
Abstract
Purpose
Increasing epidemiological and biological evidence suggests that radiation exposure enhances cancer risk in a dose-dependent manner. This can be attributed to the ‘dose-rate effect,’ where the biological effect of low dose-rate radiation is lower than that of the same dose at a high dose-rate. This effect has been reported in epidemiological studies and experimental biology, although the underlying biological mechanisms are not completely understood. In this review, we aim to propose a suitable model for radiation carcinogenesis based on the dose-rate effect in tissue stem cells.
Methods
We surveyed and summarized the latest studies on the mechanisms of carcinogenesis. Next, we summarized the radiosensitivity of intestinal stem cells and the role of dose-rate in the modulation of stem-cell dynamics after irradiation.
Results
Consistently, driver mutations can be detected in most cancers from past to present, supporting the hypothesis that cancer progression is initiated by the accumulation of driver mutations. Recent reports demonstrated that driver mutations can be observed even in normal tissues, which suggests that the accumulation of mutations is a necessary condition for cancer progression. In addition, driver mutations in tissue stem cells can cause tumors, whereas they are not sufficient when they occur in non-stem cells. For non-stem cells, tissue remodeling induced by marked inflammation after the loss of tissue cells is important in addition to the accumulation of mutations. Therefore, the mechanism of carcinogenesis differs according to the cell type and magnitude of stress. In addition, our results indicated that non-irradiated stem cells tend to be eliminated from three-dimensional cultures of intestinal stem cells (organoids) composed of irradiated and non-irradiated stem cells, supporting the stem-cell competition.
Conclusions
We propose a unique scheme in which the dose-rate dependent response of intestinal stem cells incorporates the concept of the threshold of stem-cell competition and context-dependent target shift from stem cells to whole tissue. The concept highlights four key issues that should be considered in radiation carcinogenesis: i.e. accumulation of mutations; tissue reconstitution; stem-cell competition; and environmental factors like epigenetic modifications.
1. Introduction
Cancer is the second leading cause of death worldwide. In 2018, an estimated 9.6 million cancer-related deaths were reported, accounting for one in six deaths (WHO Report on Cancer Citation2020). Cancer is characterized by the autonomous expansion and spread of aberrant somatic cell clones. The abnormal cancerous clone establishes a local microenvironment suitable for the proliferation and development of the ability to defy the mechanisms that control normal cell growth. Cancer cells grow in an uncontrolled manner and metastasize to other organs while evading immune surveillance. Thus, cancer is a major disease that can develop in almost any organ or tissue of the body (Hanahan and Weinberg Citation2011). Metastasis is the major cause of cancer-related deaths. There is no definitive cellular program that dictates the behavior of cancer clones, and individual cancers circumstantially arise from a large pool of potential pathogenic aberrations (Campbell et al. Citation2020). Mutations in somatic cells are the most common cause of cancer. Whether the acquisition of mutations directly causes carcinogenesis has long been debated; however, the parallelism between the two is clear, implying that there is a common denominator (Strong Citation1949). Thus, the theory on the somatic mutation origin of carcinogenesis remains the prevailing paradigm in cancer research.
Conversely, cancer susceptibility varies according to tissue type. For instance, lung, prostate, colorectal, stomach, and liver cancers are more common in men, whereas breast, colorectal, lung, cervical, and thyroid cancers are more common in women (WHO Report on Cancer Citation2020). Some organs are more prone to malignancy than others; in addition, certain types of cancers are more prevalent in children than adults, and vice versa (Howlader et al. Citation2018). The spatiotemporal bias in cancer formation can be partly explained by organ-specific susceptibility to carcinogens and oncogenic mutations; however, it is unclear how these and other factors contribute to the cancer risk of organs (Futreal et al. Citation2004; Danaei et al. Citation2005).
Cells undergo DNA damage caused by intrinsic factors and also have the propensity to stochastically miscopy during DNA replication. Ionizing radiation is a physical stressor that can induce DNA damage. Cells have DNA repair mechanisms in response to such intrinsic and exogenous DNA damage (Chatterjee and Walker Citation2017). However, some DNA repair mechanisms have an inherently error-prone DNA repair pathway, and in rare cases, the DNA sequence may not be restored to its original state. Thus, radiation exposure may induce mutations in DNA in a dose-dependent manner in addition to the naturally induced frequency.
Overall, the process of human carcinogenesis due to radiation exposure (herein, radiation carcinogenesis) has been investigated in cohorts of atomic bomb survivors exposed to high dose-rate radiation (Ozasa et al. Citation2012; Grant et al. Citation2017), and their specific organ cancers (Cahoon et al. Citation2017; Sadakane et al. Citation2019; Sakata et al. Citation2019; Sugiyama et al. Citation2020; Grant et al. Citation2021). Furthermore, protracted/chronic exposure studies, the International Nuclear Workers Study (INWORKS) (Laurier et al. Citation2017; Richardson et al. Citation2018), and epidemiological studies surveying the health effects of environmental releases of radionuclides from nuclear facility accidents, such as the Techa River cohort and children after Fukushima Daiichi Nuclear Power Plant accident (UNSCEAR Citation2017, Annex B, UNSCEAR Citation2020, Citation2021, Annex B), are also ongoing. Genomic analysis of thyroid carcinogenesis after the Chernobyl accident (Morton et al. Citation2021) has revealed that radiosensitivity and frequency of cancer vary among tissues and that cancer risk varies with age at exposure and total dose.
The epidemiological studies on cohorts of residents in high natural background radiation areas (HBRA) reported that chronic exposure to extremely low dose rates does not increase the risk of cancer (Nair et al. Citation2009; Tao et al. Citation2012; Jayalekshmi et al. Citation2021), suggesting that the risk of cancer may differ according to dose rate when compared to the equivalent accumulated dose. This can be interpreted as a ‘dose-rate effect’ for radiation carcinogenesis, in which animal experiments for understanding biological mechanisms from various doses and dose-rates (Paunesku et al. Citation2021), and its contribution to their incorporation into radiation protection (Rühm et al. Citation2016) has been reviewed. However, risks at low dose rates are still controversial due to the wide confidence intervals of low-dose-rate epidemiological studies which have limitations such as uncertainties of doses and confounding factors (UNSCEAR Citation2017, Annex B). Therefore, animal and molecular biological experiments employing a variety of experimental designs should clarify the mechanisms underlying dose-rate effects on carcinogenesis. Recently, the importance of tissue stem cells as targets of radiation-induced cancer risk and the supporting findings on molecular mechanisms are comprehensively updated in the latest UNSCEAR report (UNSCEAR Citation2020, Citation2021, Annex C).
This review summarizes the current findings on the mechanisms of cancer development and radiation carcinogenesis. In addition, molecular biological methods for the mechanistic elucidation of dose-rate effects in radiation carcinogenesis and future perspectives for estimating the risk of radiation carcinogenesis in humans are discussed. Note that we categorized low-doses as doses of less than 100 mGy and low-dose-rates as dose-rates less than 5 mGy/h, following the definition for the radiological protection purpose from stochastic effects of low linear energy transfer radiation (Harrison et al. Citation2021). The goal of this review is to propose a scheme based on our results and recent findings on the dose-rate-dependent response of intestinal stem cells, thus contributing to the estimation of cancer risk by suggesting key issues for future study.
2. Role of tissue stem cells in carcinogenesis
2.1. Tissue stem cells as origin of cancer
In the 1950s, Armitage and Doll proposed a multi-stage carcinogenesis model, which states that the incidence of cancer is proportional to the sixth to seventh power of age (Armitage and Doll Citation1954, Citation1957). The multi-stage carcinogenesis model was later supported by the molecular analysis of human colon cancer. The transition from normal epithelial cells to cancer cells by transformation was accompanied by the stepwise acquisition of mutations in oncogenes and tumor-suppressor genes (driver genes) (Vogelstein et al. Citation1988). In recent pan-cancer analyses of whole genomes (Campbell et al. Citation2020), an average of four to five driver mutations was found in most tumors. Thus, with respect to multi-stage carcinogenesis due to environmental factors, the correlation between the number of driver mutations and tumorigenesis remains largely unchanged.
In multi-stage carcinogenesis, the probability of simultaneous acquisition (accumulation) of multiple driver mutations in the same cell is extremely low and depends on the dose of the environmental mutagens to which the cell is exposed. Therefore, an extremely long period is required to acquire multiple mutations through natural processes. As most cancers in adulthood can be detected in the late stage, carcinogenesis is not an acute reaction as the latency stage before cancer onset can last for years to decades. Moreover, the cells that may cause carcinogenesis are not functional (i.e. terminally differentiated) cells or amplifying progenitors having a short mitotic lifespan, but rather tissue stem cells with a long mitotic lifespan or their direct daughter cells (i.e. early progenitor cells) (Reya et al. Citation2001; Clarke and Fuller Citation2006; Visvader Citation2011).
