
Abstract
Purpose
123I-MAPi, a novel PARP1-targeted Auger radiotherapeutic has shown promising results in pre-clinical glioma model. Currently, 123I-MAPi is synthesized using multistep synthesis that results in modest yields and low molar activities (MA) that limits the ability to translate this technology for human studies where high doses are administered. Therefore, new methods are needed to synthesize 123I-MAPi in high activity yields (AY) and improved MA to facilitate clinical translation and multicenter trials.
Materials and methods
123I-MAPi was prepared in a single step via 123I-iododetannylation of the corresponding tributylstannane precursor. In vitro internalization assay, subcellular fractionation and confocal microscopy where used to evaluate the performance of 123I-MAPi in a small cell lung cancer model.
Results
123I-MAPi was synthesized in a single step from the corresponding stannane precursor in AY of 45 ± 2% and MA of 11.8 ± 4.8 GBq µmol−1. In vitro in LX22 cells showed rapid internalization (5 min) with accumulation found predominantly in the membrane, nucleus and chromatin of the cell as determined by subcellular fractionation.
Conclusions
Here, we have developed an improved radiosynthesis of 123I-MAPi, an Auger theranostic agent. This process was achieved using a single step, 123I-iododestannylation reaction from the corresponding stannane precursor in good AY and MA. 123I-MAPi was evaluated in vitro in a small cell lung cancer model with high PARP expression, rapid internalization and high nuclear uptake shown.
Introduction
Targeted radionuclide therapy (TRT) uses cancer-targeting molecules to shuttle cytotoxic radiation to the tumor. TRT is an excellent therapeutic option for inoperable or disseminated disease (Pouget et al. Citation2015; Aghevlian et al. Citation2017; Gill et al. Citation2017; Malcolm et al. Citation2019), however, curative potential can vary between and within patients. As such, novel approaches can improve the potential of this therapeutic strategy and reduce side effect toxicity. Therapeutics that are targeted against a tumor-specific biomarker have attracted significant interest for their ability to specifically act against tumor cells without the systemic toxicity and side effects of chemo or radiotherapy, as shown in recent successful clinical applications (Larson et al. Citation2015; Pouget et al. Citation2015; Aghevlian et al. Citation2017; Rahbar et al. Citation2017; Strosberg et al. Citation2017; von Eyben et al. Citation2018). TRT consists essentially of a tumor-specific molecular vehicle and a radioactive payload delivered to the tumor. In the proposed research strategy, a molecularly targeted PARP1 inhibitor will be used for delivery of high-LET Auger radiation.
Auger emitters are an extremely potent radioactive source for targeted radiotherapy, and are characterized by their greater linear energy transfer, incredibly short range and ability to cause more complex, lethal DNA damage as compared to traditional X-rays or β-particles (Daghighian et al. Citation1996; Welt et al. Citation1996; Buchegger et al. Citation2006; Kiess et al. Citation2015; Gill et al. Citation2017; Bavelaar et al. Citation2018). Previous attempts to use Auger emitters as cancer therapies have not been successful (Lee et al. Citation2020), due to the limited range of the radiation emitted and the difficulty of reliably delivering the lethal electrons close enough to the DNA target (<100 Å) (Hofer and Hughes Citation1971; Kassis Citation2003; Bavelaar et al. Citation2018). Iodine-123 is a particularly suitable Auger-emitter because it also emits a 159 keV γ-ray, which can be used for SPECT/CT imaging and disease monitoring. We recently developed and characterized 4-(4-fluoro-3-(4-(3-[123I]iodobenzoyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one – which, for simplicity, we named 123I-MAPi, as Iodine-123 Meitner–Auger PARP inhibitor – the first auger-emitting PARP inhibitor that shows improved survival without toxicity in glioblastoma therapy (Pirovano et al. Citation2020).
Beyond the published benefit for glioblastoma therapy, 123I-MAPi could represent a tissue-agnostic anti-cancer agent, and its curative potential in other forms of cancer expressing PARP1 is under investigation. It could be used in small-cell lung cancer (SCLC), which of all tissues has the highest PARP1 expression (Tang et al. Citation2017). SCLC represents 13% of lung cancer cases, with a 5-year survival rate of only 5%. Due to the aggressive nature of the disease, together with its acquired resistance to chemotherapy, the standard care for advanced SCLC has essentially remained stagnant for more than 30 years, until only very recently with the introduction of immunotherapy (Poirier et al. Citation2020).
