are error-prone, meaning insertions and deletions are sometimes formed due to the DSBs being repaired imperfectly (Thurtle-Schmidt and Lo Citation2018). However, alt-NHEJ is considered more error-prone than C-NHEJ, as studies have shown that it more often leads to chromosomal aberrations (Zhu et al. Citation2002; Guirouilh-Barbat et al. Citation2007; Simsek and Jasin Citation2010). The roles of various DNA repair proteins in the context of DSBs are highlighted in reviews by Chang et al. (Citation2017) and van Gent et al. (Citation2001), with discussions focusing on the various effects of losing some of these proteins in cells, mice and humans (van Gent et al. Citation2001).
There is evidence in the literature of dose/incidence concordance between the occurrence of DSBs and the incidence of inadequate DNA repair upon exposure to radiation. DNA repair is tightly coupled to DSBs. Visually, immunofluorescence has demonstrated a co-localization of DNA repair proteins with DSB foci in response to a radiation stressor (Paull et al. Citation2000; Asaithamby and Chen Citation2009; Dong et al. Citation2017). In studies examining cellular responses to increasing doses of radiation, which is known to evoke DNA DSBs (Dikomey and Brammer Citation2000; Lobrich et al. Citation2000; Rothkamm and Lobrich Citation2003; Kuhne et al. Citation2005; Asaithamby and Chen Citation2009; Bracalente et al. Citation2013), there were resulting dose-dependent increases in DSB repair foci (Asaithamby and Chen Citation2009), non-repaired DSBs (Dikomey and Brammer Citation2000), DSB misrepair rates (McMahon et al. Citation2016), and misrejoined DSBs (Kuhne et al. Citation2005; Rydberg et al. Citation2005), as well as a dose-dependent decrease in the total DSB rejoining (Lobrich et al. Citation2000). Furthermore, only 50% of the rejoined DSBs were found to be correctly repaired after high doses (Lobrich et al. Citation2000). When an inhibitor of a DNA repair protein was added to cells prior to exposure to a radiation stressor, DNA repair foci were not formed post-irradiation (Paull et al. Citation2000), and there were significant increases in DSBs at 6 hours and 12 hours after the radiation treatment (Dong et al. Citation2017). Similarly, there have been several knock-out/knock-down studies in which cells lacking a DNA repair protein have been exposed to a radiation stressor. As a result, DSBs were found to persist in these cells longer than in the wild-type cells (Rothkamm and Lobrich Citation2003; Bracalente et al. Citation2013; McMahon et al. Citation2016; Dong et al. Citation2017), and there was an increase in incorrectly rejoined DSBs (Lobrich et al. Citation2000). In one striking example, a human cell line lacking DNA ligase IV had DSBs that were still present approximately 240–340 hours post-irradiation (McMahon et al. Citation2016).
Complete recovery or repair may require days or weeks and data suggests that, even at moderately high doses, if the dose rate is low enough or the time interval between doses is adequate, the DNA will be fully repaired. In general, it seems that most DSB repair occurs within the first 3–6 hours after irradiation, based on expression of DNA repair proteins (Asaithamby and Chen Citation2009; Dong et al. Citation2017) and the observed fraction of unrepaired DSBs (Rothkamm and Lobrich Citation2003; Pinto and Prise Citation2005). A summary of the studies contributing to this KER is shown in .
Table 3. Summary of articles forming the empirical evidence for KER2 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
KER3: inadequate repair (KE2) to mutations (KE3)
The preferred method for DSB repair is NHEJ in human somatic cells (Petrini et al. Citation1997; Mao et al. Citation2008). However, this mechanism is error-prone and has the potential to create mutations from DNA lesions (Little Citation2000). NHEJ is considered error-prone because it does not use a homologous template to repair the DSB. The NHEJ mechanism involves many proteins that work together to bridge the DSB gap by overlapping single-strand termini that are usually less than 10 nucleotides long (Anderson Citation1993; Getts and Stamato Citation1994; Rathmell and Chu Citation1994). Inherent in this process is the introduction of errors. These errors can be retained resulting in damaged DNA that is then used as a template during DNA replication. There is evidence in the literature suggesting a dose/incidence concordance between inadequate DNA repair and increases in mutation frequencies. In response to increasing doses from a radiation stressor, dose-dependent increases in both measures of inadequate DNA repair and mutation frequency have been found. In an analysis that combined results from several different studies conducted using in vitro cell-lines, the rate of DSB misrepair was revealed to increase in a dose-dependent fashion from 0 to 80 Gy, with the mutation rate also similarly increasing from 0–6 Gy (McMahon et al. Citation2016). Additionally, using a plant model, it was shown that increasing radiation dose from 0–10 Gy resulted in increased DNA damage as a consequence of inadequate repair. Mutations were observed 2–3 weeks post-irradiation (Ptacek et al. Citation2001). Moreover, increases in mutation densities were found in specific genomic regions of human cancer samples (namely promoter DNAse I-hypersensitive sites (DHS) and 100 bp upstream of transcription start sites (TSS)) that were also found to have decreased DNA repair rates attributable to inadequate nucleotide excision repair (NER) (Perera et al. Citation2016).
Interestingly, mutation rates have been shown to increase as the required DNA repair becomes more complex. Upon completion of DSB repair in response to radiation and treatment with restriction enzymes, more mutations were found in cases where the ends were non-complementary and thus required more complex DNA repair (1–4% error-free) relative to cases where ends were complementary (34–38% error-free) (Smith et al. Citation2001). Knock-out/knock-down studies suggest that there is a strong relationship between the adequacy of DNA repair and mutation frequency. In all examined cases, deficiencies in proteins involved in DNA repair resulted in altered mutation frequencies relative to wild-type cases. There were significant decreases in the frequency and accuracy of DNA repair in cell lines deficient in key proteins involved in DNA repair, including LIG4 (Smith et al. Citation2003) and Ku80 (Feldmann et al. Citation2000); rescue experiments performed with these two cell lines further confirmed that inadequate DNA repair proteins was the cause of the observed decreases in repair frequency and accuracy (Feldmann et al. Citation2000; Smith et al. Citation2003). In primary Nibrin-deficient mouse fibroblasts, there was increased spontaneous DNA damage relative to wild-type controls, suggestive of inadequate DNA repair. Using the corresponding Nibrin-deficient and wild-type mice, in vivo mutation frequencies were also found to be elevated in the Nibrin-deficient animals (Wessendorf et al. Citation2014). Furthermore, mutation densities were differentially affected in specific genomic regions in cancer patients depending on their XPC status. Specifically, mutation frequencies were increased in XPC-wild-type patients at DNase I-hypersensitive sites and 100 bp upstream of transcriptional start site relative to cancer patients lacking functional XPC (Perera et al. Citation2016). There is evidence in the literature suggesting a time concordance between the initiation of DNA repair and the occurrence of mutations. For simple ligation events, mutations were not evident until 12–24 hours, whereas DSB repair was evident at 6–12 hours. For complex ligation events, however, mutations and DSB repair were both evident at 12–24 hours. As the relative percent of DNA repair increased over time, the corresponding percent of error-free rejoining decreased over time in both ligation cases, suggesting that overall DNA repair fidelity decreases with time (Smith et al. Citation2001). A summary of the studies discussed for KER3 is provided in .