Stem cells have self-renewal capacity and multipotency, and tissue stem cells can continue to produce tissue-specific lineages throughout life (Reya et al. Citation2001). Tissue stem cells are classified as cycling stem cells, which routinely divide to maintain tissue homeostasis, and quiescent stem cells, which are at the G0 phase of the cell cycle and whose main function is to rescue a pool of cycling stem cells when they are lost due to injury. The strict regulation of cell proliferation is critical for tissue homeostasis, and its disruption can lead to diseases such as cancer and cellular senescence (Rossi et al. Citation2008).
In human leukemia, some tissue stem cell populations have been shown to contain cancer stem cells that are capable of self-renewal and can initiate tumorigenesis upon transplantation into an appropriate host (Lapidot et al. Citation1994; Bonnet and Dick Citation1997). Since then, cancer stem cells have been detected in other types of cancer, including the brain (Hemmati et al. Citation2003; Singh et al. Citation2004), mammary gland (Crabtree and Miele Citation2018), pancreas (Hermann et al. Citation2007), lungs (Ho et al. Citation2007), liver (Roskams Citation2006), and prostate (Lawson and Witte Citation2007). They are similar to normal tissue stem cells as they both exhibit side population traits (Ho et al. Citation2007) and resistance to chemotherapeutic agents. In contrast to rapidly cycling non-cancer stem cells, cancer stem cells are often quiescent in vivo (Urbán and Cheung Citation2021), because their damage checkpoint is consistently elevated (Bao et al. Citation2006).
Studies have also been conducted to understand the risk of carcinogenesis based on the properties of tissue stem cells. For instance, one study analyzed the number of stem cell divisions that occur in tissues during a person’s lifetime and the risk of developing cancer. Thus, the ‘bad-luck hypothesis’ was initially formulated, stating that endogenous factors, such as DNA replication errors, account for approximately 70% of the risk (Tomasetti and Vogelstein Citation2015). Another mathematical model suggests that intrinsic risk factors have a negligible effect on cancer development (<10–30% of lifetime risk) and that cancer risk is heavily influenced by environmental factors (Wu et al. Citation2016). However, It remains unclear whether the endogenous or environmental factors are the driving force of cancer development.
A study on leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5), an intestinal stem cell marker, provided direct evidence that tissue stem cells are the origin of tumors (Barker et al. Citation2009). It also reported that adenomas in the mouse intestine originate from Lgr5+ cells with specifically induced adenomatous polyposis coli, WNT signaling pathway regulator (Apc) mutations by the Cre-loxP system and not from progenitor cells with Apc mutations. Radiation effects, including mutations and tissue responses, on the gastrointestinal tract have been extensively studied (Hendry and Otsuka Citation2016). In the following section, results of this study and the latest findings on the carcinogenic mechanisms in the gastrointestinal tract are briefly summarized.
2.2. Gastrointestinal tract: intestinal stem cells and their radiation sensitivity
The mammalian intestinal epithelium is one of the fastest regenerating tissues (4–5 days in adults) (van der Flier and Clevers Citation2009). Previous studies have identified the structure and functional units of intestinal tissues. There are four differentiated cell types in the intestinal epithelium: enterocytes, mucosecreting or goblet cells, enteroendocrine cells, and Paneth cells in the small intestine (Potten et al. Citation2006). Their common multipotent progenitors, including intestinal stem cells, have been found at the bottom of the crypt (Potten et al. Citation2006). Intestinal crypts are concave in shape, located in the lower intestinal mucosa, and are covered with a monolayer of epithelial cells. During fetal to adult intestinal development, all cells of the mouse fetal intestinal epithelium contribute to the adult intestinal stem cell pool. The dynamics of the intestinal tract are largely reconstructed during fetal life, migrating from the non-proliferative villi to the interstitium, thus establishing the adult stem-cell niche (Guiu et al. Citation2019). Although the mechanism by which intestinal architecture is established during development is unclear, Krndija et al. (Citation2019) reported that active migration driven by actin-related protein (Arp2/3) occurs along the villi and that migratory forces are generated by apicobasal and front-back bipolarity, characterized by actin-rich basal protrusion forces as a key component of the homeostatic turnover of the adult intestinal epithelium. Epithelial regeneration needs to be tightly regulated to maintain a functional barrier in the intestinal structure (Blander Citation2016). Uncontrolled proliferation, migration, or extrusion of cells can lead to various pathologies, such as inflammatory diseases and tumorigenesis (Fearon and Vogelstein Citation1990). Tissue expansion during growth is supported by crypt fission, in which the crypt branches to form two daughter crypts. In adult tissues, very low levels of crypt fission (approximately 3.5%) (Bruens et al. Citation2017), have been observed (Cheng et al. Citation1986; Totafurno et al. Citation1987; Wong WM et al. Citation2002; Yamazaki et al. Citation2020). Quantitative analysis of genetic lineage tracing studies in mice revealed that the low basal rate of crypt fission in the normal murine colon is increased by 30-fold in the precancerous state due to Kirsten rat sarcoma viral oncogene homolog (Kras) mutations (Snippert et al. Citation2014). Thus, the abnormal growth of the differentiated components does not drive the early growth of colorectal adenomas, but rather the division of transformed crypts by branching. Eventually, polyclonal tumors are formed, and these clones compete and crowd each other out (Wong WM et al. Citation2002; Preston et al. Citation2003; Thirlwell et al. Citation2010). Therefore, it is conceivable that excessive crypt fission that occurs with tissue repair after irradiation or tissue injury may play a role in fixing clones that can become the origin of cancer later.
The identification of tissue stem cells in the intestine has been advanced by the studies of two characteristic cell populations. First, 3H-thymidine labeling experiments revealed mitotically active cells near the fourth or fifth position from the crypt base (Cairnie et al. Citation1965), which were termed ‘Position 4′ (+4) cells (Potten et al. Citation2009). Second, electron microscopy images of intestinal tissue showed cone-shaped unique cells in the crypt base (Cheng and Leblond Citation1974), which were termed crypt-base columnar cells (CBCs). The spatial location of +4 cells had fluctuations in the coordinates of their migration direction. Thus, +4 cells exhibit distinguishing characteristics such as label-retention, slower cell cycling time, and extreme radiosensitivity. Nowadays, quiescent stem cells are frequently observed at the +4 position (van Velthoven and Rando Citation2019).
In the screening of Wnt target genes, Lgr5 was detected as a molecular marker expressed in CBCs (Barker et al. Citation2007). Genetic lineage tracing in mice characterized cells expressing the Lgr5 marker as cycling stem cells. These cells frequently self-renew in an equipotent manner to produce transient-amplifying and functional cells that occupy the middle and upper parts of the crypt, respectively (Barker et al. Citation2007, Citation2008, Citation2010). Furthermore, the induction of Apc mutation into cells expressing Lgr5 resulted in tumorigenesis, providing the first experimental evidence that tissue stem cells are the cell-of-origin in cancer (Barker et al. Citation2009).
To understand and predict the dynamics of stem cells under steady-state conditions and in response to stress, it is critical to quantify stress sensitivity and the functional number of stem cells. Moreover, cycling Lgr5 stem cells could maintain a privileged position in the cryptic basal niche by undergoing neutral competition. On average, half of the stem cells lose their direction of differentiation with each division of the stem cell lineage, thereby continuously maintaining the total number of cells (Lopez-Garcia et al. Citation2010; Snippert et al. Citation2010; Kozar et al. Citation2013). Studies in the mouse intestine have averaged 5 stem cells, which was much lower than that of Lgr5+ CBCs (Kozar et al. Citation2013).
Many studies have focused on the radiosensitivity of intestinal stem cells. In models of intestinal damage recovery that evaluate the viability of microcolonies and other crypts, high doses of radiation >10 Gy, close to the so-called intestinal death dose, have been used (Hendry et al. Citation1984). The recovery of tissue stem cells after high-dose irradiation remains an important research topic and ultimately aims to reduce the damage of radiotherapy to normal tissues. Studies assessing the radiosensitivity of Lgr5+ stem cells to radiation at such lethal doses have reported that irradiation induces cell death and reduces the number of tissue stem cells showing different radiosensitivity; the small intestine is radiosensitive, whereas the large intestine is radioresistant (Hua et al. Citation2017; Martin et al. Citation2020).
