
Abstract
Purpose
Ionizing radiation is a well-known carcinogen, and epidemiologic efforts have been made to evaluate cancer risks following a radiation exposure. The basic approach has been to estimate increased levels of cancer mortality resulting from exposures to radiation, which is consistent with the somatic mutation theory of cancer. However, the possibility that an irradiation might cause an earlier onset of cancer has also been raised since the earliest days of animal studies. Recently, the mutation induction model has been challenged because it is unable to explain the observed dose-related parallel shift of entire mouse survival curves toward younger ages following an irradiation. This is because if it is assumed that only a fraction of the irradiated individuals are affected, the irradiated population would consist of two subpopulations with different mean lifespans, which makes the overall distribution of individual lifespans broader, and hence the slope of the survival curves shallower. To explain this parallel shift, it is necessary to assume that all individuals of a population are affected. As a result of these observations, possible mechanisms which could account for the parallel shift of mouse survival curves were sought by examining the radiation induction of various types of tissue damage which could facilitate an earlier onset of spontaneously arising cancers.
Conclusion
A proposed mechanism postulates that a radiation exposure leads to tissue inflammation which subsequently stimulates spontaneously arising cancers and allows them to appear earlier than usual. This notion is not unprecedented, and because the background incidence of cancer increases exponentially with an increase in age, a slight shift of the onset age toward younger ages may make it appear as if the risk is increased. In this scenario, a radiation exposure induces DNA damage, cell death, chromosome aberrations etc., which leads to the multi-pathway responses including activation of stromal fibroblasts, macrophages and various inflammatory factors. Such an inflamed microenvironment favors the growth of spontaneously arising tumor cells although currently, the sequential order or relative importance of the individual factors remains to be known.
Background
It has been established that somatic mutations are a driving force for the development of cancer: namely, cancer cells contain a number of mutations in various tumor-related genes, and the germline-derived genetic alterations of such genes may cause animals to be susceptible to the development of cancers (e.g., Walrath et al. Citation2010; Alexandrov et al. Citation2020). In accordance with this notion, it has been suggested that some types of cancers are caused by the induction of inactivating mutations at relevant genes; e.g., the loss of heterozygosity at the Znfn1a1 (Ikaros) locus in mouse thymic lymphoma (Shimada et al. Citation2000), Tsc2 mutations in renal cell carcinomas in Tsc2 heterozygous rats (Kokubo et al. Citation2010), and PTCH1 mutations in medulloblastoma in PTCH1 heterozygous mice (Tsuruoka et al. Citation2016). However, an alternative possibility has also been raised since early animal studies of radiation carcinogenesis; namely, a hypothesis proposing that there is an earlier onset of the disease following an exposure to radiation (Upton et al. Citation1960; Albert and Altshuler Citation1976; Shellabarger et al. Citation1980; Fry Citation1981). Upton et al. wrote in 1960 that “at each radiation dose level, nearly all diseases were advanced in time of onset to about the same extent” and that “radiation may merely create conditions favorable for the selection of spontaneous carcinogenic mutants, possibly by excessive growth stimulation through homeostatic mechanisms” (Upton et al. Citation1960). However, possible mechanisms were not proposed.
Unfortunately, following the discovery of activated oncogenes and inactivated tumor suppressor genes in human cancers, the mutation induction hypothesis of radiation prevailed and was used to explain the carcinogenic action of radiation. Also, this notion fits the relative risk estimates derived from epidemiologic follow-up studies of atomic-bomb survivors. In both instances, a radiation exposure is thought to affect a fraction of the exposed individuals which increases with an increase in the dose. It was only recently that the mutation-induction theory was challenged based on mouse survival data: since the mutation-induction theory assumes that only a fraction of the irradiated individuals are affected, it would not be able to explain the observed parallel shift of entire mouse survival curves toward younger ages following an exposure to radiation (e.g., Kohn and Guttman Citation1963; Storer Citation1965; Upton et al. Citation1967; Tanaka et al. Citation2003). This is because if it is assumed that only a fraction of the irradiated individuals are affected, the irradiated population would consist of two subpopulations with different mean lifespans, which would result in a broader distribution of individual lifespans, and hence a survival curve with a shallower slope after an exposure to radiation.
To explain this parallel shift, it is necessary to assume that all irradiated individuals were affected (Nakamura Citation2020). The most important advantage of this model is that an appropriate shift in time of the background mortality rate toward younger ages could faithfully reproduce the past mysterious declining trend of relative risk for cancer with an increase in post-exposure time in atomic-bomb survivors (Ozasa et al. Citation2012) and in mice (Nakamura Citation2020). To account for this shift, widely occurring and dose-related events are necessary. This leads one to imagine the possibility that the carcinogenic effects of radiation could be due to the creation of tissue damage which leads to the development of microenvironments favorable to the growth of spontaneously arising tumor cells (e.g., Tlsty and Gascard Citation2019). Specifically, an exposure to radiation would induce DNA damage, cell death, chromosome aberrations etc., which leads to the activation of stromal fibroblasts and various inflammatory factors (TGF-β, NF-κB, COX-2, PGE2 etc.), all of which could lead to inflamed tissue conditions which would favor tumor growth (e.g., Fishbein, Hammock, et al. Citation2020). This model is in accord with the well-known fact that irradiated feeder cells can support the growth of primary cultured cells and the idea of a coevolution of the stroma and cancer during carcinogenesis (Littlepage et al. Citation2005). In recent years, it has become apparent that the tumor microenvironment plays important roles, not only in tumor growth but also in chemo- and radiotherapy (e.g., Hirata and Sahai Citation2017). Thus, it seems reasonable to expect that a similar scenario may apply to the processes involved in tumorigenesis.
Major constituents in the cancer microenvironment
The cancer microenvironment consists of various types of cells, collectively termed stroma cells, and various types of cytokines/chemokines described below.
Fibroblasts and myofibroblasts (activated fibroblasts)
Fibroblasts form the connective tissues and are characteristically motile. When a tissue is damaged, these cells move rapidly to the damage site and repair the damage by secreting extra cellular matrix (ECM) materials such as collagen. They can be activated to become myofibroblasts or stellate fibroblasts through various mechanisms and show a phenotype between fibroblasts and smooth muscle cells. They express smooth muscle actin (αSMA), ECM, and PDGFR (fibrogenic cytokines), etc.
Cancer associated fibroblasts (CAFs)
These cells are activated fibroblasts or myofibroblasts which surround a tumor, may contribute to the formation of various types of cancers, and consist of a heterogeneous population of cells (Yoshida et al. Citation2019; Sahai et al. Citation2020). They are thought to contribute significantly to the outcome of cancer treatments; e.g., CAFs may increase the frequency of tumor-initiating cells with the aid of TGF-β (Calon et al. Citation2015) or promote an immunosuppressive microenvironment via the induction of pro-tumorigenic macrophages (e.g., Takahashi et al. Citation2017). They may be derived not only from local stroma cells, but also from mesenchymal stem cells migrating from the bone marrow, although their origins are not clearly understood due to ill-defined markers for fibroblasts and CAFs (Sahai et al. Citation2020).