Table 4. Summary of articles forming the empirical evidence for KER3 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
KER4: inadequate repair (KE2) to chromosomal aberrations (KE4)
Chromosomal aberrations are physical changes in the structure of a chromosome that may or may not lead to downstream effects (reviewed in Nikjoo et al. Citation1999). They can be the result of terminal and interstitial deletions and inversions or rearrangements between two or more chromosomes. It is generally accepted that mechanisms involved in the repair of DSBs are highly correlated with the formation of chromosomal aberrations. Through NHEJ, the ends of chromosomes can be ligated together; this ligation may cause translocations (van Gent et al. Citation2001). If the ends of two different chromosomes are ligated, this has the potential to fuse genes, including genes that are critical to normal tissue function or oncogenes. It has also been observed that incorrect joining of the exchanged ends of two or more DSBs leads to a loss of the DNA sequences of varied size at the breakpoints (Povirk Citation2006). Characteristic hallmarks of NHEJ include the high frequency of small terminal deletions, the apparent splicing of DNA ends at micro-homologies, and gap-filling on aligned DSB ends (Povirk Citation2006). The most convincing support for this is derived from the analysis of chromosome instability syndromes, rare human autosomally inherited disorders which are associated with dramatically increased frequency of chromosomal aberrations. In these syndromes, components of NHEJ repair protein complexes have been shown to be mutated (Pfeiffer et al. Citation2000). Other phenomena, such as chromothripsis, also highlight the role of NHEJ in chromosomes being joined randomly through incorrect repair of DNA DSBs on a single chromosome (Leibowitz et al. Citation2015; Rode et al. Citation2016). There is also the possibility that some oncogenes can fuse due to incorrect repair of DSBs, which amplifies chromosome instability (Seki et al. Citation2015). Furthermore, this seemingly desperate ligation of DSB ends tends to lead to the pairing of two incompatible ends from different chromosomes, causing genomic rearrangements known as translocations (Wang-Michelitsch and Michelitsch Citation2007). In human cells, NHEJ has been shown to be directly responsible for the generation of translocations (Rothkamm et al. Citation2001; Weinstock et al. Citation2006; Lieber et al. Citation2010; Ghezraoui et al. Citation2014). Interestingly, in mice classical non-homologous end joining (C-NHEJ) has been shown to suppress translocations, suggesting that errors arise from alternative non-homologous end joining (alt-NHEJ) (Simsek and Jasin Citation2010). Despite these negative consequences, it has been suggested that from a biological perspective, it is more advantageous for the cell to rejoin DSBs and risk chromosomal aberrations rather than not ligating the DSBs at all; non-ligated DSBs are more toxic and could easily lead to cell death via apoptosis (Hoeijmakers Citation2001).
Studies have also shown that the loss of core NHEJ-associated proteins leads to an increased occurrence of chromosomal aberrations and translocations in human and rodent cells (Karanjawala et al. Citation1999; Rothkamm et al. Citation2001; Jeggo and Lobrich Citation2015). Such inhibitions or defects in DSB repair can increase the sensitivity of the cells to stressors such as ionizing radiation and chemical genotoxicants, as reviewed by Rassool and Tomkinson (Citation2010). Rejoining of DSBs in cells deficient in key components of the NHEJ complex (e.g. ligase 4 or X-ray cross-complementing protein 4) has shown an increase in the number of translocations (Ghezraoui et al. Citation2014). Evidence has also emerged demonstrating that deregulated NHEJ may promote carcinogenesis by increasing genomic instability due to inappropriate repair (reviewed in Lin et al. Citation2014 and Sishc and Davis Citation2017). Micronuclei and translocation formation have also been shown to increase when DSB repair is inhibited (Chernikova et al. Citation1999; McMahon et al. Citation2016). Treatment of human cells with methylating agents also has been shown to trigger a significant increase in chromosomal abnormalities due to an increased frequency of incorrect DSB repair (Galloway Citation1995). This trend was also observed with other DSB-inducing agents when specific cell cycle checkpoints were inhibited (Janssen et al. Citation2011). Active overexpression of core NHEJ factors may also lead to chromosomal instability (Sishc and Davis Citation2017). This can arise if DSBs occurring on genes that produce core NHEJ factors are improperly repaired, leading to mutations which can either inactivate or overexpress these genes. In many kinds of myeloid leukemias, large deletions were found to be associated with an increased activity of NHEJ proteins (Gaymes et al. Citation2002). It is thought that Ku, a key protein of DNA repair, drives the overactive expression of the NHEJ pathway. Thus aberrant Ku activity is a candidate mechanism for inducing chromosomal instability (Gaymes et al. Citation2002). A full summary of studies is provided in .
Table 5. Summary of articles forming the empirical evidence for KER4 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
KER5 and KER6: mutations (KE3)/chromosomal aberration (KE4) leading to cell proliferation (KE5)
Clastogenic lesions such as mutations and chromosome aberrations can occur in genes regulating cell cycling processes. If this occurs, it can lead to unnatural and potentially detrimental levels of increased cell proliferation. Persistent clastogenic lesions can induce cell proliferation through three different mechanisms (Vogelstein and Kinzler Citation2004). The first is through activation of proto-oncogenes via gain of function (GOF) mutations. This activation can occur through gene amplification or subtle intragenic mutations that alter regulatory sites within the gene, thereby placing these genes under the regulation of constitutively activated genes, such as MYC (Vogelstein and Kinzler Citation2004; Varella-Garcia Citation2009) that regulate cellular proliferation. The second mechanism involves the inactivation of tumor suppressor genes (TSGs) through a loss of function (LOF) mutation. These TSG inactivations usually occur due to epigenetic events such as methylation of promoter regions, missense mutations at residues essential for protein function, or point mutations resulting in the formation of truncated proteins (Vogelstein and Kinzler Citation2004). Unlike proto-oncogenes, which only require mutations in one copy of the gene to activate the oncogene, TSGs require mutations in both copies of the gene for inactivation (Vogelstein and Kinzler Citation2004). The final mechanism for induction of cell proliferation is the alteration of a stability gene, which directly or indirectly affects the activation status of proto-oncogenes or TSGs. When these stability genes, also known as caretakers, are mutated, they promote tumorigenesis in a manner that differs from that of oncogenes and TSGs. Genes involved in mismatch repair, nucleotide-excision repair, and base-excision repair, are prominent examples of stability genes. These stability genes are responsible for repairing subtle mistakes in critical genes made during normal DNA replication and through exposure to mutagens such as ionizing radiation or chemical agents (Vogelstein and Kinzler Citation2004). A few of the most important critical genes in terms of controlling cell proliferation are KRAS, EGFR, and TP53. Empirical data obtained from a number of direct sources strongly support the idea that an increase in mutations or chromosomal aberrations in these specific genes increases cell proliferation. The majority of the evidence has been derived from studies that have examined lung tissue from patients with lung cancer. The studies either used genotyping or knock-in and knock-out constructs that manipulate TP53, p53 proteins, or other genes and proteins that have an influence on p53 pathways. Thus, the evidence primarily takes the form of essentiality studies rather than dose, incidence and temporal concordance. For both KER5 and KER6, a summary of the studies and evidence to support mutations and chromosomal aberrations as indicators later cell proliferation is provided in and respectively.