Regarding radiation protection, assessing the susceptibility of intestinal stem cells to low-dose radiation can reveal the carcinogenic risk; therefore, the effects of low-dose radiation on tissue stem cells have been widely explored. Results showed that the number of Lgr5+ stem cells in the colon is substantially reduced after exposure to 1 Gy irradiation at a high dose rate (30 Gy/h) and that they are more radiosensitive than those in the duodenum (Otsuka et al. Citation2013). The effects of radiation on Lgr5+ stem cells were further investigated using a tamoxifen-driven Cre-loxP recombination technique for tracking the Lgr5 lineage. A marked loss of LacZ+ crypts was observed in Lgr5+ stem cells after high-dose-rate irradiation (30 Gy/h) compared to low-dose rate (0.003 Gy/h). Moreover, the replenishment of the Lgr5+ stem cell pool by unlabeled cells, whose theoretical source is speculated to be quiescent stem cells, was reported (Otsuka et al. Citation2013). However, under low dose-rate irradiation conditions, the rate of replenishment of stem cells decreased, indicating a dose–rate effect (Otsuka and Iwasaki Citation2015). The dynamics of DNA repair and tissue response were also analyzed by quantifying the number of cells expressing the 53BP1 foci, which is a surrogate marker for DNA double-strand breaks (DSBs), and Ki-67+ and phosphorylated histone H3, which are markers for proliferating and mitotic cells. Under high dose-rate radiation conditions, the number of 53BP1 foci immediately increased in Lgr5+ stem cells of the colon, but they were efficiently diminished. In addition, the number of cells in the colonic mucosa was greatly reduced, and mitosis was dramatically induced, which may promote the replenishment of the stem cell pool (Otsuka and Suzuki Citation2016). Subsequently, the replenishment may contribute to the accumulation of genetic mutations in tissue stem cells. These results were the first to reveal the dose–rate effect on the turnover of tissue stem cells in normal tissues, suggesting that different dose rates have different effects on cancer risk. Even in the absence of cell death, high dose-rate irradiation damages all tissue stem cells; DNA damage subsequently causes senescence and depletes stem/progenitor cells within the tissue stem cell pool (Blasco Citation2007; Tümpel and Rudolph Citation2012). In addition to cycling stem cells, tissues retain quiescent stem cells that are resistant to radiation, and rescue tissue reconstruction in case cycling stem cells were drastically depleted (Tian et al. Citation2011). Indeed, the proliferation of quiescent stem cells is induced by high doses of radiation (Montgomery et al. Citation2011; Powell et al. Citation2012).
In the murine multiple intestinal neoplasia (Min) model harboring mutant allele of the Apc locus (ApcMin mice), which is predisposed to intestinal adenoma formation (Moser et al. Citation1995), the downregulation of B cell leukemia/lymphoma 11B (Bcl11b) gene in Lgr5+ stem cells induces proliferation after irradiation and the specific activation of β-catenin target genes in tissue stem cells, leading to further oncogenic events (Sakamaki et al. Citation2015). Cells expressing the quiescent stem cell marker leucine-rich repeats and immunoglobulin-like domains 1 (Lrig1) in the intestine, which is important for rescue from radiation injury, are oncogenic due to Apc mutations (Powell et al. Citation2012). We have also confirmed that gene expression of mouse intestinal crypts irradiated with 10 Gy showed a rapid upregulation of quiescent stem cell markers such as telomerase reverse transcriptase (Tert) and Lrig1, whereas cycling stem cell markers such as Lgr5 and achaete-scute family bHLH transcription factor 2 (Ascl2) were extremely reduced using RNA-Seq (data not shown). Therefore, the replenishment of tissue stem cells from the undamaged stem cell pool without the stimulation of quiescent stem cell proliferation is important for the maintenance of tissue health. It is speculated that the rebuilding of the pool through stem cell replenishment in the recovery from injury in healthy tissues will not be observed in tissues such as ApcMin, which have already been predisposed to cancer in principle, resulting in the absence of dose–rate effect.
3. Reconsidering the pathway of radiation carcinogenesis
After energy deposition by ionizing radiation, the biological mechanism leading to the development of cancer involves the following steps:
DNA damage – Genomic DNA, one of the biological macromolecules in the cell, is directly or indirectly damaged by ionizing radiation.
DNA repair – The damaged DNA is repaired by DNA-repair proteins through various DNA repair pathways.
Genetic mutation – In correlation with the number of DNA lesions, DNA repair rarely causes errors. DNA misrepair may result in a nucleic acid sequence that differs from the original DNA.
Radiation-induced mutagenesis – A mutation in a cancerous driver gene of tissue cells can be the origin of cancer.
Radiation carcinogenesis – The accumulation of more mutations in the initiated cells leads to the progression of cancer and eventually to malignant neoplasms.
The current paradigm of radiation carcinogenesis is summarized in . The first process is a physicochemical event. Therefore, it is reasonable to assume that the occurrence of DNA damage, including the detection of alkali-labile sites (Sakai and Okada Citation1981; Otsuka et al. Citation2006), γ-H2AX focus formation (Rothkamm and Löbrich Citation2003), and chromosome aberrations (Iwasaki et al. Citation2011), has a linear dose relationship in low-dose range. Considering this, there is a proportional risk increment of mutagenesis due to the dose of radiation exposure. Therefore, there is no gap with the paradigm underlying risk assessment in the current radiation protection system, represented by the linear non-threshold model in low-dose range.
Figure 1. Pathways to radiation carcinogenesis in the current paradigm and a revised concept proposed in this review. In the current paradigm, radiation causes DNA damage in normal cells in the initiation process. DNA damage is repaired with the arrest of the cell cycle. Cells that are not repaired properly can go to a pathway to cell death (e.g. apoptosis) and are eliminated from the tissue. Moreover, when the damaged DNA is repaired but fixed as genetic mutations, especially as driver mutations, the cells shift to a precancerous state. Following activation of the promotion and the progression of carcinogenesis, tissues will be cancerized. In the revised concept, the initial process is the same as above, but the important target for mutation is the tissue stem cells or progenitor cells that can repopulate stem-cell population. Tissues then become populations containing pre-cancer cells, but there are pathways by which pre-cancer cells can be eliminated by cell competition. If the majority of stem cells are lost, e.g. due to high-dose exposures, stem-cell replenishment occurs and tissue reconstruction derived from damaged cells is induced. Furthermore, cells that escaped from cell death or cell competition expand in a cancerized field, where microenvironmental changes are enhanced by inflammation and provide a scaffold for their expansion. Such field cancerization presumably supports the abnormal growth of stem cells that survive during mutagenesis. Some of these cells may evolve into non-cancerous and aging diseases, but a population of cells will emerge from those fixed as the predominant clone in the tissue, which will eventually develop into cancer. The schema was adapted from ‘Chemical Carcinogenesis’, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
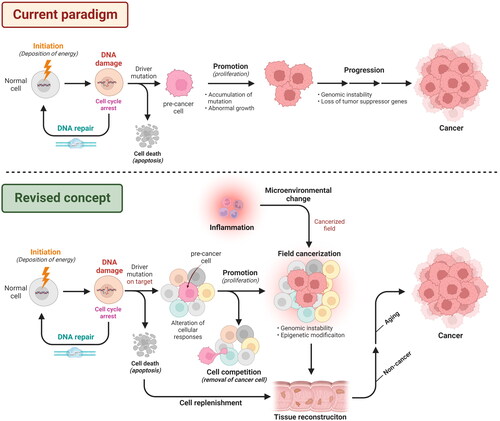
However, biological processes followed by DNA repair suggest that cancer risk does not simply have a linear relationship with input dose. In particular, at low doses and low dose rates, which are the major targets of radiation protection, the contribution of biological responses is significant due to the low physiological stress. In the next section, we would like to clarify the gap with the current paradigm and reconsider the process of radiation carcinogenesis by summarizing our results and new findings.
3.1. DNA repair works precisely under a controlled cell cycle
DSBs are the most severe DNA damage, and the correct repair of genomic information by DNA repair proteins, such as the MRN complex (Tauchi et al. Citation2002) in homologous recombination (HR) and DNA-PKcs/Artemis complex (Chang and Lieber Citation2016) in non-homologous end joining (NHEJ), is crucial for maintaining cell health. To ensure the completion of DSB repair, the cell-cycle checkpoint in proliferating cells is activated, and cell-cycle arrest occurs (Dasika et al. Citation1999). Using a live-cell time-lapse imaging technique, the dose-dependent aggregation of the focus-forming domain of 53BP1, a DSB repair protein, the concurrent induction of cell-cycle arrest, and the release of cell-cycle arrest upon DSB repair were observed (Otsuka and Tomita Citation2018). These events suggest that DNA damage in normal cells is prevented from being carried over to cell division. However, it should be considered that there is a probability of error in DSB repair itself, and induction of DSB repair does not always guarantee genomic fidelity. If errors are stochastic events, there is no gap with existing paradigms regarding DNA repair. However, it is conceivable that the probability may vary with dose and dose-rate. To date, HR, which works when there is a copy allele after DNA synthesis to refer to genetic information complement with the lost sequence at the damaged site, has been associated with very few errors, and NHEJ, which works throughout the cell cycle without referring to any complemental sequence, is considered error-prone. In several decades, the knowledge of NHEJ repair has been updated. In addition, a ‘transcription-coupled DSB repair’ mechanism, in which NHEJ is not performed in the transcriptionally active region of DSBs but is instead switched to HR, was proposed and represents a more accurate DSB repair pathway (Marnef et al. Citation2017; Shibata and Jeggo Citation2020).