Macrophages and neutrophils
Macrophages undertake phagocytosis, antigen presentation, and cytokine production. Tumor-associated macrophages, which are known to contribute to cancer progression (Weitzman et al. Citation1985), have attracted attention recently since they may also contribute to the induction of therapeutic resistance (Ruffell and Coussens Citation2015) and local inflammation (Sulciner et al. Citation2018). Neutrophils produce reactive oxygen species (ROS) and reactive nitrogen species (NO: nitric oxide) which can act as bactericides. However, the secreted radicals may produce a mutagenic milieu which can lead to DNA damage in neighboring cells.
Transforming growth factor beta (TGF-β)
TGF-β was isolated as a polypeptide which can lead to the colony formation of immortalized rodent fibroblasts in soft agar (Roberts et al. Citation1980). Anchorage-independent growth may correspond to malignant transformation in vivo. TGF-β is a multifunctional cytokine, produced by various types of cells, and stored in a latent form in the ECM. Once activated by various types of tissue stress, TGF-β exerts its biologic effects by binding to its receptors on the cell surface, which is followed by the activation of transcription factors via the Smad4 pathway (Zhao et al. Citation2018). Although TGF-β is known to have dual functions during tumorigenesis, namely suppressive in the early stages and pro-oncogenic in the later stages (e.g., Calon et al. Citation2014), most of the tumors which arise in vivo are defective in TGF-β, its receptor, or Smad4 genes, and are resistant to the cytocidal effects of TFG-β (Bauer Citation1995).
Other inflammatory factors
Additional factors that are involved in pro-oncogenic inflammation include nuclear factor-kappa B (NF-κB), inducible nitric oxide synthetase (iNOS), cyclooxygenase 2 (COX-2 or prostaglandin-endoperoxide synthase 2) and prostaglandin E2 (PGE2) etc. These molecules can also be activated by radiation (Aggarwal et al. Citation2009). NF-κB is a transcription factor located in the cytoplasm in a latent form, and after being activated it is translocated into the nucleus and induces various genes including iNOS, COX-2, IL-1, IL-2, IL-6, CXCL8 (IL-8), IL-12 etc. COX-2 is involved in conversion of arachidonic acid to PGE2 which has two facets which exhibit pro- and anti-inflammatory functions. STAT3 is a pro-oncogenic transcription factor, and is activated by various growth factors (EGF, IGF, FGF), and induces genes associated with cell proliferation (e.g., MYC), angiogenesis (e.g., VEGF) and immunologic suppression (e.g., IL-6, IL-10) (Yu et al. Citation2007). NF-κB and STAT3 are activated by most carcinogens and control the expression of many cancer-related genes (Aggarwal et al. Citation2009).
Experimental observations
In the following sections, multiple pathways which may be related to the radiation-induced advancement of cancer onset are briefly described, although their sequential order of occurrence or relative impact on tumor formation is not understood. It is possible that these mechanisms may differ among different tissues.
A glimpse of the tumor supporting nature of an irradiated stroma
There are reports which indicated that irradiated tissues support the growth of transplanted tumor cells. For example, irradiating the hind limbs of mice with doses ≤ 5 Gy improved the growth of transplanted cancer cells. Furthermore, the growth supporting effect was more pronounced when tumor cells were transplanted on day 3 instead of day 1 after an irradiation (). The expression level of TGF-β mRNA was highly elevated, not only in the tumors, but also in the tumor bed (skin and muscle) (Lee et al. Citation2014). Similarly, mouse brains which were exposed to 20 to 60 Gy of radiation supported not only better growth of glioblastoma cells transplanted 6 weeks after the irradiation, but also the progression of transplanted tumor cells toward more malignant forms (Duan et al. Citation2019). Another example is that of irradiated mouse lungs which facilitate artificial lung metastasis when tumor cells were administered via the tail vein (Withers and Milas Citation1973). Even senescent fibroblasts (myofibroblasts) (Krtolica et al. Citation2001) or dead cell debris (Sulciner et al. Citation2018) can support the growth of pre-malignant or malignant cells () through the induction of local inflammation.
Figure 1. The growth of implanted tumor cells is influenced by their microenvironment. (A) Tumor growth becomes more robust when mouse hind limbs were irradiated prior to the transplantation of Hca-1 tumor cells. In this experiment, a 5 Gy-exposure which was given 3 days earlier yielded better growth when compared with an irradiation given one day earlier (Lee et al. Citation2014). (B) Chemotherapy-generated tumor cell debris stimulates tumor growth. A subthreshold inoculum (1 × 104) of Lewis lung carcinoma (LLC) cells were co-transplanted along with variable numbers of cisplatin-induced LLC debris (Sulciner et al. Citation2018).
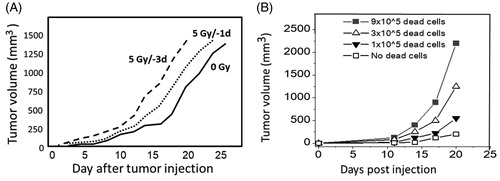
These observations remind us anew of an invisible potential of tissues during the recovery processes from different kinds of damage such as recurrent breast cancers tended to occur near the incisional scar for surgical removal of the primary cancer (Reid et al. Citation1996). In this context, it should be kept in mind that ionizing radiation is not a potent, but rather a relatively weak carcinogen (Little Citation2000), because irradiated mice or rats develop solid cancers many months after an irradiation, and hence entire life span observations are necessary to evaluate the carcinogenic effect of radiation. In contrast, potent chemical carcinogens such as 7,12-dimethyl-benz[α]anthracene can induce mammary cancers in 100% of the animals within a few months after its administration (Harris et al. Citation2000).
Roles of myofibroblasts/CAFs in tumorigenesis
The stroma is a tissue which supports the normal functions of epithelial cells. However, cancers (malignancies derived from epithelial tissue) often accompany the overgrowth of fibroblasts (CAFs) (Shimoda et al. Citation2010), especially in cancers of the breast, colon, stomach, and pancreas, and the stroma may comprise over 90% of the tumor mass (Dvorak Citation1986). Interestingly, normal fibroblasts are activated when incubated in the presence of culture supernatants from cancer cells and become myofibroblasts (i.e., they undergo epigenetic changes). Similar changes are also observed following the incubation of fibroblasts with TGF-β, PDGF, FGF2 etc. (Shimoda et al. Citation2010; Kalluri Citation2016). Furthermore, not only CAFs but also senescent appearing flat fibroblasts (stellate fibroblasts which may contain unrepaired DNA damage) can support the growth of pre-malignant or malignant, but not normal epithelial cells when these cells are co-cultured in vitro, or co-transplanted into host animals (Krtolica et al. Citation2001; Orimo et al. Citation2005; Kojima et al. Citation2010; Yoshida et al. Citation2019). The growth stimulation effect in vitro was attributed at least partly to soluble and insoluble factors derived from senescent cells (Krtolica et al. Citation2001). In this context, it is noted that TGF-β is released not only from myofibroblasts to support their own growth in an autocrine fashion, but also by cancer cells to support the growth of CAFs (Kojima et al. Citation2010). Cancer cells and CAFs exist in a mutually beneficial relationship.