Table 6. Summary of articles forming the empirical evidence for KER5 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
Table 7. Summary of articles forming the empirical evidence for KER6 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
Knock-in and knock-out chromosomal translocation studies that manipulate cells to become genetically unstable and oncogenic have shown unrestricted proliferation. The two most extensively studied translocations are (1) the juxtaposition of the MYC gene to sequences from one of three immunoglobulin loci in Burkitt’s lymphoma, mouse plasmacytoma, and rat immunocytoma (reviewed in both Cory Citation1986 and Mes-Masson and Witte Citation1987), and (2) the fusion of the c-ABL oncogene with the BCR sequence in Philadelphia chromosome-positive chronic myelogenous leukemia (reviewed in Mes-Masson and Witte Citation1987). In general, the MYC family of cellular oncogenes encodes transcription regulators that are associated with cell proliferation and death regulation (Gostissa et al. Citation2009). Translocations involving MYC genes thus ultimately lead to uncontrolled proliferation. The association between increased CAs and unrestrained cell growth is a common theme among a variety of tumorgenic pathways involving the liver, breasts, and lungs, as evidenced by empirical data from transgenic mouse studies (reviewed in both Fowlis and Balmain Citation1993 and Cory Citation1986).
KER7: cell proliferation (KE5) leading to lung cancer (AO)
Uncontrollable growth and division of cells can lead to hyperplasia and the formation of neoplasms, which will continue to grow and ultimately form tumors. As thoroughly discussed in a review by Eymin and Gazzeri (Citation2010), there are many different factors and signals that must be considered when examining tumorigenesis. A full description of this KER is provided in the AOPwiki, including components that occur in parallel to cell proliferation that are considered hallmarks of cancer (Boss et al. Citation2014). For example, balanced structural changes in genetic material (when the genetic material is exchanged evenly) are thought to initiate pathogenetic changes in cancer, while unbalanced structural changes (when genetic material is not exchanged evenly) may be causing secondary changes in cancer progression. Both of these processes involve uncontrollable cell proliferation (Mitelman Citation2005), which is the cornerstone of tumor development. Of note, is that many stimuli that are hyperplastic and toxic are not carcinogenic, (Hoel et al. Citation1988). Once cell proliferation accelerates and the abnormal growth pattern of cells continues, the formation of neoplasms ensues. As tumors develop and mature, they have the potential to become malignant and propagate throughout the body in a process known as metastasis. This relationship is still being studied. This uncontrollable and ultimately pathogenic cell proliferation may occur in any cells of the body; when these processes occur in cells specific to the lung, the result is lung cancer. There are multiple types of lung cancers that can be classified by histology. The two overarching types, non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), are very invasive diseases with high metastatic potential (Hirsch et al. Citation1982; Kelly and Bunn Citation1998). The progression of the premalignant lesions to these different lung cancer types differs slightly, but the general overall process is similar (Wistuba et al. Citation1999; Gradowski et al. Citation2007). A summary of the studies supporting a link between cell proliferation and lung cancer is provided in .
Table 8. Summary of articles forming the empirical evidence for KER7 which is derived using dose concordance (EE:DC), time scale (EE:TS) and essentiality studies (Ess).
Overall assessment of the AOP
A full detailed summary of the overall assessment of the AOP, including inconsistencies is provided in the AOP-wiki (https://aopwiki.org/aops/272), and the reader is directed there for further details. Overall, decades of scientific radio-biological research provide data to support a strong biological plausibility for the KERs outlined in this AOP. Research has shown that radiation induced carcinogenesis can evolve from clastogenic lesions in the form of chromosomal aberrations and mutations leading to cell differentiation/proliferation and eventually neoplastic transformation. Through construction of this AOP, KERs that are less supported than others were identified (). A general trend was identified: there was more support of upstream KERs (e.g. KER1, KER2 and KER3) over downstream KERs (e.g. KER5, KER6 and KER7). These downstream KERs are, by their nature, more difficult to assess given the need for well-designed long-term studies to characterize them, limiting the amount of available data. Indeed, in the case of KER7 (increased cell proliferation leading to lung cancer) this time period can extend beyond several decades. The distribution of support between biological plausibility and empirical evidence for each of the KERs is also highlighted and shows that in the case of KER3 and KER6 there is an apparent deficiency of empirical evidence necessary to complement the respective level of biological plausibility. As such, the AOP framework informs us that this particular AOP would benefit from further study of inadequate repair leading to mutations (KER3) and the impact of varying levels of chromosomal aberrations on cell proliferation (KER6).
Figure 2. Bar chart illustrating the number of articles contributing to the support metrics: biological plausibility and empirical evidence for each adjacent KER in the AOP presented here.
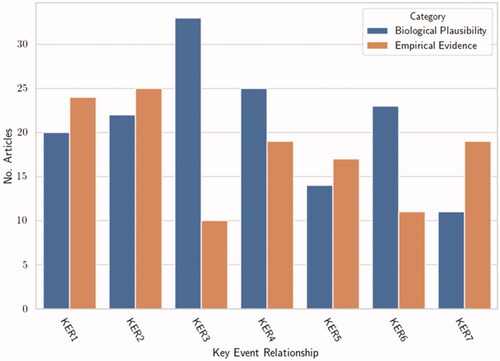
There was no single study that provided evidence to support all the KERs. For several of the KERs, there were limited data to directly demonstrate the impact of KEupstream to KEdownstream. This was particularly evident for inadequate repair leading to the generation of chromosomal aberrations and mutations leading to cell proliferation. Essentiality studies, using knock out/in and inhibitors were generally moderate/weak in level across the KERs. The majority of the upstream KERs had sufficient data to show dose-response concordance, however studies to strongly support time- and incidence-concordance were generally lacking and provided inconsistent results. In addition, throughout the KERs, discrepancies in data to support the KERs were evident, particularly for studies using low doses. There was also a lack of consistent studies that allowed for a quantitative understanding of this AOP, highlighting a clear knowledge gap worth exploring through directed research initiatives.