In our experimental model for live-cell imaging of DSBs (Otsuka and Tomita Citation2018), a peak in DSB foci during S-phase (DNA replication) was found in normal cells, whereas it remained low during other cell cycle phases (Otsuka and Fujimichi Citation2021). Furthermore, the number of background 53BP1 foci in cancer cells was substantially higher throughout the cell cycle than that in normal cells (unpublished data). These results suggest that the high number of DSBs may reflect genomic instability in cancer cells, which subsequently lead to the induction of mutation. In conclusion, the induction of mutations is itself suppressed by an accurate DNA repair mechanism in healthy cells, and the high number of mutations observed in cancer cells does not explain the cause of cancer.
3.2. Dose-response in genetic mutations
With advancements in next-generation sequencing (NGS) technology, it is now possible to obtain high-quality genomic information at the single-nucleotide level for genomic mutation analysis, such as whole-exome sequencing and whole-genome sequencing. Thus, screening of chemicals that cause various types of mutations has been conducted, focusing on environmental mutagens (Totsuka et al. Citation2021). This NGS technique also revealed a steady accumulation of approximately 40 new mutations per year in all tissue types in humans (Blokzijl et al. Citation2016).
A landmark large-scale study in radiation biology was conducted between the 1960s and 1970s. Radiation-induced mutations were evaluated at seven loci that could affect mouse phenotype. In total, 28 spontaneous mutations (7.5 × 10−6 per locus) were induced in the spermatogonia of 531,500 mice, whereas 111 mutations (13.29 × 10−5 per locus) were found in 119,326 mice irradiated with 600 R (approximately 5.2 Gy) X-rays (Searle Citation1974). Although this study did not report dose response of somatic mutations as it reflected the germline mutations and the survival bias of the mice, the mutations that might be detected by whole-exome sequencing were estimated as 5 events out of 2.1 × 105 exons per Gy, according to our calculation. Satoh et al. (Citation2020) studied the radiation signature in the germline of mice irradiated with 4 Gy and found 9.6 and 4.7 insertion/deletions (indels) in spermatocytes and oocytes, respectively, and 2.5 and 4.7 small clusters of radiation-specific multisite mutations, respectively.
When considering cancer risk, it is important to assess the number of mutations in the somatic cells as a target of cancer. The dose-dependency of somatic mutations was examined by whole-exome sequencing of intestinal organoids established from a single intestinal stem cell. A marked increase in single-nucleotide variants/indels was not detected in clones that could form organoids after irradiation compared to non-irradiated clones. High-dose (4 or 7 Gy) radiation causes loss of organoid-formation potential of intestinal stem cells (manuscript under review). This suggests that it is important to assess mutation frequency in relation to stemness. Interestingly, although radiation accelerated the development of thymic lymphoma in mice with a DNA mismatch repair deficiency, whole exome analysis revealed that the number of radiation-derived mutations in the lymphoma did not increase and the mutation spectrum did not change (Daino et al. Citation2019). This result suggests that the causal relationship between radiation-induced mutations and radiation carcinogenesis needs to be reconsidered.
Considering that the calculated DNA damage induced by 1 Gy is equivalent to the level of endogenous damage that occurs per cell cycle (Vilenchik and Knudson Citation2006; Sage and Shikazono Citation2017), it is not plausible that the radiation-induced mutations are more frequent than naturally occurring mutations. With the rough estimation on the overestimation side of 10 mutations per Gy, spontaneous genomic mutations (40 per year) correspond to 4 Gy per year, supporting the reason why the contribution of mutations to radiation carcinogenesis needs to be reconsidered. This contribution should be even smaller when it takes a longer time, or more cell cycles, at lower dose rates.
3.3. Cell competition is a key process for eliminating aberrant cells
Mutations in driver genes in a population of tissue cells can be the origin of cancer. However, as in the case of low dose-rate radiation exposure, stochastic driver mutations are extremely rare. If the dose rate is sufficiently low, the driver mutations, if any, will be spatiotemporally discrete among cells. Sporadically generated mutant cells can overcome normal cell populations and propagate throughout the tissue depending on their degree of adaptation within the cell community. Interesting findings are accumulating in the field of cellular competition.
Cellular competition is a process in which cells of the same lineage but with different properties compete for survival and space, with the less adapted cells being eliminated as losers, whereas the more adapted populations survive as winners and expand their territories (Clavería and Torres Citation2016; Bowling et al. Citation2019). Multicellular organisms are presumed to have acquired cell competition for quality control during embryogenesis; this homeostatic mechanism is believed to eliminate cancerous transformed cells from the epithelium and thereby to prevent cancer (Martins et al. Citation2014; Merino et al. Citation2016; Vishwakarma and Piddini Citation2020; Kon and Fujita Citation2021).
There are two known modes of eliminating loser cells (Fadul and Rosenblatt Citation2018; Ohsawa et al. Citation2018). The first involves cellular senescence or cell death via apoptosis or necrosis. The induction of cell death-dependent cell competition can be triggered by myc (Clavería et al. Citation2013; Sancho et al. Citation2013; Ellis et al. Citation2019), and apicobasal polarity defective caspase-dependent induction of apoptosis by Scribble and Mahjong knockdown (Tamori et al. Citation2010; Norman et al. Citation2012). The second mode is cell extrusion, in which loser cells are ejected from the epithelium by the physical pressure exerted by the surrounding winner cells. This mode can be initiated by upregulation of Ras mutants (Hogan et al. Citation2009), increased expression of Src (Kajita et al. Citation2010; Anton et al. Citation2018) or ErbB2 (Leung and Brugge Citation2012), and Yes-associated protein (YAP) activation (Chiba et al. Citation2016). The function of most of the identified genes has been studied in cultured cells; however, their roles have also been observed at the tissue and organ levels. For instance, the majority of RasV12 cells were speculated to be apically extruded into the lumen of the mouse intestine to prevent cancer progression (Kon et al. Citation2017).
In the establishment of cellular competition, environmental factors also critically affect the frequency of the elimination of transformed cells. In mice fed with a high-fat diet, the exclusion efficiency of RasV12 transformed cells was reduced due to altered lipid metabolism and inflammatory changes (Sasaki et al. Citation2018). Conversely, caloric restriction increased the elimination of mutated stem cells from the crypt by reducing interaction with the mutated stem cell niche, as demonstrated by the higher number of stem cells in the intestinal crypt than under normal conditions (Igarashi and Guarente Citation2016; Bruens et al. Citation2020). Moreover, new findings have been reported that intestinal stem cells with mutations in Apc, Kras, or Pik3ca win the stem-cell competition by stimulating differentiation through secretion of bone morphogenetic protein (BMP) activators and Wnt inhibitors without increasing cell death of wild-type stem cells in the intestinal crypts (Flanagan et al. Citation2021; van Neerven et al. Citation2021; Yum et al. Citation2021). The fact that the expression of some of these genes increases with age has led to the notion that changes in the environment surrounding stem cells can promote cancer, and the possibility that long-term changes in the microenvironment, such as aging (Chia and DeGregori Citation2021), may act on stem cells should be considered (see below).
3.4. New perspectives based on field cancerization
Although multiple driver mutations can be found in many cancerous tissues, recent evidence demonstrated that driver mutations do exist in normal tissues. Driver mutations in somatic cell carcinoma were present in the lesions of deep invasive endometriosis with low malignancy risk (Anglesio et al. Citation2017). Martincorena et al. (Citation2018) observed NOTCH1 mutations, one of the driver mutations in the esophagus, in 25–42% of esophageal tissues of healthy individuals showing somatic clonal expansion. Similarly, Yokoyama et al. (Citation2019) reported the accumulation of driver mutations during postnatal development in a healthy human esophagus. In mechanistic studies using mouse models, a large number of mutant clones with a positive selection of multiple genes, such as Notch1, Notch2, and Trp53, were detected in normal esophageal tissues, indicating that the dominance of mutant clones depends on the nature of the neighboring cells. The competition between clones with similar adaptations has also been reported to revert to homeostasis (Murai et al. Citation2018; Colom et al. Citation2020). In a study of the human colon, the accumulation of driver mutations was not directly related to carcinogenesis, and the loss of IL-17 was proposed as a mechanism for the expansion of mutant clones by inducing cell death in a niche environment (Nanki et al. Citation2020). These findings suggest that the accumulation of driver mutations in normal tissues is not a direct cause of carcinogenesis but is just a ‘necessary condition’ in carcinogenesis. Recent report demonstrated that accumulation of mutations in the intestinal crypts in various animal species was inversely correlated with the length of life span (Cagan et al. Citation2022). One study also revealed that the mutational spectrum in colorectal cancer (CRC) is derived from the gut microbiota (Pleguezuelos-Manzano et al. Citation2020), indicating that the origin of the mutations plays a critical role in carcinogenesis, whereas the number of mutations is not important. Consequently, how the tissue responds to microenvironmental factors, such as inflammation, may contribute to cancer development.