The processes of activating TGF-β
TGF-β is stored in a latent form in the ECM and requires activation through proteolytic digestion to exert its biologic functions (Jenkins Citation2008). Activation of latent TGF-β or its biosynthesis and release is reported to increase with radiation doses either in vivo (Ehrhart et al. Citation1997) or in vitro (Carl et al. Citation2016) while the underlying mechanisms are not clearly defined because there are different ways in activating latent TGF-β. For example, thrombin and plasmin, both of which are serine proteases and involved in blood coagulation and thrombolysis, respectively, can activate latent TGF-β (Hirata and Sahai Citation2017). Also, activated metalloproteinases (MMP-2, -9, -13, and -14) are reported to activate latent TGF-β (Conlon and Murray Citation2019). These MMPs are stored in extra-cellular spaces in latent forms and need to be activated by various types of inflammatory stimuli such as proteases, integrins (cell-extracellular adhesion molecules), different types of shock (heat, pH, ROS, and hence probably ionizing radiation) (Jenkins Citation2008) and activated Ras (Ballin et al. Citation1988). In this context, it is interesting to note that tumors lacking both MMP-2 and MMP-9 do not grow well in wild type (WT) host animals, and WT tumors also do not grow well in host animals lacking both MMP-2 and MMP-9 (Hingorani et al. Citation2018). These results seem to indicate that the formation of a relaxed microenvironment which is aided by MMPs helps promote the growth of tumor cells.
How an exposure to radiation causes tissue inflammation
An exposure to radiation can lead to creating pro-tumorigenic conditions. These include transformation of normal fibroblasts into a CAF-like phenotype (Legrand et al. Citation2018), activation of TGF-β (Carl et al. Citation2016), NF-κB and interleukins in an ATM dependent manner (Habraken and Piette Citation2006; Nikitaki et al. Citation2016), and induction of apoptosis which leads to cell debris-derived local inflammation (Huang et al. Citation2011). NF-κB induces the iNOS and COX-2 genes, which leads to an increased production of NO and PGE2 (Hei et al. Citation2008; Lemay et al. Citation2017). All of these events will also lead to local inflammation ().
Figure 2. A simplified schema depicting radiation-induced damage and the subsequent development of an inflammatory microenvironment. Circled factors indicate major contributing factors to inflammation. IR: ionizing radiation; iNOS: inducible NO synthetase; NO: nitric oxide; casp3: caspase 3; iPLA2: inducible plasminogen activator 2; COX-2: cyclooxygenase 2; PGE2: prostaglandin E2; cGAS: cyclic GMP-AMP synthetase; STING: stimulator of interferon genes.
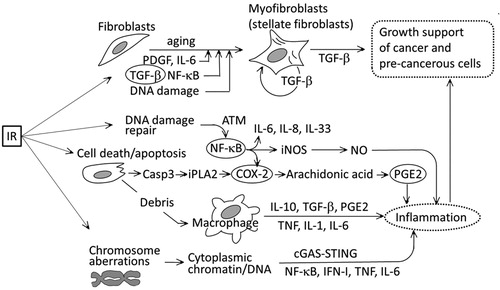
Cells bearing unstable-type chromosome aberrations would remain quiescent and may not affect the microenvironment until they undergo their first post-exposure mitosis where a dicentric chromosome causes a mechanical problem during the separation of the two daughter nuclei by forming an inter-nucleus chromosome bridge, which may lead to either cell death or polyploidization. Likewise, cells bearing an acentric chromosome fragment may result in a release of free chromatin or DNA (uncoated with nuclear membrane) into the cytoplasm after a cell division, which triggers the activation of IFN-I, MAPK, and NF-κB (Emming and Schroder Citation2019), or may be detected by the cGAS (cyclic GMP-AMP synthetase)-STING (stimulator of interferon genes) system, which leads to an induction of inflammatory responses (Dou et al. Citation2017; Emming and Schroder Citation2019).
Although one might think that the frequency of chromosome aberrations is not high enough to contribute to radiation carcinogenesis, it probably does contribute since the frequency of total lethal aberrations (dicentrics, rings and fragments) per cell attains to as high as 17% after an exposure to 1 Gy of X rays in density-inhibited normal human fibroblasts (Cornforth and Bedford Citation1987). This means that following a whole-body exposure to 1 Gy, there would be an enormous number of cells that are destined to die. Specifically, among the approximately 106 cells which are present in one cubic millimeter of a tissue, there may be around 105 lethally damaged cells following an exposure to 1 Gy. This type of situation may aid spontaneous carcinogenesis since a sub-threshold inoculum of tumor cells (which by itself does not form a tumor in mice) may form tumors when they are co-transplanted with an order of 105 dead cells (, Sulciner et al. Citation2018). In short, failure to clear the cell debris may lead to cancer progression (Fishbein, Hammock, et al. Citation2020).
The strength and duration of inflammatory conditions
It is mainly radiation oncologists who were concerned with residual radiation damage to normal tissues after radiotherapy for cancer. Consequently, the strength and duration of inflammatory conditions following an exposure to radiation doses below, say, 2 Gy which we are concerned have not been well studied. Nevertheless, it seems probable that the strength of inflammatory conditions is dose dependent. This is because an exposure to radiation causes DNA damage in a dose-dependent manner, and if the damage remains unrepaired, cells may die either through apoptosis which requires activated macrophages for removal of the cell debris (Rastogi et al. Citation2013) and leads to activation of the COX-2-PGE2 cascade or remain quiescent and express a senescence-associated secretory phenotype, and either path can lead to inflammation. If it is assumed that macrophages can clear low levels of cell debris without causing persistent inflammation, one may expect for the presence of a threshold dose. However, at least for radiation pneumonitis, it is reported that there is no evident threshold among lung cancer patients although the dose response curve for the probability of pneumonitis is upward bending (Marks et al. Citation2010). The duration of the inflammatory condition in relation to the dose that we are concerned remains unclear. In a few long-term studies, it is suggested that full recovery from radiation-induced inflammation would take a long time. Examples are the thoracic irradiation of radiation fibrosis-prone C57BL/6 mice (5 or 12.5 Gy) which caused cyclic changes in IL-1α and TGF-β levels etc. in the lung for at least 6 months (Rubin et al. Citation1995); bone marrow cells derived from whole-body irradiated mice with 4 Gy exhibited inflammatory conditions for 15 months after an irradiation (Rastogi et al. Citation2012); intercellular bridges (a manifestation of cell divisions in dicentric-bearing cells) were observed in human thyroid biopsy samples about 20 years after the Chernobyl accident (estimated mean thyroid dose was 2.5 Gy: Nadyrov et al. Citation2012); and finally the presence of increased levels of serum inflammatory markers was suggested in atomic-bomb survivors 50 years after their exposure to radiation (Hayashi et al. Citation2012). In short, it seems possible that any step in the multistep processes involved in carcinogenesis may be affected by radiation-induced tissue injuries depending on the severity of the damage and its duration.
shows a simplified schema which shows associations between radiation-induced damage and inflammation.