Discussion
Considerable mechanistic dose–response data have been generated in the radiation field, particularly in the area of clastogenic lesions. These data have been compiled and captured in an AOP to the most simplified, modular path to lung cancer from a molecular initiating event of deposited energy on DNA. This AOP is supported through KERs for which there is biological plausibility and available empirical evidence. Although it is clear that our proposed AOP is not the only route to the AO, it does represent a classic targeted response of radiation insult on a cell. The empirical evidence to support this pathway is strong and probabilistic. As per AOP conventions, the pathway does not describe every mechanism and alteration that is ultimately involved in radiation-associated carcinogenesis. Instead, KEs that are routinely measured using modern and conventional assays are described. For this reason, not all of the KEs that are hallmarks of cancer, that is, evasion, angiogenesis etc. are mapped out, but as they are critical events they can be developed separately. This AOP will be the first to use a MIE that is radiation-specific and therefore can act as a foundational AOP to build networks of radiation-specific responses. Networks can evolve to multiple AOs with additional KEs that incorporate nontargeted effects, immune and adaptive responses, in parallel.
While this AOP is applicable to other types of radiation-induced cancers, lung cancer was selected as the AO due to its relevance to radon risk assessment and its broader applicability to the chemical field. Lung cancer is a major public health problem world-wide, killing an estimated 1.5 million people annually (https://www.wcrf.org/dietandcancer/cancer-trends/worldwide-cancer-data). Although smoking is the leading cause of lung cancer, numerous environmental sources are also important contributors including radon, asbestos, air pollution and arsenic (Hubaux et al. Citation2012). Some of these stressors can act synergistically to increase risk, particularly among smokers. It has been shown that the histological lung profile of individuals that are smokers is quite different from nonsmokers exposed to high radon levels (Egawa et al. Citation2012). This is in part due to the complexity of each stressor, in terms of its interaction with cells at the molecular level. The AOP concept may help better understand the mode of action of different stressors at the cellular level that have a common adverse outcome.
Despite the decades of research in the area of radiation and DNA damage, a major challenge in developing this AOP was in finding the required components (i.e. essentiality, temporal, incidence and dose concordance) to provide strong empirical evidence to help support the KERs. Across all KERs, studies were lacking that used a broad dose-range. Most studies conducted analysis at one time-point and there were limited studies that supported the essentiality criteria. This was particularly evident for the KERs of inadequate repair to mutations/CA and mutations/CA to cellular proliferation. The non-adjacent KERs (i.e. DDoE to CA or DDoE to mutations), were generally more well supported. Furthermore, no single study encompassed all the KERs proposed in this AOP. In addition, there were considerable discordant results across KEs simply due to the MIE as its outcome is dependent on factors such as cell type, dose, dose-rate, and radiation quality. These factors can influence the amount and type of damage, which in turn can affect the probability to drive a path forward to cancer. The principle knowledge gap arose from the lack of data in the form of essentiality studies, using inhibitors and knock-in/out genes. As well for a number of KERs, there were minimal dose-response and temporal response data in well-conducted animal studies. There is also a range of uncertainty on how confounders such as lifestyle, health status, and radiosensitivities affect an individual’s path to an AO. Additional KEs may need to be added in parallel as our knowledge in these areas improves. These challenges can drive research priorities in the future.
An overall assessment of this AOP shows that there is strong biological plausibility and moderate empirical evidence to suggest a qualitative link between deposition of energy on DNA to the final AO of lung cancer. This evidence has been derived predominately from decades of research using laboratory studies and through mathematical simulations of cell-based models. These studies have shown both dose- and temporal-response relationships for select KEs. The quantitative thresholds to initiate each of the KEs have been shown to vary with factors such as the cell type, dose rate of exposure and radiation quality. Thus, an absolute amount of deposited energy (MIE) to drive a KE forward to a path of cancer is not yet definable. This is particularly relevant to low doses and low dose-rates of radiation exposure where the biology is interplayed with conflicting concepts of hormesis, hypersensitivity and the linear no threshold theory. Furthermore, due to the stochastic nature of the MIE, it remains difficult to identify specific threshold values of DSBs needed to overwhelm the DNA repair machinery to cause ‘inadequate’ DNA repair leading to downstream genetic abnormalities and eventually cancer. With a radiation stressor, a single hit to the DNA molecule could drive a path forward to lung cancer; however, this is with low probability. Conversely, at much higher doses, a cell will induce apoptosis and may not be driven to cancer induction. Although empirical modeling of cancer probability vs. mean radiation dose and time to lethality, does provide a good visualization of the effective thresholds (Raabe Citation2011), practically, there is still considerable uncertainty surrounding the connection of biologically contingent observations and stochastic energy deposition. Future work may focus on developing more precise quantitative and predictive models to help address these types of uncertainties.
This foundational AOP will initiate the building of networks and feedback loops that will provide further evidence linking the essential events to lung cancer, including genome alterations, oxidative stress, and metabolomics effectors. This will require efforts from the larger radiation community. As the empirical evidence to support these areas becomes stronger, a better representation of events to lung cancer will emerge. By identifying uncertainties and inconsistencies in the literature, research can be directed to address knowledge gaps, which can later help refine the pathway. It is our goal, with this AOP to motivate radiation researchers to use this framework for bringing together research data, exchanging knowledge and identifying research priority areas in the low-dose ionizing radiation field. Long-term, this AOP alongside others in the radiation field will help to identify key events common to chemical stressors and multiple adverse outcomes, which will be important to help refine risk assessment. In all, by building more radiation-relevant AOPs, the AOP framework will have a bigger role in supporting the practice of radiation protection.
| Abbreviations | ||
| AOPs | = | Adverse outcome pathways |
| MIE | = | Molecular Initiating Event |
| KEs | = | Key Events |
| KERs | = | Key Event Relationships |
| AO | = | Adverse Outcome |
| OECD | = | Organisation for Economic Cooperation and Development |
| NHEJ | = | non-homologous end-joining |
| HR | = | homologous recombination |
| alt-EJ | = | alternative end joining |
| DSBs | = | double strand breaks |
| CNV | = | copy number variants |
| LET | = | linear energy transfer |
| TSG | = | tumor suppressor genes |
| LOF | = | loss of function |
Acknowledgments
The authors would like to acknowledge Ruth Wilkins, Sami Qutob and Daniel Beaton for critical review and Baki Sadi, Fatemeh Nabavi, Sandra Noble, Nadine Adam, and Deepti Bijlani for reference checking and final editing of the AOP.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Notes on contributors
Vinita Chauhan
Vinita Chauhan is a research scientist at Health Canada.
Samantha Sherman
Samantha Sherman was a research assistant from the University of Ottawa.
Zakaria Said
Zakaria Said was a fourth year Honor student from the University of Ottawa.
Carole L. Yauk
Carole L. Yauk is a research scientist at Health Canada.
Robert Stainforth
Robert Stainforth was a post-doctoral fellow at Health Canada.
Related Research Data
References
- Amundson SA, Chen DJ. 1996. Ionizing radiation-induced mutation of human cells with different DNA repair capacities. Adv Space Res. 18(1–2):119–26.