Materials and methods
General
Unless specified otherwise, all reagents were purchased from Sigma-Aldrich and used as received. 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazine-1(2H)-one was purchased from Synthonix (Wake Forest, NC). 123I-Na in 0.1 N NaOH was purchased from GE Healthcare. Olaparib (AZD-2281) was purchased from LC Laboratories (Woburn, MA). Water (18.2 MΩcm−1 at 25 °C) was obtained from an Alpha-Q Ultrapure water system (Millipore, Burlington, MA) and acetonitrile (MeCN) as well as ethanol (EtOH) were of high-performance liquid chromatography (HPLC) grade purity. HPLC purification and analysis were performed on a Shimadzu UFLC HPLC system equipped with a DGU-20A degasser, a SPD-M20A UV detector, a LC-20AB pump system, and a CBM-20A Communication BUS module. A LabLogic Scan-RAM radio-thin layer chromatography/HPLC detector was used to detect activity.
Chemical synthesis
Synthesis of 127I-PARPi: 4-(4-fluoro-3-(4-(3-iodobenzoyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one, here called 127I-PARPi, was synthesized according to a previously published procedure (Jannetti et al. Citation2018).
Synthesis of 2,5-dioxopyrrolidin-1-yl 3-bromobenzoate
To a flame dried round bottom flask under an atmosphere of argon was added 3-bromobenzoic acid (3.00 g, 14.9 mmol), N,N′-disuccinimidyl carbonate (5.73 g, 22.4 mmol), trimethylamine (3.12 mL, 22.4 mmol) and anhydrous acetonitrile (200 mL). The reaction was stirred overnight before the excess solvent was removed in vacuo. The crude mixture was extracted with DCM (100 mL) and washed with Brine (3 × 100 mL). The aqueous layers were combined and extracted with DCM (2 × 100 mL). The organic layers were combined, and the excess solvent removed in vacuo. The crude material was purified by flash column chromatography (n-Hex:EtOAc 20–80%) affording the title compound was a white powder (3.81 g, 12.8 mmol, 86%); 1H NMR (500 MHz, Chloroform-d) δ 8.28 (t, J = 1.8 Hz, 1H), 8.07 (dt, J = 7.9, 1.2 Hz, 1H), 7.81 (ddd, J = 8.0, 2.0, 1.1 Hz, 1H), 7.41 (t, J = 7.9 Hz, 1H), 2.92 (s, 4H); 13C NMR (151 MHz, CDCl3) δ 169.0, 160.7, 137.9, 133.4, 130.5, 129.1, 127.0, 122.9 and 25.7. The data was found to be in accordance with the literature (Lv et al. Citation2016).
2,5-Dioxopyrrolidin-1-yl 3-(tributylstannyl)benzoate (1)
To a flame dried round bottom flask connected to reflux condenser under an atmosphere of argon was added 2,5-dioxopyrrolidin-1-yl 3-bromobenzoate (1.50 g, 5.05 mmol) and palladium tetrakis triphenylphosphine (1.17 g, 1.01 mmol). The mixture was heated to 100 °C before adding 1,4-dioxane (51 mL) and bis(tributyltin) (7.66 mL, 15.15 mmol). The reaction was then stirred at 100 °C until the solution went completely black (4 h approx.). The reaction was cooled and filtered over celite, the excess solvent removed in vacuo and the crude mixture columned directed using flash column chromatography (n-Hex:EtOAc 5–40%). The purification process was repeated until a clean (purity > 99%) product was observed by NMR. After final purification, the title compound was isolated as a brown oil (623 mg, 1.22 mmol, 24%); 1H NMR (500 MHz, Chloroform-d) δ 8.25 − 8.14 (m, 1H), 8.05 (dt, J = 7.9, 1.7 Hz, 1H), 7.75 (dt, J = 7.2, 1.2 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 2.91 (s, 4H), 1.59 − 1.49 (m, 6H), 1.33 (h, J = 7.4 Hz, 6H), 1.13 − 1.06 (m, 6H), 0.89 (t, J = 7.3 Hz, 9H); 13C NMR (151 MHz, CDCl3) δ 169.39, 162.43, 143.59, 143.01, 138.20, 130.06, 128.08, 124.40, 29.01, 27.33, 25.72, 13.68, 9.71. The data was found to be in accordance with the literature (Salinas et al. Citation2015).