The tumor forming microenvironment is mutagenic
In the 1970s, it was found that non-transformed NIH3T3 or C3H10T1/2 cells did not form tumors following subcutaneous transplantation, whereas they did form tumors if the cells were attached to plastic plates or glass beads before transplantation (Boone Citation1975). The result raised the possibility that a tumor forming microenvironment in vivo might be mutagenic. However, those cell lines were not suited for mutagenesis studies. Later, cultured cells derived from a mouse mammary tumor was used to show that mutation frequencies at the HPRT gene could be increased by a factor of 5 to 10 when the cells were co-cultured with macrophages (Yamashina et al. Citation1986). In another study, cultured cells derived from a mouse fibrosarcoma were grown in vivo and in vitro to show that the mutation frequencies were nearly 4 times higher in the former (Wilkinson et al. Citation1995). These results indicate that inflammatory microenvironments are formed following transplantations of plastic plates or during tumor cell growth in vivo, and that TGF-β released from monocytes, epithelial and stroma cells, and ROS/NO released from macrophages and neutrophils could have led to increased mutation rates in neighboring cells, thereby stimulating malignant progressions (Okada Citation2014). It was also found that MMP-9 released from infiltrating neutrophils could contribute to the activation of TGF-β (Germann et al. Citation2020). Therefore, tumor forming conditions can be mutagenic and it is not necessary to assume that an irradiation directly causes oncogenic mutations which then lead to an elevated risk for cancer.
Dead cells may contribute to the creation of a microenvironment favorable for tumor cell growth
In cancer therapy, massive cell death occurs. Removal of cell debris requires phagocytic macrophages while persisting debris may affect the local microenvironment. An example is shown in which indicates that cisplatin-induced dead cell debris stimulates tumor growth when co-injected with a subthreshold inoculum of live tumor cells (Sulciner et al. Citation2018). This cell debris-derived escape from tumor dormancy is partly caused by attracted macrophages which produce a proinflammatory eicosanoid and cytokine storm [activations of soluble epoxide hydrolase (sEH) and COX-2 are involved]. And a dual COX-2/sEH inhibitor 4-(5-phenyl-3-{3-[3-(4-trifluoromethyl-phenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (PTUPB) is shown to prevent debris-stimulated eicosanoid and cytokine storm both in vitro and in vivo and is implicated as an approach to prevent carcinogen-induced cancer (Fishbein, Wang, et al. Citation2020). In this context, tumor associated macrophages are noted to change their functions during tumor progression: i.e., from production of TNF, IL-1, IL-6 etc. at an initiation/promotion phase to production of COX-2, PGE2, IL-10, VEGF etc. in later stages (Mantovani and Sica Citation2010). Another effect of apoptotic cell death results because apoptosis occurs through the activity of caspase 3 which activates iPLA2 (which catalyzes the release of fatty acids from phospholipids) and leads to the activation of the COX-2-PGE2 cascade and the development of inflammation (PGE2 causes vasodilation, fever and pain) (Li et al. Citation2010; Huang et al. Citation2011) ().
Stroma may determine the fate of epithelial cells
It has been thought that an accumulation of oncogenic mutations in epithelial stem cells leads to the development of a cancer. However, there are experimental evidences which do not fit the dogma. For example, in a study which used transgenic mice whose TGF-β type II receptors were knocked out in fibroblasts (Tgfbr2fspKO), stromal hyper-proliferation and epithelial transformation (prostate intraepithelial neoplasia, a possible precursor of prostate cancer, and invasive squamous cell carcinomas in the forestomach) were observed (Bhowmick, Chytil, et al. Citation2004).
In another example, familial juvenile polyposis is a dominant hereditary disease in humans and is caused by a mutation in the BMPR1A or SMAD4 gene. Patients develop numerous benign polyps and show an increased risk for colon cancer. These patients develop stromal abnormalities prior to the development of epithelial aberrations, and all patients showed elevated expression levels of MMP activity (Jacoby et al. Citation1997; Howe et al. Citation1998). Curiously, in a mouse model of this disease developed by knocking out the Smad4 gene, selective inactivation of the gene in T cells caused epithelial cancers throughout the digestive tract, but not when the gene was inactivated in epithelial cells (Kim et al. Citation2006). It appears that the intestinal microenvironment requires the normal functioning of T cells to maintain its homeostasis.
Finally, there are reports which show primary roles for irradiated stroma in the process of malignant transformation. For example, thymic lymphomas may arise from non-treated thymic grafts when they were transplanted into thymectomized irradiated mice (Kaplan and Brown Citation1954; Muto et al. Citation1983); non-irradiated mammary epithelial cells can transform and become tumorigenic more easily when transplanted onto irradiated stroma (Barcellos-Hoff and Ravani Citation2000); and FDC-P1 pre-leukemic cells can transform into a malignant phenotype more rapidly when they are transplanted into irradiated syngeneic hosts, and U16.6 cells can transform into leukemic cells only when transplanted into irradiated hosts (Dührsen and Metcalf Citation1990).
In summary, at least in some instances, an abnormal stroma may lead to tumorigenesis in vivo at the same time as, or prior to the hyper-proliferation of malignant cells (Bhowmick, Neilson, et al. Citation2004; Kalluri and Zeisberg Citation2006). This brings us an old argument about what will promote tumor metastasis: the seed or the soil (the cells or their environment) (Paget Citation1889).
Dormant cancer cells are not rare
It has been recognized that microscopic colonies of cancer cells, termed dormant, latent, occult or in situ cancers, are found frequently in autopsied cases of individuals who did not have cancers during their lifetime. Specifically, over 30% of women had occult breast cancers at ages 40 to 50 years (Folkman and Kalluri Citation2004), and similar frequencies were reported for occult prostate cancers in Caucasian males aged over 60 (Jahn et al. Citation2015) and occult papillary thyroid cancers (OPC) (Harach et al. Citation1985). In the latter OPC study, since sectioning was done at 2- to 3-mm intervals while the majority of OPCs were below 1.0 mm in diameter, the number of OPCs over 0.15 mm in diameter was estimated to be about 300 among 36 thyroid samples. Those occult tumors may be activated by different mechanisms, one of which is neovascularization by VEGF (Udagawa et al. Citation2002) whose expression is dependent on NF-κB (Kiriakidis et al. Citation2003). Since we now know that a radiation exposure can activate NF-κB, microscopic tumors which had existed at the time of an exposure or arise after an exposure may have more opportunities to start growing earlier than usual if radiation-induced inflammatory conditions could last for a sufficiently long time.