- Anderson CW. 1993. DNA damage and the DNA-activated protein kinase. Trends Biochem Sci. 18(11):433–437.
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, et al. 2010. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 29(3):730–741.
- Antonelli AF, Campa A, Esposito G, Giardullo P, Belli M, Dini V, Meschini S, Simone G, Sorrentino E, Gerardi S, et al. 2015. Induction and repair of DNA DSB as revealed by H2AX phosphorylation foci in human fibroblasts exposed to low- and high-LET radiation : relationship with early and delayed reproductive cell death induction and repair of DNA DSB as revealed by H2AX phosphor. Radiat Res. 183(4):417–431.
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. 2013. Copy number variants are produced in response to low-dose ionizing radiation in cultured cells. Envrion Mol Mutagen. 55(2):103–113.
- Asaithamby A, Chen DJ. 2009. Cellular responses to DNA double-strand breaks after low-dose c -irradiation. Nucleic Acids Res. 37(12):3912–3923.
- Balajee AS, Bertucci A, Taveras M, Brenner D. 2014. Multicolour FISH analysis of ionising radiation-induced micronucleus formation in human lymphocytes. Mutagenesis. 29(6):447–455.
- Barron CC, Moore J, Tsakiridis T, Pickering G, Tsiani E. 2014. Inhibition of human lung cancer cell proliferation and survival by wine. Cancer Cell Int. 14(1):6.
- Beels L, Bacher K, De Wolf D, Werbrouck J, Thierens H. 2009. Gamma-H2AX foci as a biomarker for patient X-ray exposure in pediatric cardiac catheterization: are we underestimating radiation risks? Circulation. 120(19): 1903–1909.
- Betermier M, Bertrand P, Lopez BS. 2014. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 10(1):e1004086.
- Boss MK, Bristow R, Dewhirst MW. 2014. Linking the history of radiation biology to the hallmarks of cancer. Radiat Res. 181(6):561–577.
- Bracalente C, Ibañez IL, Molinari B, Palmieri M, Kreiner A, Valda A, Davidson J, Durán H. 2013. Induction and persistence of large g H2AX foci by high linear energy transfer radiation in DNA-dependent protein kinase e deficient cells. Int J Radiat Oncol Biol Phys. 87(4):785–794.
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double – strand break repair. Nat Rev Mol Cell Biol. 18(8):495–506.
- Charlton DE, Nikjoo HH, Humm JL. 1989. Calculation of initial yields of single- and double-strand breaks in cell nuclei from electrons, protons and alpha particles. Int J Radiat Biol. 56(1):1–19.
- Chauhan V, Said Z, Daka J, Sadi B, Bijlani D, Marchetti F, Beaton D, Gaw A, Li C, Burtt J, et al. 2019. Is there a role for the adverse outcome pathway framework to support radiation protection? Int J Radiat Biol. 95(2):225–232.
- Chernikova SB, Wells RL, Elkind MM. 1999. Wortmannin sensitizes mammalian cells to radiation by inhibiting the dna-dependent protein kinase-mediated rejoining of double-strand breaks. Radiat Res. 151(2):159–166.
- Cornforth M, Bedford J. 1985. On the nature of a defect in cells from individuals with ataxia-telangiectasia. Science. 227(4694):1589–1591.
- Cortot AB, Younes M, Martel-Planche G, Guibert B, Isaac S, Souquet PJ, Commo F, Girard P, Fouret P, Brambilla E, et al. 2014. Mutation of TP53 and alteration of p14(arf) expression in EGFR- and KRAS-mutated lung adenocarcinomas. Clin Lung Cancer. 15(2):124–130.
- Cory S. 1986. Activation of cellular oncogenes in hemopoietic cells by chromosome Translocation Review. Adv Cancer Res. 47:189–234.
- de Lara CM, Hill MA, Jenner TJ, Papworth D, O'Neill P. 2001. Dependence of the yield of DNA double-strand breaks in Chinese hamster V79-4 cells on the photon energy of ultrasoft X rays. Radiat Res. 155(3):440–448.
- Desouky O, Ding N, Zhou G. 2015. Targeted and non-targeted effects of ionizing radiation. J Radiat Res Appl Sci. 8(2):247–254.
- Dikomey E, Brammer I. 2000. Relationship between cellular radiosensitivity and non-repaired double-strand breaks studied for di Ú erent growth states, dose rates and plating conditions in a normal human broblast line. Int J Radiat Biol. 76(6):773–781.
- Dong J, Zhang T, Ren Y, Wang Z, Ling CC, He F, Li GC, Wang C, Wen B. 2017. Inhibiting DNA-PKcs in a non-homologous end-joining pathway in response to DNA double-strand breaks. Oncotarget. 8(14):22662–22673.
- Duan W, Gao L, Jin D, Otterson GA, Villalona-Calero MA. 2008. Lung specific expression of a human mutant p53 affects cell proliferation in transgenic mice. Transgenic Res. 17(3):355–366.
- Eymin B, Gazzeri S. 2010. Role of cell cycle regulators in lung carcinogenesis. Cell Adhes Migrations. 4(1):114–123.
- Egawa H, Furukawa K, Preston D, Funamoto S, Yonehara S, Matsuo T, Tokuoka S, Suyama A, Ozasa K, Kodama K, et al. 2012. Radiation and smoking effects on lung cancer incidence by histological types among atomic bomb survivors. Radiat Res. 178(3):191–201.
- Fedak KM, Bernal A, Capshaw ZA, Gross S. 2015. Applying the Bradford Hill criteria in the 21st century: how data integration has changed causal inference in molecular epidemiology. Emerg Themes Epidemiol. 12(1):14.
- Feldmann E, Schmiemann V, Goedecke W, Reichenberger S, Pfeiffer P. 2000. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells : implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res. 28(13):2585–2596.
- Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, Jones PH. 2019. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell. 25(3):329–341.e6.
- Flegal M, Blimkie MS, Wyatt H, Bugden M, Surette J, Klokov D. 2015. Measuring DNA damage and repair in mouse splenocytes after chronic in vivo exposure to very low doses of beta- and gamma-radiation. J Vis Exp. 3:e52912.
- Fowlis DJ, Balmain A. 1993. Oncogenes and tumour suppressor genes in transgenic mouse models of neoplasia. Eur J Cancer. 29(4):638–645.
- Galloway MS. 1995. A role for mismatch repair in production of chromosome aberrations by methylating agents in human cells. Mutat Res. 346:231–245.
- Gaymes TJ, Mufti GJ, Rassool FV. 2002. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 62(10):2791–2797.
- Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J, Foley C, Dong B, Zhang L, Kown O-J, et al. 2017. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 36:4767–4777.
- Getts RC, Stamato TD. 1994. Absence of a Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J Biol Chem. 269(23):15981–15984.