Synthesis of 4-(4-fluoro-3-(4-(3-(tributylstannyl)benzoyl)piperazine-1-carbonyl)benzyl) phthalazin-1(2H)-one (5)
To a flame dried round bottom flask under an atmosphere of argon was added, anhydrous DCM (10 mL), 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (64 mg, 0.17 mmol), HBTU (88 mg, 0.24 mmol), DIPEA (42 µL, 0.12 mmol) and N-succinimidyl-4-(tributylstannyl)benzoate (100 mg, 0.19 mmol). The reaction mixture was stirred overnight before removing the excess solvent in vacuo. The reaction was extracted with EtOAc (30 mL) and washed with Brine (3 × 30 mL). The organic phase was collected, and the excess solvent removed in vacuo. The crude mixture was purified by flash column chromatography (DCM:MeOH 0–10%) affording the title compound as a white solid (37.1 mg, 0.049 mmol, 29%); 1H NMR (500 MHz, Chloroform-d) δ 10.20 (bs, 1H), 8.46 (dt, J = 6.9, 3.8 Hz, 1H), 7.79 − 7.67 (m, 3H), 7.48 (d, J = 31.8 Hz, 2H), 7.79 − 7.67 (m, 4H), 7.03 (s, 1H), 4.28 (s, 2H), 3.94 − 3.23 (m, 8H), 1.50 (h, J = 9.1, 8.3 Hz, 6H), 1.31 (h, J = 7.3 Hz, 6H), 1.14 − 0.97 (m, 6H), 0.86 (t, J = 7.3 Hz, 9H); 13C NMR (101 MHz, CDCl3) δ: 171.2, 165.2, 160.0, 157.0 (d, J = 250.6 Hz, 1 C), 145.5, 143.1, 138.2, 134.5, 134.4, 134.3, 133.7, 131.8, 131.7, 129.5, 129.3, 128.4, 127.9, 127.3, 126.6, 125.0, 123.7 (d, J = 17.8 Hz, 1 C), 116.3 (d, J = 25.2 Hz, 1 C), 47.4 (2 C), 42.3 (2 C), 37.7, 29.0 (3 C), 27.3 (3 C), 13.7 (3 C), 9.6 (3 C); 19F(Yang et al. Citation2012) NMR (376 MHz, CDCl3) δ: −117.5 (s); LC-ESI-MS (ES+), m/z calculated for [C39H50FN4O3Sn]+ 761.29 found 761.30 [M + H]+.
Radiosynthesis of 123I-MAPi ([123I]4)
To a 1.5 mL microcentrifuge tube was added 4-(4-fluoro-3-(4-(3-(tributylstannyl)benzoyl)piperazine-1-carbonyl)benzyl) phthalazin-1(2H)-one (100 µg, 0.132 µmol) in anhydrous MeCN (10 µL), N-Chlorosuccinimide (NCS) (50 µg, 0.37 µmol) in a solution of anhydrous methanol (25 µL), acetic acid (10 µL), anhydrous acetonitrile (20 µL) and 123I-NaI in NaOH 0.1 M (0.34–0.41 GBq, 9.29–11.04 mCi). After addition, the reaction was stirred at 650 rpm for 15 min at 25 °C. The crude reaction was purified by RP-HPLC (C18 Waters Atlantis T3) column (C18-RP, 5 µm, 6 × 250 mm) Solvent A: H2O, Solvent B: MeCN; flow rate: 1 mL/min; gradient: 0–5 min 5% B; 5–15 min 5–95% B; 15–18 min 95% B; 18–20 min 95 − 5% B). Under these conditions, 123I-MAPi has retention time of about 15 min (activity yield (AY): 45 ± 2% (n = 7)), which was collected and concentrated to dryness in vacuum. The compound has radiochemical purity > 99% and molar activity (MA) of 11.8 ± 4.8 GBq µmol−1 (EOS) and was formulated in 30% PEG300/70% saline (0.9% NaCl) and used for in vitro assays. The overall synthesis time was about 45 min.
Cell culture
FaDu and U251 cell lines were purchased from ATCC, Manassas, VA. LX22 was generously donated by Lewis lab at MSKCC. Cells were grown in a monolayer culture at 37 °C in a 5% CO2 humidified atmosphere. FaDu and U251 cell lines were maintained in MEM medium containing 10% (v/v) FBS and 1% PenStrep. LX22 were grown in RPMI containing 10% (v/v) FBS and 1% PenStrep. All cell lines were tested negative for mycoplasma infection.