In addition, not only the accumulation of driver mutations but also the microenvironment where the mutated cells reside is known to be important in cancer development. In 1953, Slaughter et al. first proposed the concept of ‘field cancerization’ to explain the statistically multifocal enrichment of oral squamous cell carcinomas (Slaughter et al. Citation1953). Later, the definition was revised by Braakhuis et al. to ‘the growth of a mutant clone to produce a field of cells predisposed to subsequent tumor growth,’ and the interaction of the mutated cells with their microenvironment determines which mutations are selected for (Braakhuis et al. Citation2003). In field cancerization, a normal cell lineage acquires tumor-promoting genetic mutations or epimutations that undergo positive selection in the microenvironment of normal organs before a malignant lesion grows. Therefore, mutant clones proliferate, creating a large patch of predisposed cells, called cancerized field, that eventually progress to neoplasia (Curtius et al. Citation2018). Curtius et al. further revised the definition of a cancerized field using an evolutionary approach. A cancerized field can be induced by intrinsic DNA replication errors associated with aging, known as ‘unfortunate mutations’ (Tomasetti et al. Citation2017), or by mutagen-induced damage. However, according to Curtius’s definition, areas of tissue with DNA damage or mutations do not yet have a cancer-related phenotype. In addition to DNA mutations, they emphasize exposure to common mutagens and promoters of clonal expansion, as well as multiple independent clonal expansions resulting from subsequent convergent evolution of the epigenome. Immediately after mutagen exposure, the field is composed of many genetically diverse clones, but over time, the clone with the most suitable phenotype is expected to dominate the cancerized field (Driessens et al. Citation2012). This is the case when mutagenic damage continues due to persistent environmental factors such as tobacco-derived carcinogens, diet, and infection (Lochhead et al. Citation2015), new clones are continually generated and the field continues to appear as a genetic mosaic. Eventually, each clone also presents the idea that the best-fitting clone will dominate the field over time through natural selection (Curtius et al. Citation2018).
In intestinal epithelial tissue, the initial neoplastic lesion is polyclonal, derived from multiple independent clones; nevertheless, for the clone to expand, mutant stem cells within the crypt must first replace all other stem cells. Next, the repopulating mutant crypt must undergo crypt fission to form a mutant crypt (Greaves et al. Citation2006; McDonald et al. Citation2008; Baker et al. Citation2014). In the colon, transformed intestinal crypts have a significant impact on adjacent wild-type crypts (Bjerknes and Cheng Citation1999; Thliveris et al. Citation2005). In a healthy colon, the fission rate of the crypts is quite low, once every few decades (Baker et al. Citation2014), and thus the ‘field cancerization drivers’ that clonally expand to form large crypt patches expand by causing a large increase in fission rate.
Although the stromal cells do not progress to cancer per se, abnormal changes in the stroma, or microenvironment, also contribute significantly to field cancerization of the epithelium. In the colon, changes in the microenvironment alter the adaptability of mutations in epithelial cells, as genome-wide association studies of CRC have shown that BMP family members, which are regulated and expressed by stromal cells, are important regulators of cancer risk (Tomlinson et al. Citation2011). Furthermore, in the inflammation of the gut, Trp53 mutations increase stem-cell replacement in a chronic inflammatory environment and play as a field cancerization driver, which means functionally important mutation for phenotypic changes that support a cancerized field, in mouse models (Vermeulen et al. Citation2013). In the absence of inflammation, Trp53 mutant clones have no advantage, and the interaction of the mutation lineage with the microenvironment determines the selective advantage of the phenotype induced by the cancerization driver mutations (Vermeulen et al. Citation2013).
As shown above, several new pieces of evidence for the field cancerization have been reported. This is because the exposure of mutagens and environmental factors act as a driving force for carcinogenesis, and mechanisms of carcinogenic process that focus only on the accumulation of mutations should be reconsidered.
Integrating this evidence, we propose a revised pathway for radiation carcinogenesis that includes cellular competition, replenishment, and changes in cellular responses stimulated by microenvironmental factors, as well as field cancerization ().
4. Dose rate-based cancer model: a context-dependent optimization
In the previous section, we summarized the conventional concept of radiation carcinogenesis and issues arising from new evidence. Here in this section, we would like to discuss how the process of radiation carcinogenesis, including new findings, should be refined in the assessment of radiation carcinogenesis risk. Radiation risk in the Life-Span Study (LSS) of atomic bomb survivors has been extensively investigated (Wakabayashi et al. Citation1983; Thompson et al. Citation1994; Grant et al. Citation2017). Descriptive models linking excess cancer risk and mortality to radiation exposure by statistical association have been the mainstay of cancer-risk assessments at low-dose rates. In addition, biologically based models have been developed to understand the mechanisms of disease progression (Armitage and Doll Citation1957; Moolgavkar et al. Citation1980; Moolgavkar and Knudson Citation1981; Heidenreich and Paretzke Citation2001) and have been applied to model radiation-induced cancer risk in epidemiological studies (Little MP et al. Citation1992; Kai et al. Citation1997; Hazelton et al. Citation2006; Rühm et al. Citation2017). Although biologically based models allow for a more comprehensive risk assessment than descriptive models, they assume biological events where linearity is maintained in the initial processes, such as DNA damage described above. Thus, Kaiser et al. (Citation2021) mentioned that it is not easy to express the risk response as a simple parametric function (Kaiser et al. Citation2021).
With recent advancements in single-cell multi-omics (Kelsey et al. Citation2017; Wagner and Klein Citation2020), the profiling of clonal expansion in normal tissues at single-cell resolution increased our understanding of the somatic evolutionary processes in multicellular human bodies (Nam et al. Citation2021). However, it is still a challenge to parametrically understand the continuum of radiation response from weak stress at low-dose/low-dose-rate to strong stress at high-dose/high-dose-rate. Therefore, it is important to evaluate the factors that are important when considering radiation-induced cancer risk by optimizing them in a situation-dependent manner, i.e. time information as described by dose rate and target shifts (or crossover) of the cells affected. Thus, utilizing experimental animals and human organ models, it is important to select which cell phenomena are important when the level of stress per unit time, such as dose-rate, varies. Based on previous findings including our studies, we propose a four-quadrant event model with two axes (), the threshold of stem-cell competition and the target-cell crossover, as the quantitative information necessary to understand the radiation carcinogenesis based on dose rate, which is important for the assessment of radiation exposure risk as follows: (a) modes of stem-cell competition; (b) the spatiotemporal dynamics of tissue remodeling; (c) mutations occurring in tissue stem cells per cell cycle; and (d) factors that bring changes in the tissue environment.
Figure 2. Cancer model based on dose-rate. (A) Recent evidence supports the idea that induction of carcinogenesis originates in tissue stem cells with driver mutations, whereas severe damage by potent stressors also induces tissue reconstitution on a whole tissue scale. (B) Our studies support the hypothesis that stem-cell competition will be dominant under low dose-rate irradiation, whereas stem-cell replenishment will be enhanced under high dose-rate irradiation. (C) Combined scheme includes two different information sets shown in (A) and (B), which pragmatically consist of two axes: dose-rate (horizontal) and cumulative dose (vertical). Dot lines are ambiguously dividing cell competition and cell replenishment (black) or stem cells and whole tissue (blue) and highlight four key issues (green letters) preferentially important for considering carcinogenesis in different dose/dose-rate situations.
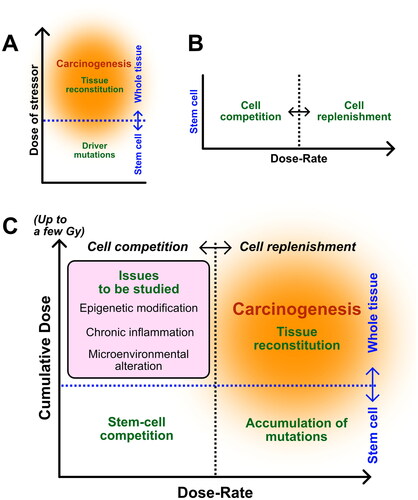
Conventionally, radiation carcinogenesis has been considered a pathway, starting from radiation-induced mutations. However, even at high-dose and high-dose-rates, the most important events would be cell death of tissue stem cells and major changes in the microenvironment that lead to stem-cell replenishment and inflammation. In this case, how the surviving cells reconstruct the tissue and how the cellular senescence of the surviving cells is distributed and developed in the tissue should be clarified.
In cases of low-dose exposure, cell death may not significantly increase because the amount of damage per unit time is small. Therefore, genetic mutations might have accumulated in cells with surviving long periods. However, when considering tissue homeostasis, studying the division period of tissue stem cells is important, as low-dose-rate radiation is sporadic and some short-life stem cells may exist as unexposed. At low-dose-rate, the epigenetic modification should also be considered as another factor that determines the fate of surviving cells. This includes not only stem cells, which are the source of radiation carcinogenesis, but also changes in niches that serve as scaffolds for such cells.