- George KA, Hada M, Jackson LJ, Elliott T, Kawata T, Pluth JM, Cucinotta FA. 2009. Dose response of gamma rays and iron nuclei for induction of chromosomal aberrations in normal and repair-deficient cell lines. Radiat Res. 171(6):752–763.
- Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA, Giovannangeli C, et al. 2014. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 55(6):829–842.
- Godwin AR, Bollag RJ, Christie DM, Liskay RM. 1994. Spontaneous and restriction enzyme-induced chromosomal recombination in mammalian cells. Proc Natl Acad Sci. 91(26):12554–12558.
- Goodhead DT. 2006. Energy deposition stochastics and track structure: what about the target? Radiat Prot Dosimetry. 122(1–4):3–15.
- Gostissa M, Ranganath S, Bianco JM, Alt FW. 2009. Chromosomal location targets different MYC family gene members for oncogenic translocations. Proc Natl Acad Sci USA. 106(7):2265–2270.
- Gradowski JF, Mantha GS, Hunt JL, Dacic S. 2007. Molecular alterations in atypical adenomatous hyperplasia occurring in benign and cancer-bearing lungs. Diagn Mol Pathol. 16(2):87–90.
- Grudzenski S, Raths A, Conrad S, Rube CE, Lobrich M. 2010. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc Natl Acad Sci USA. 107(32):14205–14210.
- Guamerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, Lo-Coco F, Tay Y, Beck AH, Pandolfi PP. 2016. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell. 165(2):289–302.
- Guirouilh-Barbat J, Rass E, Plo I, Bertrand P, Lopez BS. 2007. Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc Natl Acad Sci USA. 104(52):20902–20907.
- Hada M, Georgakilas AG. 2008. Formation of clustered DNA damage after high-LET irradiation: a review. J Radiat Res. 49(3):203–210.
- Hirsch FA, Paulson OB, Hansen HH, Vraa-Jensen J. 1982. Intracranial metastases in small cell carcinoma of the lung: correlation of clinical and autopsy findings. Cancer. 50(11):2433–2437.
- Hoeijmakers JH. 2001. Genome maintenance mechanisms for preventing cancer. Nature. 411(6835):366–374.
- Hoel DG, Haseman JK, Hogan MD, Huff J, McConnell EE. 1988. The impact of toxicity on carcinogenicity studies: implications for risk assessment. Carcinogenesis. 9(11):2045–2052.
- Hubaux R, Becker-Santos DD, Enfield KS, Lam S, Lam WL, Martinez VD. 2012. Arsenic, asbestos and radon: emerging players in lung tumorigenesis. Environ Health. 11(1):89.
- Hundley JE, Koester SK, Troyer DA, Hilsenbeck SG, Subler MA, Windle JJ. 1997. Increased tumor proliferation and genomic instability without decreased apoptosis in MMTV-ras mice deficient in p53. Mol Cell Biol. 17(2):723–731.
- Iliakis G. 1991. The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells. BioEssays. 13:641–648.
- Irwin ME, Nelson LD, Santiago-O’Farrill JM, Knouse PD, Miller CP, Palla SL, Siwak DR, Mills GB, Estrov Z, Li S, et al. 2013. Small molecule ErbB inhibitors decrease proliferative signalling and promote apoptosis in philadelphia chromosome-positive acute lymphoblastic leukemia. PLOS One. 8(8):e70608.
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. 2011. Chromosomal segregation errors cause of DNA damage and structural chromosomal aberrations. Science. 333(6051):1895–1898.
- Jarvis EM, Kirk JA, Clarke CL. 1998. Loss of nuclear BRCA1 expression in breast cancers is associated with a highly proliferative tumor phenotype. Cancer Genet Cytogenet. 101(2):109–115.
- Jeggo P. A, Lobrich M. 2015. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem J. 471(1):1–11.
- Jeggo PA. 1998. 5 DNA breakage and repair. Adv Gen. 38:185–218.
- Joiner MC, Van Der Kogel A. 2009. Basic clinical radiobiology. 4th ed. London, UK: Hodder Arnold.
- Kaczmarska M, Żydek D, Wilkłacz-Potoczny J, Fornal M, Grodzicki T, Kochowska E, Kozak K, Gocal Ł, Pohorecki W, Matlak K, et al. 2017. The influence of very small doses of alpha radiation on the stability of erythrocytes. Microsc Res Tech. 80(1):131–143.
- Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. 1999. The non-homologous DNA end joining pathway is important for chromosome stability in primary fibrobalasts. Curr Biol. 9(24):1501–1504.
- Kassie F, Matise I, Negia M, Upadhyaya P, Hecht SS. 2008. Dose-dependent inhibition of tobacco smoke carcinogen-induced lung tumorigenesis in A/J mice by indole-3-carbinol. Cancer Prev Res (Phila). 1(7):568–576.
- Kelly K, Bunn PA. Jr. 1998. Is it time to reevaluate our approach to the treatment of brain metastases in patients with non-small cell lung cancer? Lung Cancer. 20(2):85–91.
- Kim HR, Shim HS, Chung JH, Lee YJ, Hong YK, Rha SY, Kim SH, Ha SJ, Kim SK, Chung KY, et al. 2012. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 118(3):729–739.
- Kuefner MA, Grudzenski S, Schwab SA, Wiederseiner M, Heckmann M, Bautz W, Lobrich M, Uder M. 2009. DNA double-strand breaks and their repair in blood lymphocytes of patients undergoing angiographic procedures. Investigat Radiol. 44(8):440–446.
- Kuhne M, Urban G, Frankenberg D, Löbrich M. 2005. DNA double-strand break misrejoining after exposure of primary human fibroblasts to CK characteristic X rays, 29 kVp X rays and 60Co g rays. Radiat Res. 164:669–676.
- Kurgan N, Tsakiridis E, Kouvelioti R, Moore J, Klentrou P, Tsiani E. 2017. Inhibition of human lung cancer cell proliferation and survival by post-exercise serum is associated with the inhibition of Akt, mTOR, p70 S6K, and Erk1/2. Cancers. 9(5)pii:E46.
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 119(6):861–872.
- Leibowitz ML, Zhang C, Pellman D. 2015. Chromothripsis: a new mechanism for rapid karyotype evolution. Annu Rev Genet. 49:183–211.
- Li H, Ma XY, Wang J, Koontz J, Nucci M, Sklar J. 2007. Effects of rearrangement and allelic exclusion of JJAZ1/SUZ12 on cell proliferation and survival. PNAS. 104(50):20001–20006.
- Li Z, Xiong Y. 2017. Cytoplasmic E3 ubiquitin ligase CUL9 controls proliferation, senescence, apoptosis and genome integrity through p53. Oncogene. 36(36):5212–5218.
- Lieber MR, Ma Y, Pannicke U, Schwarz K. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 4(9):712–720.
- Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. 2010. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem. 50:279–296.