In this context, it is worth mentioning that an experimental approach to the study of dormant conditions was developed by transplanting a subthreshold inoculum of, for example, breast cancer cells into mouse fat pads (Krall et al. Citation2018). Interestingly, the dormant tumors’ condition was overridden by introducing a wound to the animals. The effect of the wound was more effective when introduced near the transplanted site of cancer cells but was systemic (even a remote wound could affect the tumor growth), and increased levels of serum IL-6, G-CSF, etc. were observed. It would be of interest to see if an exposure to radiation can mimic the effect of a wound. The results indicated a pre-operative administration of non-steroid anti-inflammatory drug (NSAID) would improve the therapeutic effectiveness, and it was indeed the case (Panigrahy et al. Citation2019).
The immunosuppressive microenvironment and IL-33
For many years, the tumor microenvironment was known to be abnormal because the local immune regulatory network is altered. Specifically, in many instances, CD8+ cytotoxic T cells are disabled and cannot eradicate tumor cells. This inability is partly caused by regulatory T cells (CD4+ CD25+ cells, T-reg cells) and TGF-β is involved in this suppressive effect (Chen et al. Citation2005; Batlle and Massagué Citation2019). On the other hand, IL-33, one of the genes downstream of NF-κB, is induced by an exposure to radiation (Ivanov et al. Citation2010) and promotes T-reg cells (Pastille et al. Citation2019). Thus, an exposure to radiation may lead to not only inflammatory conditions, but also to the creation of an immunosuppressive microenvironment by activating TGF-β and NF-κB which lead to promoting T-reg cells.
The presumed final step: the dynamics of tissue remodeling may facilitate the start of tumor growth
The avian sarcoma virus can induce fibrosarcomas in chickens and induce malignant transformations in various avian cells cultured in vitro, which is attributed to the function of v-jun gene. Transgenic mice that carry the H2 promoter-associated v-jun gene may look normal but they form wound-related fibrosarcomas after a long delay in repairing an induced wound (Schuh et al. Citation1990). These results are reminiscent of the results observed when using the Rous sarcoma virus (RSV). The infection of chick embryos with RSV does not affect normal development and leads to the formation of chickens, but fibrosarcomas can be induced at the location of a wound in the RSV-bearing chickens (Dolberg et al. Citation1985). Interestingly, injection of TGF-β can mimic the conditions present in an induced wound (Sieweke et al. Citation1990). These results indicate that tissue dynamics can override dormant or quiescent conditions.
Discussion
Reasons for the age-at-exposure effect
It is well established that those who were at younger ages at the time of an exposure are at a higher risk of developing cancer according to observations from human epidemiology and animal studies. The underlying mechanism is not understood, however. According to the theory of oncogenic mutation induction by radiation, older individuals are expected to be more responsive (susceptible) to a radiation-induced insult because there may be many somatic cells which have already accumulated some mutations, and a final additional mutation then satisfies the requirements necessary for the development of cancer. Contrary to this expectation, there is no clear experimental evidence that supports this hypothesis while a few studies indicated the opposite results; i.e., the carcinogenic effect of radiation was undetectably small following radiation exposures to old animals (Kohn and Guttman Citation1963; Di Majo et al. Citation1990).
Here, a new model is proposed which postulates that with advancing age, the strength or robustness of tissue responses to injuries may decrease due to decreased levels of the amounts of induced inflammatory factors, and/or to decreased numbers of tissue stem cells. Consequently, older individuals would be likely to show weaker responses following an exposure to radiation than would younger subjects. In fact, it was reported that the induced levels of inflammatory cytokines, adhesion molecules, chemotactic cytokines, and MMP-9 were lower in the brains of older rats after an irradiation (Lee et al. Citation2010).
Radiation-associated carcinogenesis should be preventable
Because the somatic mutation theory has serious difficulties in explaining the mouse survival data, the stromal theory of radiation carcinogenesis is attractive and provides a path to try to explain experimental and epidemiologic observations. In this context, it is interesting to note that when 10T1/2 cells were treated with the protease inhibitor antipain for 10 days after an irradiation, the cells lost their sensitivity to post-irradiation treatments with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA); that is, the cells reverted to a normal stable condition which existed before an irradiation (Kennedy Citation1985). This suggests that radiation-induced unstable conditions (e.g., an increased sensitivity to TPA) may only be temporary and can be suppressed by antipain.
Such a result indicates that a path may exist leading to the prevention of developing radiation-related cancers (Barcellos-Hoff and Nguyen Citation2009). Indeed, treatment of animals after radiation exposures with curcumin (an anti-inflammatory polyphenol: Inano and Onoda Citation2002; Dange et al. Citation2007), an inhibitor of iNOS (Inano and Onoda Citation2005), or a vitamin-E derivative (Ueno et al. Citation2009) was able to reduce the carcinogenic effects of radiation. Efforts have also been made in preventing chemical carcinogenesis: e.g., in a rat model of mammary carcinogenesis by dimethylbenz[a]anthracene, post treatment with a NSAID ibuprofen or celecoxib could not only delay the occurrence of the tumor but also reduce the tumor incidence by half or more (Harris et al. Citation2000). In another rat model of colon carcinogenesis by azoxymethane (AOM), curcumin (Kawamori et al. Citation1999), or a NSAID piroxicum (Reddy et al. Citation1987), sulindac (Rao et al. Citation1995), celecoxib (Reddy et al. Citation2000, Citation2005), or naproxen in combination with atorvastatin (Suh et al. Citation2011) were able to reduce the tumor incidence when they were administered 1 day to more than 10 weeks after the AOM treatment. More recently, in a mouse model of tumor therapy, it was reported that a pre-operative administration of NSAID and/or resolvins could attenuate surgery- or chemotherapy-induced dormancy escape and increases the survival time of the animals (Panigrahy et al. Citation2019; Fishbein, Hammock, et al. Citation2020, Fishbein, Wang, et al. Citation2020). Since a dual COX-2/sEH inhibitor PTUPB was shown to be effective in attenuating the debris-stimulated eicosanoid/cytokine storm (Fishbein, Wang, et al. Citation2020), it is important to know if it works on preventing radiotherapy-induced dormancy escape and radiation-associated cancer development as well.
Cancer risks at low doses
The present study indicates that cancer risks following an exposure to radiation may be better described as an individual’s “time lost in one’s life” rather than as a collective “fold increase”. For example, assuming a linear-non-threshold response at low doses, a 1 mGy acute exposure would cause the loss of one to five days in one’s lifespan if one died from cancer, and there would be no excess cases of cancer patients (Nakamura Citation2020) except for some types of malignancies that are more likely to be induced by radiation (see below). Such a specific length of time may facilitate a better understanding of low dose risks, while it should be kept in mind that there exists a large inter-individual variation in individual lifespans.
Exceptions
It should be kept in mind that some types of malignancies with a baseline incidence which does not increase exponentially with an increase in age (e.g., childhood cancers, leukemia etc.) do not fit the present model.