Immunoblotting
Cell pellets obtained from 80% confluent flasks were washed twice with PBS and lysed with 200 µL ice-cold RIPA buffer with Triton X100 (Boston BioProducts, Ashland, MA) and one Mini Complete Protease Inhibitor Cocktail tablet (Roche, Indianapolis, IN) The samples were spun at 4 °C at 12,000 rpm for 10 min and the supernatant was collected. Protein quantification was performed using bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL). For PARP expression, 20 µg of protein was loaded on the gel. Proteins were separated with SDS/PAGE gel electrophoresis and transferred to a Nitrocellulose membrane. Proteins were detected using antibodies specific for PARP1 (1:1000, Invitrogen, Carlsbad, CA; PA5-16452) and ß-actin (1:40,000; Sigma, St. Louis, MO; A-3854) with a corresponding horseradish peroxidase (HRP) conjugated secondary antibody (1:20,000, ab6721, Abcam, Eugene, OR). Detection was performed using a chemiluminescent substrate (Thermo Scientific #34077, Super Signal West Pico, Waltham, MA). The bands were visualized using an automated blot processing machine (Ewen-Parker X-Ray Corporation, New York, NY) with a light sensitive clear blue x-ray film (Thermo Scientific, 24 × 30 cm, SB2324231, Geel, Belgium) with 1 min exposure time.
Uptake assay
Uptake of 123I-MAPi was tested in vitro (three replicates) as previously described (Pirovano et al. Citation2020). 5 × 105 LX22 cells were plated 24 h prior to the experiment (n = 3). Media was changed and 1 h later 3.7 kBq of 123I-MAPi were added to the cells. For blocking, cells were incubated with a 100-fold molar excess of olaparib 1 h before adding 123I-MAPi. Media was removed, and cells were washed with PBS and lysed (1 M NaOH) at different time points. The lysate was collected, and uptake was determined by radioactivity on a γ-counter.
Subcellular fractionation
The subcellular distribution of 123I-MAPi was determined with a commercially available assay kit ‘Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific Product #78840)’ following manufacturer’s protocol. Briefly, cells were seated in six-well plates at concentrations of 1 × 106 cells/well. Plates were incubated overnight to allow cells to attach to well plates. Each well was treated with 3.7 kBq of 123I-MAPi and returned to the incubator for 4 h. Media was removed and cells were washed with cold PBS before trypsinization and transfer to 1.5 mL microcentrifuge tubes. Cells were spun down, supernatant was removed, and cells were washed with cold PBS. Individual cellular compartments with their contents were then separated through a series of incubations with various detergents, centrifugation and removal of supernatants. Finally, each supernatant which contained the contents of a separate cellular compartment and the radioactivity was measured in an automated ɣ-counter.
Microscopy
Cells were plated on a coverslip slide on the bottom of a 6-well plate and incubated overnight. 123I-MAPi, 127I-PARPi or vehicle control was added to the media and cells were returned to the incubator overnight. Cells were then fixed in 4% paraformaldehyde/PBS, permeabilized and stained with anti γ-H2AX antibody (Millipore Sigma, 05-636, Burlington, MA) and anti PARP1 antibody (Invitrogen, Carlsbad, CA, PA5-16452). DAPI was used to localize nuclei. Coverslips were mounted on slides for microscopy imaging. Microscopy was acquired using a confocal microscope at the MSK Molecular Cytology Core Facility.
Results
Chemistry and radiochemistry
Conjugation of Sn-NHS ester 1 and 3 afforded precursor 5 in 29%. 123I-iododestannylation of 5 using NCS as an oxidant, afforded 123I-MAPi in an activity yield (AY) of 45 ± 2% (n = 7), MA of 11.8 ± 4.8 GBq µmol−1 and synthesis time of 45 min (; Supplementary Figure 1). Radiochemical purity was >99% for all prepared compounds (Supplementary Figure 2).