- Lim EH, Zhang SL, Li JL, Yap WS, Howe TC, Tan BP, Lee YS, Wong D, Khoo KL, Seto KY, et al. 2009. Using whole genome amplification (WGA) of low-volume biopsies to assess the prognostic role of EGFR, KRAS, p53, and CMET mutations in advanced-stage non-small cell lung cancer (NSCLC). J Thorac Oncol. 4(1):12–21.
- Lin YF, Nagasawa H, Little JB, Kato TA, Shih HY, Xie XJ, Wilson Jr PF, Brogan JR, Kurimasa A, Chen DJ, et al. 2014. Differential radiosensitivity phenotypes of DNA-PKcs mutations affecting NHEJ and HRR systems following irradiation with gamma-rays or very low fluences of alpha particles. PLoS One. 9(4):e93579.
- Little JB. 2000. Radiation carcinogenesis. J Toxicol Environ Health. 6(5–6):51–56.
- Liu M, Cai X, Yu W, Lv C, Fu X. 2015. Clinical significance of age at diagnosis among young non-small cell lung cancer patients under 40 years old: a population-based study. Oncotarget. 6(42):44963–44970.
- Lloyd SM, Lopez M, El-Zein R. 2013. Cytokinesis-blocked micronucleus cytome assay and spectral karyotyping as methods for identifying chromosome damage in a lung cancer case-control population. Genes Chromosomes Cancer. 52(7):694–707.
- Lobrich M, Kühne M, Wetzel J, Rothkamm K. 2000. Joining of correct and incorrect DNA double-strand break ends in normal human and ataxia telangiectasia fibroblasts. Genes Chromosomes Cancer. 68:59–68.
- Lomax ME, Folkes LK, O'Neill P. 2013. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol. 25(10):578–585.
- Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. 2012. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLOS One. 7(4):e35065.
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 7(18):2902–2906.
- Mcmahon SJ, Scheumann J, Paganetti H, Prise KM. 2016. Mechanistic modelling of DNA repair and cellular survival following radiation-induced DNA damage. Sci Rep. 6:33290.
- Mes-Masson A, Witte N. O. 1987. Role of the abl oncogene in chronic myelogenous leukemia. Adv Cancer Res. 49:53–74.
- Minina VI, Soboleva OA, Glushkov AN, Voronina EN, Sokolova EA, Bakanova ML, Savchenko YA, Ryzhkova AV, Titov RA, Druzhinin VG, et al. 2017. Polymorphisms of GSTM1, GSTT1, GSTP1 genes and chromosomal aberrations in lung cancer patients. J Cancer Res Clin Oncol. 143(11):2235–2243.
- Mitelman F. 2005. Cancer cytogenetics. Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2:342–346.
- Mosconi M, Giesen U, Langner F, Mielke C, Dalla Rosa I, Dirks WG. 2011. 53BP1 and MDC1 foci formation in HT-1080 cells for low- and high-LET microbeam irradiations. Radiat Envrion Biophys. 50(3):345–352.
- Nikjoo H, Munson JR, Bridges AB. 1999. RBE-LET relationships in mutagenesis by ionizing radiation. J Radiat Res. 40(Suppl):85–105.
- OECD. 2018a. Revised guidance document on developing and assessing adverse outcome pathways. Available from: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO%282013%296&doclanguage=en.
- OECD 2018b. Users' Handbook supplement to the Guidance Document for developing and assessing Adverse Outcome Pathways, OECD Series on Adverse Outcome Pathways. Paris: OECD Publishing.
- Paik PK, Johnson ML, D'Angelo SP, Sima CS, Ang D, Dogan S, Miller VA, Ladanyi M, Kris MG, Riely GJ, et al. 2012. Driver mutations determine survival in smokers and never-smokers with stage IIIB/IV lung adenocarcinomas. Cancer. 118(23):5840–5847.
- Pal HC, Sharma S, Strickland LR, Agarwal J, Athar M, Elmets CA, Afaq F. 2013. Delphinidin reduces cell proliferation and induces apoptosis of non-small-cell lung cancer cells by targeting EGFR/VEGFR2 signalling pathways. PLOS One. 8(10):e77270.
- Pannunzio NR, Watanabe G, Lieber MR. 2018. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. 293(27):10512–10523.
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 10(15):886–895.
- Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. 1998. Involvement of Brca2 in DNA repair. Mol Cell. 1(3):347–357.
- Perera D, Poulos RC, Shah A, Beck D, Pimanda JE, Wong JW. 2016. Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature. 532(7598):259–263.
- Petrini JHJ, Bressan DA, Yao MS. 1997. The RAD52 epistasis group in mammalian double strand break repair. Semin Immunol. 9(3):181–188.
- Pfeiffer P, Goedecke W, Obe G. 2000. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Annu Rev Genet. 47:433–455.
- Pinto M, Prise K. 2005. Evidence for complexity at the nanometer scale of radiation-induced DNA DSBs as a determinant of rejoining kinetics evidence for complexity at the nanometer scale of radiation-induced DNA DSBs as a determinant of rejoining kinetics. Radiat Res. 164:73–85.
- Povirk LF. 2006. Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair. 5(9-10):1199–1212.
- Ptacek O, Stavreva DA, Kim JK, Gichner T. 2001. Induction and repair of DNA damage as measured by the Comet assay and the yield of somatic mutations in gamma-irradiated tobacco seedlings. Mutat Res. 491(1–2):17–23.
- Raabe OG. 2011. Toward improved ionizing radiation safety standards. Health Phys. 101(1):84–93.
- Rassool FV, Tomkinson AE. 2010. Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci. 67(21):3699–3710.
- Rathmell WK, Chu G. 1994. Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc Natl Acad Sci. 91(16):7623–7627.
- Raynard S, Niu H, Sung P. 2017. DNA double-strand break processing: the beginning of the end. Genes Dev. 22:2903–2907.
- Rode A, Maass KK, Willmund KV, Lichter P, Ernst A. 2016. Chromothripsis in cancer cells: an update. Int J Cancer. 138(10):2322–2333.
- Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 146(5):905–916.
- Rothkamm K, Kühne M, Jeggo PA, Löbrich M. 2001. Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks. Cancer Res. 61(10):3886–3893.
- Rothkamm K, Lobrich M. 2003. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 100(9):5057–5062.
- Rube CE, Grudzenski S, Kühne M, Dong X, Rief N, Löbrich M, Rübe C. 2008. Cancer therapy : preclinical DNA double-strand break repair of blood lymphocytes and normal tissues analysed in a preclinical mouse model : implications for radiosensitivity testing. Clin Cancer Res. 14(20):6546–6556.
- Rydberg B, Cooper B, Cooper PK, Holley WR, Chatterjee A. 2005. Dose-dependent misrejoining of radiation-induced DNA double-strand breaks in human fibroblasts : experimental and theoretical study for high- and low-LET radiation. Radiat Res. 534:526–534.
- Samet JM, Eradze GR. 2000. Radon and lung cancer risk: taking stock at the millenium. Environ Health Perspect. 108(Suppl 4):635–641.