We believe that we can understand the cancer risk at different dose rates when sufficient quantitative information about the underlying stochastic process can be distinguished independently and how each information is connected to the pathway (). In the following section, we discuss the importance of the response of tissue stem cells to different doses and dose rates, as well as the challenges in radiation protection.
4.1. Modes of cell competition determining the fate of tissues
As a quality control mechanism at the tissue level, stem-cell competition has been proposed as a mechanism that contributes to the dose–rate effect in the publication 131 of the International Commission on Radiological Protection (ICRP) (Niwa et al. Citation2015). Quantitative studies of the mode and dose/dose-rate dependence of radiation-induced cellular competition revealed important findings. Particularly, the role of p53 is a noteworthy indicator of radiation effects as a stress response. DNA damage induced by high doses of radiation causes high levels of p53 activity and oscillation (Stewart-Ornstein et al. Citation2017), leading to cell cycle arrest and apoptosis. However, cellular response to mild p53 activity induced by low levels of stress in vivo remains largely unexplored. Bondar and Medzhitov (Citation2010) reported the cell type-dependent threshold for DNA damage levels and observed that p53-mediated cell competition occurs at low DNA damage levels. Furthermore, double heterozygous mice for a murine double minute 2 (MDM2) and MDM4, two major negative regulators of p53, showed mild p53 activation in vivo. Although it was associated with normal embryonic development, mild activation was detrimental to growth. Genetic mosaic embryos containing wild-type cells competed during development based on their p53 levels (Zhang et al. Citation2017). Moreover, the knowledge of radiation-induced cell competition is very limited, these studies on p53-dependent activation indicate that cell competition depends on the strength of the stress response. Upon the transplantation of a population of cells containing irradiated hematopoietic stem cells (HSCs) with non-irradiated cells, the chimerism of irradiated transplanted cells decreased in a dose-dependent manner; the competition for the HSC niche was associated with oxidative stress state (Otsuka et al. Citation2010). Oxidative stress in cells is directly related to the level of DNA damage, which can be reduced by caloric restriction (Qiu et al. Citation2010). In a mouse esophageal model, the combination of antioxidant treatment and low-dose irradiation eliminated wild-type cell proliferation and p53 mutant clones from tissues by differentiation. This suggests that interventions such as reducing external oxidative stress can alter the mutational status of aging tissues (Fernandez-Antoran et al. Citation2019). In some cases, low-dose radiation activated antioxidant enzymes in the hematopoietic system (Otsuka et al. Citation2006), implying that cell competition likely occurs in low-dose exposure environment. In addition, an experimental model of stem-cell competition depicting the competition among tissue stem cells was developed and proved to be the possible origin of cancers in the intestinal epithelium of mice. When irradiated Lgr5+ cells were mixed with non-irradiated ones, the constituent organoids were more likely to exclude previously irradiated stem cells (Fujimichi et al. Citation2019). In a mathematical modeling approach, the ‘strength of competition’ was proposed to play a key role in radiation-induced stem-cell competition (Uchinomiya et al. Citation2020), supporting molecular mechanisms that can explain the strength of competition would fill a necessary gap in our understanding of the dose-rate effect.
As discussed in the previous section, there are two modes of cell competition: one involving cell death and the other involving apical extrusion. Among the cellular responses induced by radiation, cell death induced by high-doses is the most mechanistically explored. However, it is probably a noncompetitive cell death triggered by intrinsic cellular damage. Even if radiation-induced cell death is caused by cell competition, the mode of cell competition in the context of cell death does not necessarily act as elimination of transformed cells. Contrary to cell competition, ‘super-competition’ is also known to occur when mutant cells further proliferate and actively eliminate normal cells (Baker Citation2020). Levayer et al. (Citation2016) proposed that independent of known markers important in cell competition, crowd-induced death leads to a mechanical super-competitor, which promotes tumor growth. In addition to cell competition, compensatory proliferation due to Jun N-terminal kinase (JNK) activation (Wong WY et al. Citation2019) and apoptotic signaling (Fan and Bergmann Citation2008) in irradiated tissues is also known to occur. The ability of radiosensitive cells to replace their population with radioresistant cells due to cell-death signals has been addressed as a challenge in radiotherapy with relatively high doses of radiation (Waghmare et al. Citation2014). Thus, if radiation can induce cell competition or compensatory proliferation in the context of cell death, the possibility of acting on cancer progression should be considered.
Moreover, how stem-cell competition based on apical extrusion works at low-doses and low-dose-rates, where cell death is not dominant, needs to be further investigated for radiation protection considering exposure in the low-doses and low-dose-rates. Molecular indicators of extrusion without cell death, such as humoral factors and cytoskeleton, are being studied. To understand what kind of factors are induced by radiation, it will be important to screen for genes that are specifically expressed in low dose-rate exposure by using next-generation sequencing (Otsuka et al. Citation2018).
In addition, cell competition triggered by intrinsic genomic stresses, such as S-phase replication stress (Dwivedi et al. Citation2021) in C. elegans, has not yet been investigated in vertebrates. As the DNA damage response to replication stress is an evolutionarily conserved mechanism and an unavoidable problem in normal tissue development, it is important to elucidate whether exogenous environmental factors and endogenous genomic stress contribute to tissue maintenance during cell competition. Currently, we are investigating the mechanisms underlying genomic stress-induced cell competition in mammalian cells and tissues using an artificial synthetic gene Focicle (Otsuka and Tomita Citation2018) that allows the simultaneous detection of replication stress and cell cycle phase.
4.2. Tissue reconstitution enhances cancer promotion
Because post-injury tissue remodeling is a further proliferative process that differs from steady-state homeostasis, excess replication stress may lead to mutation accumulation and cancer progression. The origin of carcinogenesis by stem cell and progenitor cell markers is summarized as follows: cells expressing proliferative and quiescent tissue stem cell markers can develop into tumor cells by the acquisition of driver mutations, whereas progenitor and functional cells do not develop into tumor cells through driver mutations alone, but through considerable inflammation (Huels and Sansom Citation2015). Therefore, when considering the risk of cancer development, it is important to evaluate the effect of inflammatory responses on tissues and the accumulation of mutations in tissue stem cells. In adults, quiescent stem cells can resist environmental and radiation stresses in various tissues, and injury can cause clonal expansion of quiescent stem cells, resulting in the reconstruction of both quiescent and proliferating tissues, such as the brain and muscle (Doetsch et al. Citation1999; der Vartanian et al. Citation2019; Scaramozza et al. Citation2019), intestinal epithelium (Montgomery et al. Citation2011), skin (Sada et al. Citation2016), and hair follicles (Matsumura et al. Citation2021). We also evaluated the dynamics of loss of the intestinal stem-cell pool and its replenishment by quiescent cells even at a relatively small dose of approximately 1 Gy (Otsuka et al. Citation2013). In many mouse models, the incidence of tumors is determined by the whole-life regeneration capacity of the mutated cells, thus strongly supporting the idea that stem cells determine the risk of organ cancers and that the risk of cancer markedly increases upon the activation of stem cell function by injury. The combination of mutagenesis of stem cells and exogenous factors that promote the proliferation of these cell populations ultimately determines the risk of organ cancer (Zhu et al. Citation2016).
The mechanisms by which tissue damage increases the risk of cancer are not well-understood (Giroux and Rustgi Citation2017). In a mouse model of pancreatic cancer, tissue damage-associated pancreatitis, along with activating mutations in the KRAS oncogene, markedly accelerated the formation of early neoplastic lesions (Guerra et al. Citation2007; Habbe et al. Citation2008). Recently, mutated KRAS and early events of epigenetic changes after tissue damage cooperatively promote a unique chromatin switch with the alarmin cytokine IL-33 in the pancreatic epithelium have been proposed as a mechanism leading to adenocarcinoma (Alonso-Curbelo et al. Citation2021). Gregorieff et al. (Citation2015) reported that Yap, a downstream transcriptional effector of Hippo signaling pathway, is important for the recovery of the intestinal epithelium after exposure to ionizing radiation. A transient reprogramming of Lgr5+ stem cells and cell survival and regeneration program were induced by activating the epidermal growth factor (EGF) pathway. The inactivation of YAP resulted in the disappearance of adenomas in an ApcMin mouse model of colorectal cancer, indicating that tissue remodeling via the activation of the Hippo pathway plays an important role in cancer development (Moya and Halder Citation2019; Hageman et al. Citation2020).
In summary, defining the ‘minimum dose rate for reconstruction’ for each organ that results in inflammation and the proliferation of quiescent stem cells associated with radiation exposure is important for determining a threshold for estimation of cancer risk.