Conclusion
Traditional theories of radiation carcinogenesis assume that radiation exposures can induce oncogenic mutations and thereby increase the risk of developing cancer. However, carcinogenesis consists of multistep changes of not only tumorigenic cells themselves but also tumor microenvironment, and each step has started to be understood at the molecular revel while their compositions and functions are complex and overlapped. In this report, an effort was made to develop a hypothesis to explain how radiation-induced tissue damage can cause a series of inflammatory events, and how the resulting microenvironment can support the growth of tumor-initiating cells. The formation of such an environment would aid tumor growth and thus permit them to appear earlier than if the tissue had not been exposed to radiation. If this hypothesis were correct, then intervention in the processes of tissue inflammation would retard or attenuate the development of radiation-related cancers.
Acknowledgement
The author is grateful to Dr. Leon Kapp for his careful reading of the manuscript and Dr. Asao Noda for his comments.
Disclosure statement
The author reports no conflict of interest involved in this work.
Additional information
Funding
Notes on contributors
Nori Nakamura
Nori Nakamura, PhD, is a Radiation Biologist and Geneticist working at the Radiation Effects Research Foundation in Hiroshima, Japan, for more than 35 years. He is also interested in biodosimetry methods which include cytogenetic methods and the electron-spin (paramagnetic)-resonance method used to examine tooth enamel.
References
- Aggarwal BB, Vijayalekshmi RV, Sung B. 2009. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 15(2):425–430.
- Albert RE, Altshuler B. 1976. Assessment of environmental carcinogen risks in terms of life shortening. Environ Health Perspect. 13:91–94.
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, PCAWG Consortium, et al. 2020. The repertoire of mutational signatures in human cancer. Nature. 578(7793):94–101.
- Ballin M, Gomez DE, Sinha CC, Thorgeirsson UP. 1988. Ras oncogene mediated induction of a 92 kDa metalloproteinase; strong correlation with the malignant phenotype. Biochem Biophys Res Commun. 154(3):832–838.
- Barcellos-Hoff MH, Nguyen DH. 2009. Radiation carcinogenesis in context: how do irradiated tissues become tumors? Health Phys. 97(5):446–457.
- Barcellos-Hoff MH, Ravani SA. 2000. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 60:1254–1260.
- Batlle E, Massagué J. 2019. Transforming growth Factor-β Signaling in Immunity and Cancer. Immunity. 50(4):924–940.
- Bauer G. 1995. Resistance to tgf-Beta-induced elimination of transformed-cells is required during tumor progression (review-hypothesis). Int J Oncol. 6(6):1227–1229.
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. 2004. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 303(5659):848–851.
- Bhowmick N, Neilson EG, Moses HL. 2004. Stromal fibroblasts in cancer initiation and progression. Nature. 432(7015):332–337.
- Boone CW. 1975. Malignant hemangioendotheliomas produced by subcutaneous inoculation of Balb/3T3 cells attached to glass beads. Science. 188(4183):68–70.
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D, et al. 2015. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 47(4):320–329.
- Calon A, Tauriello DV, Batlle E. 2014. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. 25:15–22.
- Carl C, Flindt A, Hartmann J, Dahlke M, Rades D, Dunst J, Lehnert H, Gieseler F, Ungefroren H. 2016. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell Mol Life Sci. 73(2):427–443.
- Chen M-L, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K. 2005. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA. 102(2):419–424.
- Conlon GA, Murray GI. 2019. Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol. 247(5):629–640.
- Cornforth MN, Bedford JS. 1987. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat Res. 111(3):385–405.
- Dange P, Sarma H, Pandey BN, Mishra KP. 2007. Radiation-induced incidence of thymic lymphoma in mice and its prevention by antioxidants. J Environ Pathol Toxicol Oncol. 26(4):273–279.
- Di Majo V, Coppola M, Rebessi S, Covelli V. 1990. Age-related susceptibility of mouse liver to induction of tumors by neutrons. Radiat Res. 124(2):227–234.
- Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. 1985. Wounding and its role in RSV-mediated tumor formation. Science. 230(4726):676–678.
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 550(7676):402–406.
- Duan C, Yang R, Yuan L, Engelbach JA, Tsien CI, Rich KM, Dahiya SM, Johanns TM, Ackerman JJH, Garbow JR. 2019. Late effects of radiation prime the brain microenvironment for accelerated tumor growth. Int J Radiat Oncol Biol Phys. 103(1):190–194.
- Dührsen U, Metcalf D. 1990. Effects of irradiation of recipient mice on the behavior and leukemogenic potential of factor-dependent hematopoietic cell lines. Blood. 75(1):190–197.
- Dvorak HF. 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 315(26):1650–1659.
- Ehrhart EJ, Segarini P, Tsang ML, Carroll AG, Barcellos-Hoff MH. 1997. Latent transforming growth factor beta1 activation in situ: quantitative and functional evidence after low-dose gamma-irradiation. Faseb J. 11(12):991–1002.
- Emming S, Schroder K. 2019. Tiered DNA sensors for escalating responses. Science. 365(6460):1375–1376.
- Fishbein A, Hammock BD, Serhan CN, Panigrahy D. 2020. Carcinogenesis: failure of resolution of inflammation? Pharmacol Ther. 218:107670.
- Fishbein A, Wang W, Yang H, Yang J, Hallisey VM, Deng J, Verheul SML, Hwang SH, Gartung A, Wang Y, et al. 2020. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc Natl Acad Sci USA. 117(35):21576–21587.
- Folkman J, Kalluri R. 2004. Cancer without disease. Nature. 427(6977):787.
- Fry RJM. 1981. Experimental radiation carcinogenesis: what have we learned? Radiat Res. 87(2):224–239.
- Germann M, Zangger N, Sauvain M-O, Sempoux C, Bowler AD, Wirapati P, Kandalaft LE, Delorenzi M, Tejpar S, Coukos G, et al. 2020. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFβ. EMBO Mol Med. 12(1):e10681.
- Habraken Y, Piette J. 2006. NF-kappaB activation by double-strand breaks . Biochem Pharmacol. 72(9):1132–1141.
- Harach HR, Franssila KO, Wasenius BV-M. 1985. Occult papillary carcinoma of the thyroid. A “normal” finding in Finland. A systematic autopsy study. Cancer. 56(3):531–538.
- Harris RE, Alshafie GA, Abou-Issa H, Seibert K. 2000. Chemoprevention of breast cancer in rats by celecoxib, a cycloocygenase 2 inhibitor. Cancer Res. 60:2101–2103.
- Hayashi T, Morishita Y, Khattree R, Misumi M, Sasaki K, Hayashi I, Yoshida K, Kajimura J, Kyoizumi S, Imai K, et al. 2012. Evaluation of systemic markers of inflammation in atomic-bomb survivors with special reference to radiation and age effects. Faseb J. 26(11):4765–4773.
- Hei TK, Zhou H, Ivanov VN, Hong M, Lieberman HB, Brenner DJ, Amundson SA, Geard CR. 2008. Mechanism of radiation-induced bystander effects: a unifying model. J Pharm Pharmacol. 60(8):943–950.
- Hingorani DV, Lippert CN, Crisp JL, Savariar EN, Hasselmann JPC, Kuo C, Nguyen QT, Tsien RY, Whitney MA, Ellies LG. 2018. Impact of MMP-2 and MMP-9 activation on wound healing, tumor growth and RACPP cleavage. PLoS One. 13(9):e0198464.