- Samet JM. 2006. Residential radon and lung cancer: end of the story? J Toxicol Environ Health A. 69(7-8):527–531.
- Sanders HR, Albitar M. 2010. Somatic mutations of signaling genes in non-small-cell lung cancer. Cancer Genet Cytogenet. 203(1):7–15.
- Sato T, Morita M, Tanaka R, Inoue Y, Nomura M, Sakamoto Y, Miura K, Ito S, Sato I, Tanaka N, et al. 2017. Ex vivo model of non-small cell lung cancer using mouse lung epithelial cells. Oncol Lett. 14(6):6863–6868
- Schabath MB, Welsh EA, Fulp WJ, Chen L, Terr JK, Thompson ZJ, Engel BE, Xie M, Berglund AE, Creelan BC, et al. 2016. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 35(24):3209–3216.
- Seki Y, Mizukami T, Kohno T. 2015. Molecular process producing oncogene fusion in lung cancer cells by illegitimate repair of DNA double-strand breaks. Biomolecules. 5(4):2464–2476.
- Shelke S, Das B. 2015. Dose response and adaptive response of non-homologous end joining repair genes and proteins in resting human peripheral blood mononuclear cells exposed to gamma radiation. Mutagenesis. 30(3):365–379.
- Shore RE, Beck HL, Boice JD, Caffrey EA, Davis S, Grogan HA, Mettler FA, Preston RJ, Till JE, Wakeford R, et al. 2018. Implications of recent epidemiologic studies for the linear nonthreshold model 879 and radiation protection. J Radiol Prot. 38(3):1217–1233.
- Shrivastav M, De Haro LP, Nickoloff JA. 2008. Regulation of DNA double-strand break repair pathway choice. Cell Res. 18(1):134–147.
- Simsek D, Jasin M. 2010. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 17(4):410–416.
- Sishc BJ, Davis AJ. 2017. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers. 9(7):pii: E81.
- Smith J, Baldeyron C, De Oliveira I, Sala-Trepat M, Papadopoulo D. 2001. The influence of DNA double-strand break structure on end-joining in human cells. Nucleic Acids Res. 29(23):4783–4792.
- Smith J, Riballo E, Kysela B, Baldeyron C, Manolis K, Masson C, Lieber MR, Papadopoulo D, Jeggo P. 2003. Impact of DNA ligase IV on the delity of end joining in human cells. Nucleic Acids Res. 31(8):2157–2167.
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S-I, Watanabe H, Kurashina K, Hatanaka H, et al. 2007. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 448:561–566.
- Stopper H, Schmitt E, Gregor C, Mueller SO, Fischer WH. 2003. Increased cell proliferation is associated with genomic instability: elevated micronuclei frequencies in estradiol-treated human ovarian cancer cells. Mutagenesis. 18(3):243–247.
- Sudprasert WP, Navasumrit P, Ruchirawat M. 2006. Effects of low-dose gamma radiation on DNA damage, chromosomal aberration and expression of repair genes in human blood cells. Int J Hyg Environ Health. 209(6):503–511.
- Sun Q, Shi R, Wang X, Li D, Wu H, Ren B. 2016. Overexpression of ZIC5 promotes proliferation in non-small cell lung cancer. Biochem Biophysi Res Commun. 479(3):502–509.
- Sutherland BM, Bennett PV, Sidorkina O, Laval J. 2000. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. PNAS. 97(1):103–108.
- Suto Y, Akiyama M, Noda T, Hirai M. 2015. Construction of a cytogenetic dose-response curve for low-dose range gamma-irradiation in human peripheral blood lymphocytes using three-color FISH. Mutat Res Genet Toxicol Envrion Mutagen. 794:32–38
- Thomas P, Umegaki K, Fenech M. 2003. Nucleoplasmic bridges are a sensitive measure of chromosome rearrangement in the cytokinesis-block micronucleus assay. Mutagenesis. 18(2):187–194.
- Thurtle-Schmidt DM, Lo TW. 2018. Molecular biology at the cutting edge: a review on CRISPR/CAS9 gene editing for undergraduates. Biochem Mol Biol Educ. 46(2):195–205.
- Tu S, Zhang XL, Wan HF, Xia YQ, Liu ZQ, Yang XH, Wan FS. 2018. Effect of taurine on cell proliferation and apoptosis human lung cancer A549 cells. Oncology. 15(4):5473–5480.
- Tucker JD, Cofield J, Matsumoto K, Ramsey MJ, Freeman DC. 2005. Persistence of chromosome aberrations following acute radiation: I, PAINT translocations, dicentrics, rings, fragments, and insertions. Envrion Mol Mutagen. 45(2–3):229–248.
- van Gent DC, Hoeijmakers JHJ, Kanaar R. 2001. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2(3):196–206.
- Varella-Garcia M. 2009. Chromosomal and genomic changes in lung cancer. Cell Adhesion Migration. 4(1):100–106.
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. 2007. Restoration of p53 function leads to tumour regression in vivo. Nature. 445(7128):661–665.
- Vogelstein B, Kinzler KW. 2004. Cancer genes and the pathways they control. Nat Med. 10(8):789–799.
- Wang-Michelitsch J, Michelitsch MT. 2007. Cell transformation in tumor-development: a result of accumulation of Misrepairs of DNA through many generations of cells. UK: Department of Medicine, Addenbrooke's Hospital, University of Cambridge.
- Ward JF. 1988. DNA damage produced by ionizing radiation in mammalian cells; identities, mechanisms of formation and repairability. Prog Nucleic Acid Res Mol Biol. 35:95–125.
- Warin RF, Chen H, DN Soroka, Zhu Y, Sang S. 2014. Induction of lung cancer cell apoptosis through a p53 pathway by [6]-shogaol and its cysteine-conjugated metabolite M2. J Agric Food Chem. 62(6):1352–1362.
- Weinstock DM, Richardson CA, Elliott B, Jasin M. 2006. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair. 5(9–10):1065–1074.
- Wessendorf P, Vijg J, Nussenzweig A, Digweed M. 2014. Mutation research/fundamental and molecular mechanisms of mutagenesis deficiency of the DNA repair protein nibrin increases the basal but not the radiation induced mutation frequency in vivo. Mutat Res. 769:11–16.
- Wilhelm T, Magalou I, Barascu A, Técher H, Debatisse M, Lopez B. 2014. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. PNAS. 111(2):763–768.
- Wistuba II, Behrens C, Milchgrub S, Bryant D, Hung J, Minna JD, Gazdar AF. 1999. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene. 18(3):643–650.
- Yamaguchi H, Uchihori Y, Yasuda N, Takada M, Kitamura H. 2005. Estimation of yields of OH radicals in water irradiated by ionizing radiation. J Radiat Res. 46(3):333–341.
- Zhong CY, Zhou YM, Douglas GC, Witschi H, Pinkerton KE. 2005. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis. 26(12):2187–2195.
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. 2002. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 109(7):811–821.