4.3. Genetic mutations: the stochasticity per cell cycle
As discussed in the previous Section 3.2, mutation is a prerequisite for cancer, and the dynamics of mutant clones in tissues are important factors that lead to increased oncogenic events. However, in parallel with the accumulation of mutations due to permanent endogenous errors, the sporadic accumulation of mutations due to low-dose exposure is dependent on the cell cycle (division frequency) of tissue stem cells; thus, dose rate is an effective factor in the expansion of mutated cell clones. When damaged cells escape apoptosis due to prolonged exposure, as in the case of low dose-rate radiation, the probability of survival of cells with accumulated damage and mutations increases, and this could lead to tumor promotion (Amundson et al. Citation2003). To address this problem, the quantitative relationship between endogenous error mutations and exogenous induced mutations should be investigated at the stem-cell level using advanced NGS technologies.
4.4. Factors that bring changes in the tissue homeostasis
In contrast to the above three factors, it is extremely difficult to consider what factors are relevant to the mechanism of carcinogenesis with respect to the effects of high-dose exposure due to long-term exposure at low dose-rate. Based on a literature survey, we raise three key topics that may make important contributions to long-term tissue homeostasis: epigenetic changes, chronic inflammation, and the microenvironment surrounding stem cells.
Recently, it has become clear that cancer is a disease caused by both genetic and epigenetic alterations, and that solid tumors and leukemias involve both genetic mutations and epigenetic changes, resulting in epigenomic dysregulation that favors cell growth at the expense of the host (Altucci et al. Citation2005; Kim et al. Citation2013; Timp and Feinberg Citation2013). More than 300 genes and gene products have been identified as epigenetically altered in various human cancer tissues (Kanwal and Gupta Citation2010). A meta-analysis of these altered genes in colorectal cancer has confirmed their involvement in tumorigenesis (Durso et al. Citation2017).
Epigenetic changes are known to regulate gene activity and expression not only during development and differentiation but also in response to stimuli from environmental factors such as ionizing radiation (Feil and Fraga Citation2012; Pacchierotti and Spanò Citation2015; Miousse et al. Citation2017; CitationBelli and Tabocchini 2020). A major epigenetic factor is the methylation state of DNA. DNA is partially modified to a high or low methylation state. DNA hypomethylation is associated with an inactive chromatin state and is almost always associated with the suppression of gene expression (Klose and Bird Citation2006; Weber and Schübeler Citation2007; Baylin and Jones Citation2016). Loss of genome-wide methylation promotes genomic instability, a major hallmark of cancer (Hoffmann and Schulz Citation2005; Rodriguez et al. Citation2006; Negrini et al. Citation2010). Conversely, hypermethylation of genes may be associated with their transcriptional repression. Loss of function of repressed genes may be an important event contributing to carcinogenesis (Baylin and Jones Citation2011); dramatic changes in DNA methylation are even more frequent in cancer than genetic mutations (Zingg and Jones Citation1997; Zhao and Epstein Citation2008) and are considered early events (Jones and Gonzalgo Citation1997).
There are conflicting reports on the effects of radiation on DNA methylation status, including reports that radiation exposure promotes DNA methylation (Goetz et al. Citation2011; Miousse et al. Citation2017) and DNA hypomethylation in a dose-dependent, sex-and tissue-specific manner (Tawa et al. Citation1998; Pogribny et al. Citation2004; Raiche et al. Citation2004; Koturbash et al. Citation2005).
Thus, there is currently no consensus regarding genetic mutations and epigenetic changes responsible for carcinogenesis. However, the following three consequences, i.e. (i) epigenetic abnormalities, in addition to genetic alterations, are involved in the development and progression of cancer (Jones and Baylin Citation2002; Sharma et al. Citation2010); (ii) γ-ray-induced initiation at a specific locus in the genome is a mutation as the first step in carcinogenesis (Kamiya et al. Citation1995; Clifton Citation1996; Feinberg et al. Citation2006); and (iii) radiation-induced frequency of genomic instability is considerably higher than the frequency of genetic mutations at comparable doses (Little JB Citation2000; Morgan Citation2003), suggesting that mutations alone are highly unlikely to initiate genomic instability. Epigenetically altered stem/progenitor cells play an important role in the initial process of carcinogenesis, which is consistent with the nature of cancer stem cells (Feinberg et al. Citation2006; Jordan et al. Citation2006).
Some biological effects of low-dose radiation classified as ‘beneficial,’ have recently been highlighted as one of the proposed hallmarks of health (López-Otín and Kroemer Citation2021). These effects are often associated with epigenetic mechanisms. Evidence for reduced incidence of naturally occurring cancers in model organisms and the involvement of epigenetic mechanisms in hormesis-like and life-extending responses has been reported (Vaiserman Citation2011). Chronic low-dose radiation exposure (ranging from µGy/h to mGy/sec) is a more potent inducer of epigenetic changes than acute exposure, e.g. DNA methylation and hypermethylation induced by chronic low-dose γ-irradiation (Kovalchuk et al. Citation2004; Taki et al. Citation2009; Ye et al. Citation2013; Horemans et al. Citation2019). Although the majority of experimental observations are due to chronic but high doses of radiation, chronic exposure at very low levels, such as in the natural background, has been speculated to trigger defense mechanisms such as activation of antioxidant enzymes via epigenetic mechanisms without genetic changes (Fratini et al. Citation2015).
Chronic inflammation is detrimental to tissue homeostasis, as shown by its involvement in the development and progression of cancer, non-cancer, and various metabolic diseases (De Nardo and Latz Citation2011). Besides, chronic inflammation affects stem-cell behavior in many chronic diseases. So, the importance of understanding the cross-talk mechanisms between the inflammatory environment and tissue stem cells is the issue (Kizil et al. Citation2015). Inflammation occurring in the microenvironment of intestinal tissues mediated by pro-inflammatory cytokines, particularly tumor necrosis factor (TNF), Wnt signaling by macrophages (Naito et al. Citation2003; Saha et al. Citation2016), Notch and Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) signaling (Taniguchi et al. Citation2015), IL-6 (Rigby et al. Citation2007; Shaker et al. Citation2010), IL-17 (Andoh et al. Citation2007; Grivennikov et al. Citation2012; Nanki et al. Citation2020), and IFN-γ (Nusse et al. Citation2018) have been reported to play important roles in the proliferation and tissue remodeling of intestinal progenitor cells. Similarly, anti-inflammatory effects mediated by IL-22 (Huber et al. Citation2012; Lindemans et al. Citation2015), defensin, and NOD2 (Netea et al. Citation2004; Ramasundara et al. Citation2009) released by paneth cells (Keshav Citation2006), and transforming growth factor (TGF-β) play important roles in tissue repair, including the microenvironment. However, understanding the correlation between pro-inflammatory and anti-inflammatory effects in the microenvironment is extremely challenging. For example, upregulation of TGF-β signaling has been shown to underlie inflammatory diseases and is involved in promoting tumor emergence, immunosuppression in the tumor microenvironment, and tumor immune evasion (Batlle and Massagué Citation2019). Interestingly, Miyoshi et al. found that Wnt5a, which is an essential ligand for stem-cell proliferation, is activated by TGF-β signaling after tissue injury in the mouse intestinal microenvironment (Miyoshi et al. Citation2012). Since ionizing radiation induces the activation of TGF-β in microenvironment (Shimura Citation2021), TGF-β may be one of the key molecules involved in the altered inflammatory state of the microenvironment following irradiation. Therefore, assessing the effects of low-dose and low-dose-rate radiation in terms of the kinetics of TGF-β expression surrounding tissue stem cells may contribute to unraveling tissue homeostasis and risk estimation toward disease.
5. Conclusions and future perspectives
In this review, we discussed key issues to be considered in radiation carcinogenesis, including mutation on tissue stem cells, microenvironmental alteration, stem-cell competition, and the concept of context-dependent tissue reconstruction, which emphasizes not only mutation accumulation but also environmental factors such as epigenetic modifications and field cancerization. Considering these key events, we proposed a unique scheme that incorporates and demonstrates a dose-rate dependent response of tissue stem cells. As the radiation response of tissues is diverse across various organs, a uniform coefficient for each individual may not always be appropriate. The variance of response may also be related to individual susceptibility, which is important to be addressed for radiological protection. Differences in radiosensitivity among individuals or groups may be related to epigenetic variation, which is affected by gender, age at exposure, health status, genetic and epigenetic modification, lifestyle, and attained age (Kreuzer et al. Citation2018; Seibold et al. Citation2020). Radiation protection studies have not explored the implications of epigenetic effects in radiation-induced cancer, and the development of mechanistic models of radiation action is required. As epigenetic changes are increasingly recognized as important factors contributing to cancer development, there is a need to construct models of radiation-induced cancer that incorporate both genetic and epigenetic effects in the estimation of radiation risk (CitationBelli and Tabocchini 2020). We believe that an organ-specific understanding of the differences in cell mutation, epigenetics, modes of stem-cell competition, and tissue remodeling among individuals in the context of dose–rate effects can guide the best management approach for personalized radiation protection.