Internalization assay
In vitro internalization was tested on LX22 cells expressing PARP1 (). 123I-MAPi (3.7 kBq/well) was added to adherent cells in monolayer and uptake was calculated by measuring γ radiation in cell lysates at different time points (5, 30 and 60 min). We confirmed rapid cellular internalization, with an uptake of 6.5 ± 0.5% being reached after 5 min post-treatment. Uptake was blockable with 100-fold excess dose of olaparib and showed significant differences at 5 (****p value < .0001, unpaired student’s t-test), 30 (***p value < .001, unpaired student’s t-test) and 60 min (****p value < .0001, unpaired student’s t-test). The final total uptake was 6.9 ± 0.5% of added activity, compared to 1.4 ± 0.2% blocked uptake. Uptake was blocked with a 100-fold excess dose of olaparib to show target specificity.
Figure 1. Summary of radiosynthesis for 123I-MAPi. (A) Two-step approach described by Pirovano and coworkers (previous work). (B) Synthesis of stannane precursor and one-pot 123I-iododestannylation (this work).
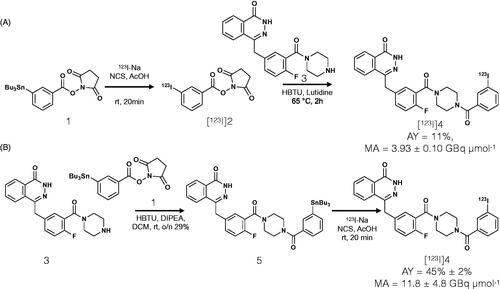
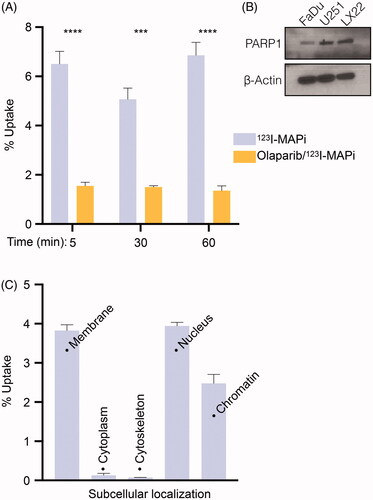
Figure 2. Subcellular localization of 123I-MAPi. (A) Internalization assay showing uptake of 123I-MAPi in LX22 cancer cells in vitro. 3.7 kBq of 123I-MAPi were added to the cells. For blocking, cells were incubated with a 100-fold molar excess of olaparib 1 h before adding 123I-MAPi. (B) Immunoblotting showing expression of PARP1 target in a set of cell lines including FaDu, U251 and LX22. (C) Subcellular binding of 123I-MAPi in LX22 in vitro. Error bars represent standard deviation.
Subcellular fractionation
In vitro subcellular accumulation of 123I-MAPi was investigated in LX22 cells (4 h post-treatment). A 3.9 ± 0.2% accumulation in cellular membrane was observed, as well as low levels of accumulation in cytoplasm and cytoskeleton (0.1 ± 0.1% uptake in both cases). A higher binding was observed in the nucleus (3.9 ± 0.1%) with a 2.5 ± 0.2% binding to the chromatin ().
Induction of DNA damage
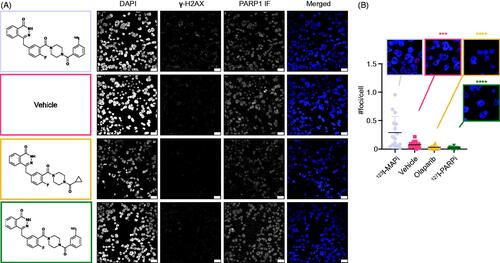
LX22 cells were treated in vitro with 123I-MAPi and levels of DNA damage were investigated by analyzing the number of γ-H2AX foci formed (). 123I-MAPi treated cells showed a significantly increased number of γ-H2AX foci when compared to vehicle (***p value < .001), olaparib (****p value < .0001) and 127I-PARPi isotopologue (****p value < .0001) (). This result confirmed the ability of 123I-MAPi to reach the DNA with cytotoxic amounts of Auger radiation.
Figure 3. DNA damage induction of 123I-MAPi. (A) Immunofluorescence showing DAPI, γ-H2AX and PARP1 staining in LX22 cells treated, respectively, with (top to bottom) 123I-MAPi, Vehicle (30% PEG/PBS), olaparib, 127I-PARPi. Panels on the left show each molecular structure. (B) Quantification of the number of γ-H2AX foci per cell induced by 123I-MAPi treatment in LX22 cells in vitro. Scalebar 25 μm. Error bars represent standard deviation.