- Hirata E, Sahai E. 2017. Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. 7(7):a026781.
- Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, et al. 1998. Mutations in the SMAD4/DPC4 Gene in Juvenile Polyposis. Science. 280(5366):1086–1088.
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan AC, et al. 2011. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 17(7):860–866.
- Inano H, Onoda M. 2002. Radioprotective action of curcumin extracted from curcuma longa Line: inhibitory effect on formation of urinary 8-hydroxy-2’-deoxyguanosine, tumorigenesis, but not mortality, induced by gamma-ray irradiation. Int J Radiat Biol Phys. 53(3):735–743.
- Inano H, Onoda M. 2005. Nitric oxide produced by inducible nitric oxide synthase is associated with mammary tumorigenesis in irradiated rats. Nitric Oxide. 12(1):15–20.
- Ivanov VN, Zhou H, Ghandhi SA, Karasic TB, Yaghoubian B, Amundson SA, Hei TK. 2010. Radiation-induced bystander signaling pathways in human fibroblasts: a role for interleukin-33 in the signal transmission. Cell Signal. 22(7):1076–1087.
- Jacoby RF, Schlack S, Cole CE, Skarbek M, Harris C, Meisner LF. 1997. A Juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 112(4):1398–1403.
- Jahn JL, Giovannucci EL, Stampfer MJ. 2015. The high prevalence of undiagnosed prostate cancer at autopsy: implications for epidemiology and treatment of prostate cancer in the Prostate-specific Antigen-era . Int J Cancer. 137(12):2795–2802.
- Jenkins G. 2008. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol. 40(6-7):1068–1078.
- Kalluri R. 2016. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 16(9):582–598.
- Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat Rev Cancer. 6(5):392–401.
- Kaplan HS, Brown MB. 1954. Development of lymphoid tumors in nonirradiated thymic grafts in thymectomized irradiated mice. Science. 119(3092):439–440.
- Kawamori T, Lubet R, Steele VE, Kelloff GJ, Kaskey RB, Rao CV, Reddy BS. 1999. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 59(3):597–601.
- Kennedy AR. 1985. The conditions for the modification of radiation transformation in vitro by a tumor promoter and protease inhibitors. Carcinogenesis. 6(10):1441–1445.
- Kim B-G, Li C, Qiao W, Mamura M, Kasprzak B, Anver M, Wolfraim L, Hong S, Mushinski E, Potter M, et al. 2006. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 441(7096):1015–1019.
- Kiriakidis S, Andreakos E, Monaco C, Foxwell B, Feldmann M, Paleolog E. 2003. VEGF expression in human macrophages is NF-kappaB-dependent: studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J Cell Sci. 116(Pt 4):665–674.
- Kohn HI, Guttman PH. 1963. Age at exposure and the late effects of X-Rays. Survival and tumor incidence in CAF, mice irradiated at 1 to 2 years of age. Radiat Res. 18:348–373.
- Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, et al. 2010. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. 107(46):20009–20014.
- Kokubo T, Kakinuma S, Kobayashi T, Watanabe F, Iritani R, Tateno K, Nishimura M, Nishikawa T, Hino O, Shimada Y. 2010. Age dependence of radiation-induced renal cell carcinomas in an Eker rat model . Cancer Sci. 101(3):616–623.
- Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, Lambert AW, Bierie B, Ploegh HL, Dougan SK, et al. 2018. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med. 10(436):eaan3464.
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. 2001. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 98(21):12072–12077.
- Lee EJ, Park HJ, Lee IJ, Kim WW, Ha SJ, Suh YG, Seong J. 2014. Inhibition of IL-17A suppresses enhanced-tumor growth in low dose pre-irradiated tumor beds. PLoS One. 9(9):e106423.
- Lee WH, Sonntag WE, Lee YW. 2010. Aging attenuates radiation-induced expression of pro-inflammatory mediators in rat brain. Neurosci Lett. 476(2):89–93.
- Legrand AJ, Poletto M, Pankova D, Clementi E, Moore J, Castro-Giner F, Ryan AJ, O'Neill E, Markkanen E, Dianov GL. 2018. Persistent DNA strand breaks induce a CAF-like phenotype in normal fibroblasts. Oncotarget. 9(17):13666–13681.
- Lemay R, Lepage M, Tremblay L, Therriault H, Charest G, Paquette B. 2017. Tumor cell invasion induced by radiation in Balb/C mouse is prevented by the Cox-2 inhibitor NS-398. Radiation Res. 188(6):685–694.
- Li F, Huang Q, Chen J, Peng Y, Roop D, Bedford JS, Li C-Y. 2010. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 3(110):ra13.
- Little JB. 2000. Radiation carcinogenesis. Carcinogenesis. 21(3):397–404.
- Littlepage LE, Egeblad M, Werb Z. 2005. Coevolution of cancer and stromal cellular responses. Cancer Cell. 7(6):499–500.
- Mantovani A, Sica A. 2010. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 22(2):231–237.
- Marks LB, Bentzen SM, Deasy JO, Kong F-MS, Bradley JD, Vogelius IS, El Naqa I, Hubbs JL, Lebesque JV, Timmerman RD, et al. 2010. Radiation dose-volume effects in the lung. Int J Radiat Oncol Biol Phys. 76(3 Suppl):S70–S76.
- Muto M, Sado T, Hayata I, Nagasawa F, Kamisaku H, Kubo E. 1983. Reconfirmation of indirect induction of radiogenic lymphomas using thymectomized, irradiated B10 mice grafted with neonatal thymuses from Thy 1 congenic donors. Cancer Res. 43(8):3822–3827.
- Nadyrov E, Rozhko A, Kravtsov V, Mabuchi K, Hatch M, Nakamura N, Nikonovich S, Aleksanin S. 2012. Karyopathological traits of thyrocytes and exposure to radioiodines in Belarusian children and adolescents following the accident at the Chernobyl nuclear power plant. Radiat Environ Biophys. 51(2):187–193.
- Nakamura N. 2020. A hypothesis: radiation carcinogenesis may result from tissue injuries and subsequent recovery processes which can act as tumor promoters and lead to an earlier onset of cancer. BJR. 93(1115):20190843.
- Nikitaki Z, Mavragani IV, Laskaratou AD, Gika V, Moskvin VP, Theofilatos K, Vougas K, Stewart RD, Georgakilas AG. 2016. Systemic mechanisms and effects of ionizing radiation: a new 'old' paradigm of how the bystanders and distant can become the players. Semin Cancer Biol. 37-38:77–95.
- Okada F. 2014. Inflammation-related carcinogenesis: Current findings in epidemiological trends, causes and mechanisms. Yonago Acta Med. 57(2):65–72.
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. 2005. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 121(3):335–348.
- Ozasa K, Shimizu Y, Suyama A, Kasagi F, Soda M, Grant EJ, Sakata R, Sugiyama H, Kodama K. 2012. Studies of the mortality of atomic bomb survivors, Report 14, 1950-2003: an overview of cancer and noncancer diseases. Radiat Res. 177(3):229–243.