Discussion
In 2020, our group reported the use of 123I-MAPi, a novel PARP1-targeted Auger radiotherapeutic. In this work, we highlight the potential of 123I-MAPi as a theranostics tool for imaging and killing cancer cells in a glioblastoma model (Pirovano et al. Citation2020). To access 123I-MAPi, a multi-step approach had been taken. Whilst this strategy was successful in providing proof-of-concept, the modest AY and MA (AY: 11%, MA: 3.93 ± 0.10 GBq µmol−1) are not only economically burdensome because iodine-123 cost per mCi is generally accessible, but present a significant barrier to clinical translation wherein high doses with higher MA are needed. Therefore, it was crucial to optimize the yields to facilitate clinical translation. Furthermore, increasing the MA of 123I-MAPi will allow for a wider investigation into whether or not, this property is crucial to the efficacy of 123I-MAPi as a theranostic agent. To this end, we set out to develop a single step approach. Such a strategy would hopefully lead to improved AY, MA as well as reducing the overall synthesis time. Furthermore, by moving from a multi-step approach to a single step, this would make the procedure more amenable to automation, a crucial juncture for any future translation of the molecule into a clinical setting.
The first and most crucial stage seen to the success of the project was to eliminate the post-labeling amide coupling step. To this end, the commercially available N-Succinimidyl-3-(tributylstannyl)benzoate and 4-(4-Fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one were conjugated to form 5. Initial investigations into the 123I-iododestannylation of 5 were promising with good AY’s and reduced synthesis times observed (45 min). Pleasingly, no protection of the phthalazinone NH group was required. However, the MA of the final compound was still modest (3.29 GBq/µmol).
We identified that one potential source of iodide that was otherwise diluting the MA of the final product was from the commercially available, N-Succinimidyl-3-(tributylstannyl)benzoate. To eradicate any unwanted source of iodide within the 123I-iododestannylation reaction, the synthesis of N-Succinimidyl-3-(tributylstannyl)benzoate was conducted in-house from the corresponding aryl bromide. Using palladium tetrakis (20 mol%) and an excess of bis(tributyltin), N-Succinimidyl-3-(tributylstannyl)benzoate 1 was isolated in 24%. Subsequent conjugation of 1–3 was performed facilitating access to the desired precursor 5 in 29%. Gratifyingly, when 5 was exposed to the standard 123I-iododestannylation conditions; a significant increase in MA was observed (11.8 ± 4.8 GBq µmol−1). With all three criteria of good to high AY, MA and short reaction times being met, we proceeded to evaluate the performance of 123I-MAPi within a small cell lung cancer cell line (LX22) in vitro.
In vitro internalization in LX22 cells demonstrated the rapid uptake of 123I-MAPi into LX22 cells. A blocking study with olaparib confirmed target specificity of 123I-MAPi for PARP1 (), a protein whose expression within LX22 cells was confirmed via western-blot () by comparison with FaDu and U251, cell lines known to express PARP1 at detectable levels (Pirovano et al. Citation2019; Kossatz et al. Citation2020; Zhu et al. Citation2020). Subcellular fractionation was performed showing uptake in the membrane, nucleus and chromatin and showed that most of the internalized compound is found in the nucleus and chromatin, with extremely small cytoplasmic localization. The high membrane portion could be due to the molecule transitioning in or out the cell through the membrane.
Confocal microscopy confirmed a higher number of DSBs as shown by intra-nuclear γ-H2AX foci. This result is in accordance with what was previously shown in glioblastoma cells and in vivo model, and confirms the DNA-damaging potential of an Auger emitter when located in proximity to the DNA.
Conclusions
In summary, we have developed a novel strategy to access 123I-MAPi, a PARP1 targeting radiotherapeutic agent, in improved yields, MA and reduced synthesis times. We have demonstrated the specific uptake of 123I-MAPi in vitro in a non-small cell lung cancer cell line, namely LX22, as well as demonstrated good uptake within the cell nucleus through subcellular fractionation. Preliminary DNA damage analysis in LX22 cells, performed via γ-H2AX induction monitoring confirmed the potential of 123I-MAPi as a PARP-targeting DNA-damaging agent with great potential for cancer theranostics. Taken together, these results set the stage for an in-depth mechanistic examination of 123I-MAPI and its wider use as an Auger theranostic agent.