- Paget S. 1889. The distribution of secondary growths in cancer of the breast. Lancet. 133(3421):571–573.
- Panigrahy D, Gartung A, Yang J, Yang H, Gilligan MM, Sulciner ML, Bhasin SS, Bielenberg DR, Chang J, Schmidt BA, et al. 2019. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J Clin Invest. 129(7):2964–2979.
- Pastille E, Wasmer MH, Adamczyk A, Vu VP, Mager LF, Phuong NNT, Palmieri V, Simillion C, Hansen W, Kasper S, et al. 2019. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 12(4):990–1003.
- Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, Steele V, Reddy BS. 1995. Chemoprevention of colon carcinogenesis by Sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res. 55:1464–1472.
- Rastogi S, Boylan M, Wright EG, Coates PJ. 2013. Interactions of apoptotic cells with macrophages in radiation-induced bystander signaling. Radiat Res. 179(2):135–145.
- Rastogi S, Coates PJ, Lorimore SA, Wright EG. 2012. Bystander-Type Effects Mediated by Long-Lived Inflammatory Signaling in Irradiated Bone Marrow. Radiat Res. 177(3):244–250.
- Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV. 2000. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 60:293–297.
- Reddy BS, Patlolla JM, Simi B, Wang SH, Rao CV. 2005. Prevention of colon cancer by low doses of celecoxib, a cyclooxygenase inhibitor, administered in diet rich in ω-3 polyunsaturated fatty acids. Cancer Res. 65(17):8022–8027.
- Reddy BS, Maruyama H, Kelloff G. 1987. Dose-related inhibition of colon carcinogenesis by dietary Piroxicam, a nonsteroidal antiinflammatory drug, during different stages of rat colon tumor development. Cancer Res. 47(20):5340–5346.
- Reid SE, Scanlon EF, Kaufman MW, Murthy MS. 1996. Role of cytokines and growth factors in promoting the local recurrence of breast cancer. Br J Surg. 83(3):313–320.
- Roberts AB, Lamb LC, Newton DL, Sporn MB, De Larco JE, Todaro GJ. 1980. Transforming growth factors: isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction. Proc Natl Acad Sci USA. 77(6):3494–3498.
- Rubin P, Johnston J, Williams JP, McDonald S, Finkelstein JN. 1995. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 33(1):99–109.
- Ruffell B, Coussens LM. 2015. Macrophages and therapeutic resistance in cancer. Cancer Cell. 27(4):462–472.
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 20(3):174–186.
- Schuh AC, Keating SJ, Monteclaro FS, Vogt PK, Breitman ML. 1990. Obligatory wounding requirement for tumorigenesis in v-jun transgenic mice. Nature. 346(6286):756–760.
- Shellabarger CJ, Chmelevsky D, Kellerer AM. 1980. Induction of mammary neoplasms in the Sprague-Dawley rat by 430keV neutrons and X-rays. J Natl Cancer Inst. 64(4):821–833.
- Shimada Y, Nishimura M, Kakinuma S, Okumoto M, Shiroishi T, Clifton KH, Wakana S. 2000. Radiation-associated loss of heterozygosity at the Znfn1a1 (Ikaros) locus on chromosome 11 in murine thymic lymphomas. Radiat Res. 154(3):293–300.
- Shimoda M, Mellody KT, Orimo A. 2010. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol. 21(1):19–25.
- Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. 1990. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science. 248(4963):1656–1660.
- Storer JB. 1965. Radiation resistance with age in normal and irradiated populations of mice. Radiat Res. 25:435–459.
- Suh N, Reddy BS, DeCastro A, Paul S, Lee H-J, Smolarek AK, So JY, Simi B, Wang CX, Janakiram NB, Steele V, et al. 2011. Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev Res. 4(11):1895–1902.
- Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, Lehner KA, Bielenberg DR, Schmidt B, Dalli J, et al. 2018. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med. 215(1):115–140.
- Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, Chikamatsu K. 2017. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget. 8(5):8633–8647.
- Tanaka S, Tanaka IB, Sasagawa S, Ichinohe K, Takabatake T, Matsushita S, Matsumoto T, Otsu H, Sato F. 2003. No lengthening of life span in mice continuously exposed to gamma rays at very low dose rates. Radiat Res. 160(3):376–379.
- Tlsty TD, Gascard P. 2019. Stromal directives can control cancer. Science. 365(6449):122–123.
- Tsuruoka C, Blyth BJ, Morioka T, Kaminishi M, Shinagawa M, Shimada Y, Kakinuma S. 2016. Sensitive detection of radiation-induced medulloblastomas after acute or protracted gamma-ray exposures in Ptch1 heterozygous mice using a radiation-specific molecular signature. Radiat Res. 186(4):407–414.
- Udagawa T, Fernandez A, Achilles E-G, Folkman J, D'Amato RJ. 2002. Persistence of microscopic human cancers in mice: alterations in the angiogenic balance accompanies loss of tumor dormancy. Faseb J. 16(11):1361–1370.
- Ueno M, Inano H, Onoda M, Murase H, Ikota N, Kagiya TV, Anzai K. 2009. Modification of mortality and tumorigenesis by tocopherol-mono-glucoside (TMG) administered after X irradiation in mice and rats. Radiat Res. 172(4):519–524.
- Upton AC, Kimball AW, Furth J, Christenberry KW, Benedict WH. 1960. Some delayed effects of atomic-bomb radiation in mice. Cancer Res. 20(Pt2):1–59.
- Upton AC, Randolph ML, Conklin JW. 1967. Late effects of fast neutrons and gamma rays in mice as influenced by the dose rate of irradiation: life shortening. Radiat Res. 32(3):493–509.
- Walrath JC, Hawes JJ, Van Dyke T, Reilly KM. 2010. Chapter 4 - Genetically engineered mouse models in cancer research. Adv Cancer Res. 106:113–164.
- Weitzman SA, Weitberg AB, Clark EP, Stossel TP. 1985. Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science. 227(4691):1231–1233.
- Wilkinson D, Sandhu JK, Breneman JW, Tucker JD, Birnboim HC. 1995. Hprt Mutants in a transplantable murine tumour arise more frequently in vivo than in vitro. Br J Cancer. 72(5):1234–1240.
- Withers HR, Milas L. 1973. Influence of preirradiation of lung on development of artificial pulmonary metastases of fibrosarcoma in mice. Cancer Res. 33(8):1931–1936.
- Yamashina K, Miller BE, Heppner GH. 1986. Macrophage-mediated induction of drug-resistant variants in a mouse mammary tumor cell line. Cancer Res. 46:2396–2401.
- Yoshida GJ, Azuma A, Miura Y, Orimo A. 2019. Activated fibroblasts program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. IJMS. 20(9):2256.
- Yu H, Kortylewski M, Pardoll D. 2007. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 7(1):41–51.
- Zhao M, Mishra L, Deng CX. 2018. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 14(2):111–123.