IJRB_123I-MAPi_Supplemntarl.docx
Download MS Word (13.3 MB)Acknowledgments
The authors thank Drs Jason S. Lewis, Brian M. Zeglis and John L. Humm for useful discussion. The authors gratefully acknowledge the Tow Foundation, Memorial Sloan Kettering Cancer Center’s Animal Imaging Core Facility, the Radiochemistry & Molecular Imaging Probes Core, the Molecular Cytology Core, the Nuclear Magnetic Analytical Core and the Memorial Sloan Kettering Center for Molecular Imaging & Nanotechnology.
Disclosure statement
T.R. is shareholder of Summit Biomedical Imaging, LLC. T.R. is a co-inventor on filed U.S. patent (WO2016164771). T.R. is a co-inventor on U.S. patent (WO2012074840) held by the General Hospital Corporation. T.R. is a co-inventor on U.S. patent (WO2016033293) held by MSK. T.R. is a paid consultant for Theragnostics, Inc.
Additional information
Funding
Notes on contributors
Thomas C. Wilson
Thomas C. Wilson, DPhil is a Research Fellow at Memorial Sloan Kettering Cancer Center.
Stephen A. Jannetti
Stephen A. Jannetti, PhD is a Postdoctoral Associate, Duke University Medical Center.
Navjot Guru
Navjot Guru is a Research Technician, Memorial Sloan Kettering Cancer Center.
Nagavarakishore Pillarsetty
Nagavarakishore Pillarsetty, PhD is an Associate Attending Radiochemist at Memorial Sloan Kettering Cancer Center.
Thomas Reiner
Thomas Reiner, PhD is an Associate Member at Memorial Sloan Kettering Cancer Center.
Giacomo Pirovano
Giacomo Pirovano, DPhil is a Research Fellow at Memorial Sloan Kettering Cancer Center.
References
- Aghevlian S, Boyle AJ, Reilly RM. 2017. Radioimmunotherapy of cancer with high linear energy transfer (LET) radiation delivered by radionuclides emitting α-particles or Auger electrons. Adv Drug Deliv Rev. 109:102–118.
- Bavelaar BM, Lee BQ, Gill MR, Falzone N, Vallis KA. 2018. Subcellular targeting of theranostic radionuclides. Front Pharmacol. 9:996.
- Buchegger F, Perillo-Adamer F, Dupertuis YM, Delaloye AB. 2006. Auger radiation targeted into DNA: a therapy perspective. Eur J Nucl Med Mol Imaging. 33(11):1352–1363.
- Daghighian F, Barendswaard E, Welt S, Humm J, Scott A, Willingham MC, McGuffie E, Old LJ, Larson SM. 1996. Enhancement of radiation dose to the nucleus by vesicular internalization of iodine-125-labeled A33 monoclonal antibody. J Nucl Med. 37(6):1052–1057.
- Gill MR, Falzone N, Du Y, Vallis KA. 2017. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 18(7):e414–e423.
- Hofer KG, Hughes WL. 1971. Radiotoxicity of intranuclear tritium, 125 iodine and 131 iodine. Radiat Res. 47(1):94–101.
- Jannetti SA, Carlucci G, Carney B, Kossatz S, Shenker L, Carter LM, Salinas B, Brand C, Sadique A, Donabedian PL, et al. 2018. PARP-1-targeted radiotherapy in mouse models of glioblastoma. J Nucl Med. 59(8):1225–1233.
- Kassis AI. 2003. Cancer therapy with Auger electrons: are we almost there. J Nucl Med. 44(9):1479–1481.
- Kiess AP, Minn I, Chen Y, Hobbs R, Sgouros G, Mease RC, Pullambhatla M, Shen CJ, Foss CA, Pomper MG, et al. 2015. Auger radiopharmaceutical therapy targeting prostate-specific membrane antigen. J Nucl Med. 56(9):1401–1407.
- Kossatz S, Pirovano G, Demétrio De Souza França P, Strome AL, Sunny SP, Zanoni DK, Mauguen A, Carney B, Brand C, Shah V, et al. 2020. Validation of the use of a fluorescent PARP1 inhibitor for the detection of oral, oropharyngeal and oesophageal epithelial cancers. Nat Biomed Eng. 4(3):272–285.
- Larson SM, Carrasquillo JA, Cheung NK, Press OW. 2015. Radioimmunotherapy of human tumours. Nat Rev Cancer. 15(6):347–360.
- Lee H, Riad A, Martorano P, Mansfield A, Samanta M, Batra V, Mach RH, Maris JM, Pryma DA, Makvandi M, et al. 2020. PARP-1-targeted Auger emitters display high-LET cytotoxic properties in vitro but show limited therapeutic utility in solid tumor models of human neuroblastoma. J Nucl Med. 61(6):850–856.
- Lv Y, Sun K, Pu W, Mao S, Li G, Niu J, Chen Q, Wang T. 2016. Metal-free intermolecular C–O cross-coupling reactions: synthesis of N-hydroxyimide esters. RSC Adv. 6(96):93486–93490.
- Malcolm J, Falzone N, Lee BQ, Vallis KA. 2019. Targeted radionuclide therapy: new advances for improvement of patient management and response. Cancers (Basel). 11(2):268.
- Pirovano G, Jannetti SA, Carter LM, Sadique A, Kossatz S, Guru N, Demétrio De Souza França P, Maeda M, Zeglis BM, Lewis JS, et al. 2020. Targeted brain tumor radiotherapy using an Auger emitter. Clin Cancer Res. 26(12):2871–2881.
- Pirovano G, Roberts S, Brand C, Donabedian PL, Mason C, de Souza PD, Higgins GS, Reiner T. 2019. [18F]FE-OTS964: a small molecule targeting TOPK for in vivo PET imaging in a glioblastoma xenograft model. Mol Imaging Biol. 21(4):705–712.
- Poirier JT, George J, Owonikoko TK, Berns A, Brambilla E, Byers LA, Carbone D, Chen HJ, Christensen CL, Dive C, et al. 2020. New approaches to small cell lung cancer therapy: from the laboratory to the clinic. J Thorac Oncol. 15(4):520–540.
- Pouget JP, Lozza C, Deshayes E, Boudousq V, Navarro-Teulon I. 2015. Introduction to radiobiology of targeted radionuclide therapy. Front Med (Lausanne). 2:12.
- Rahbar K, Ahmadzadehfar H, Kratochwil C, Haberkorn U, Schäfers M, Essler M, Baum RP, Kulkarni HR, Schmidt M, Drzezga A, et al. 2017. German multicenter study investigating 177Lu-PSMA-617 radioligand therapy in advanced prostate cancer patients. J Nucl Med. 58(1):85–90.
- Salinas B, Irwin CP, Kossatz S, Bolaender A, Chiosis G, Pillarsetty N, Weber WA, Reiner T. 2015. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 5(1):123.
- Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, Mittra E, Kunz PL, Kulke MH, Jacene H, et al. 2017. Phase 3 trial of 177Lu-dotatate for Midgut neuroendocrine tumors. N Engl J Med. 376(2):125–135.
- Tang J, Salloum D, Carney B, Brand C, Kossatz S, Sadique A, Lewis JS, Weber WA, Wendel HG, Reiner T, et al. 2017. Targeted PET imaging strategy to differentiate malignant from inflamed lymph nodes in diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 114(36):E7441–E7449.
- von Eyben FE, Roviello G, Kiljunen T, Uprimny C, Virgolini I, Kairemo K, Joensuu T. 2018. Third-line treatment and 177Lu-PSMA radioligand therapy of metastatic castration-resistant prostate cancer: a systematic review. Eur J Nucl Med Mol Imaging. 45(3):496–508.
- Welt S, Scott AM, Divgi CR, Kemeny NE, Finn RD, Daghighian F, Germain JS, Richards EC, Larson SM, Old LJ, et al. 1996. Phase I/II study of iodine 125-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 14(6):1787–1797.
- Yang W, Qin W, Hu Z, Suo Y, Zhao R, Ma X, Ma W, Wang T, Liang J, Tian J, et al. 2012. Comparison of Cerenkov luminescence imaging (CLI) and gamma camera imaging for visualization of let-7 expression in lung adenocarcinoma A549 Cells. Nucl Med Biol. 39(7):948–953.
- Zhu W, Pirovano G, O’Neal PK, Gong C, Kulkarni N, Nguyen CD, Brand C, Reiner T, Kang D. 2020. Smartphone epifluorescence microscopy for cellular imaging of fresh tissue in low-resource settings. Biomed Opt Express. 11(1):89–98.