Acknowledgments
The authors would like to thank members of our laboratory in Central Research Institute of Electric Power Industry for their helpful comments and suggestions in the preparation of this manuscript.
Disclosure statement
The authors declare that there are no conflicts of interest regarding the publication of the paper.
Additional information
Funding
Notes on contributors
Kensuke Otsuka
Kensuke Otsuka, Ph.D., is a Senior Research Scientist of Central Research Institute of Electric Power Industry (CRIEPI). He is a molecular biologist working on the radiation responses of tissue stem cells using various approaches including animal models, live-cell imaging, and organoids.
Toshiyasu Iwasaki
Toshiyasu Iwasaki, Ph.D., is an Associate Vice President and Program Manager of Nuclear Energy (Radiation Safety) of CRIEPI. He is a molecular biologist and currently overseeing the in-house programme on radiation effects and radiological protection.
References
- Alonso-Curbelo D, Ho YJ, Burdziak C, Maag JLV, Morris JP, Chandwani R, Chen HA, Tsanov KM, Barriga FM, Luan W, et al. 2021. A gene–environment-induced epigenetic program initiates tumorigenesis. Nature. 590(7847):642–648.
- Altucci L, Clarke N, Nebbioso A, Scognamiglio A, Gronemeyer H. 2005. Acute myeloid leukemia: Therapeutic impact of epigenetic drugs. Int J Biochem Cell Biol. 37(9):1752–1762.
- Amundson SA, Lee RA, Koch-Paiz CA, Bittner ML, Meltzer P, Trent JM, Fornace AJ. 2003. Differential responses of stress genes to low dose-rate gamma irradiation. Mol Cancer Res. 1(6):445–452.
- Andoh A, Ogawa A, Bamba S, Fujiyama Y. 2007. Interaction between interleukin-17-producing CD4+ T cells and colonic subepithelial myofibroblasts: what are they doing in mucosal inflammation? J Gastroenterol. 42(S17):29–33.
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, et al. 2017. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 376(19):1835–1848.
- Anton KA, Kajita M, Narumi R, Fujita Y, Tada M. 2018. Src-transformed cells hijack mitosis to extrude from the epithelium. Nat Commun. 9(1):4695.
- Armitage P, Doll R. 1954. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 8(1):1–12.
- Armitage P, Doll R. 1957. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 11(2):161–169.
- Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, et al. 2014. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 8(4):940–947.
- Baker NE. 2020. Emerging mechanisms of cell competition. Nat Rev Genet. 21(11):683–697.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444(7120):756–760.
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 449(7165):1003–1007.
- Barker N, Wetering M v d, Clevers H. 2008. The intestinal stem cell. Genes Dev. 22(14):1856–1864.
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 457(7229):608–611.
- Barker N, Bartfeld S, Clevers H. 2010. Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell. 7(6):656–670.
- Batlle E, Massagué J. 2019. Transforming growth factor-β signaling in immunity and cancer. Immunity. 50(4):924–940.
- Baylin SB, Jones PA. 2011. A decade of exploring the cancer epigenome-biological and translational implications. Nat Rev Cancer. 11(10):726–734.
- Baylin SB, Jones PA. 2016. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 8(9):a019505.
- Belli M, Tabocchini MA. 2020. Ionizing radiation-induced epigenetic modifications and their relevance to radiation protection. IJMS. 21(17):5934–5993.
- Bjerknes M, Cheng H. 1999. Clonal analysis of mouse intestinal epithelial progenitors. Gastroenterology. 116(1):7–14.
- Blander JM. 2016. Death in the intestinal epithelium—basic biology and implications for inflammatory bowel disease. FEBS J. 283(14):2720–2730.
- Blasco MA. 2007. Telomere length, stem cells and aging. Nat Chem Biol. 3(10):640–649.
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. 2016. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 538(7624):260–264.
- Bondar T, Medzhitov R. 2010. p53-Mediated hematopoietic stem and progenitor cell competition. Stem Cell. 6(4):309–322.
- Bonnet D, Dick JE. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 3(7):730–737.
- Bowling S, Lawlor K, Rodríguez TA. 2019. Cell competition: the winners and losers of fitness selection. Development. 146(13):dev167486.
- Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. 2003. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 63(8):1727–1730.
- Bruens L, Ellenbroek SIJ, van Rheenen J, Snippert HJ. 2017. In vivo imaging reveals existence of crypt fission and fusion in adult mouse intestine. Gastroenterology. 153(3):674–677.e3.
- Bruens L, Ellenbroek SIJ, Suijkerbuijk SJE, Azkanaz M, Hale AJ, Toonen P, Flanagan DJ, Sansom OJ, Snippert HJ, van Rheenen J. 2020. Calorie restriction increases the number of competing stem cells and decreases mutation retention in the intestine. Cell Rep. 32(3):107937.
- Cahoon EK, Preston DL, Pierce DA, Grant E, Brenner AV, Mabuchi K, Utada M, Ozasa K. 2017. Lung, laryngeal and other respiratory cancer incidence among Japanese atomic bomb survivors: an updated analysis from 1958 through 2009. Radiat Res. 187(5):538–548.
- Cairnie AB, Lamerton LF, Steel GG. 1965. Cell proliferation studies in the intestinal epithelium of the rat. I. Determination of the kinetic parameters. Exp Cell Res. 39(2):528–538.
- Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, Perry MD, Nahal-Bose HK, Ouellette BFF, Li CH, et al. 2020. Pan-cancer analysis of whole genomes. Nature. 578:82–93.
- Chang HHY, Lieber MR. 2016. Structure-specific nuclease activities of artemis and the artemis: DNA-PKcs complex. Nucleic Acids Res. 44(11):4991–4997.
- Chatterjee N, Walker GC. 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 58(5):235–263.
- Cheng H, Leblond C. 1974. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. I. Columnar cell, II. mucous cell, III. entero-endocrine cells, IV. Am J Anat. 141(4):461–479.
- Cheng H, Bjerknes M, Amar J, Gardiner G. 1986. Crypt production in normal and diseased human colonic epithelium. Anat Rec. 216(1):44–48.
- Chia SB, DeGregori J. 2021. Cancer stem cells in the gut have a bad influence on neighbouring cells. Nature. 594(7863):340–341.
- Chiba T, Ishihara E, Miyamura N, Narumi R, Kajita M, Fujita Y, Suzuki A, Ogawa Y, Nishina H. 2016. MDCK cells expressing constitutively active Yes-associated protein (YAP) undergo apical extrusion depending on neighboring cell status. Sci Rep. 6:28383.
- Clarke MF, Fuller M. 2006. Stem cells and cancer: two faces of Eve. Cell. 124(6):1111–1115.
- Clavería C, Torres M. 2016. Cell competition: mechanisms and physiological roles. Annu Rev Cell Dev Biol. 32:411–439.
- Clavería C, Giovinazzo G, Sierra R, Torres M. 2013. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 500(7460):39–44.
- Clifton KH. 1996. Comments on the evidence in support of the epigenetic nature of radiogenic initiation. Mutat Res. 350(1):77–80.
- Colom B, Alcolea MP, Piedrafita G, Hall MWJ, Wabik A, Dentro SC, Fowler JC, Herms A, King C, Ong SH, et al. 2020. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat Genet. 52(6):604–614.
- Cagan A, Baez-Ortega A, Brzozowska N, Abascal F, Coorens THH, Sanders MA, Lawson ARJ, Harvey LMR, Bhosle S, Jones D, et al. 2022. Somatic mutation rates scale with lifespan across mammals. Nature. 604(7906):517–524.
- Crabtree JS, Miele L. 2018. Breast cancer stem cells. Biomedicines. 6(3):77.
- Daino K, Ishikawa A, Suga T, Amasaki Y, Kodama Y, Shang Y, Hirano-Sakairi S, Nishimura M, Nakata A, Yoshida M, et al. 2019. Mutational landscape of T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1: no synergism with ionizing radiation. Carcinogenesis. 40(2):216–224.
- Curtius K, Wright NA, Graham TA. 2018. An evolutionary perspective on field cancerization. Nat Rev Cancer. 18(1):19–32.
- Danaei G, Vander Hoorn S, Lopez AD, Murray CJL, Ezzati M. 2005. Causes of cancer in the world: Comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 366(9499):1784–1793.
- Dasika GK, Lin SCJ, Zhao S, Sung P, Tomkinson A, Lee E. 1999. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene. 18(55):7883–7899.
- De Nardo D, Latz E. 2011. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 32(8):373–379.
- der Vartanian A, Quétin M, Michineau S, Auradé F, Hayashi S, Dubois C, Rocancourt D, Drayton-Libotte B, Szegedi A, Buckingham M, et al. 2019. PAX3 confers functional heterogeneity in skeletal muscle stem cell responses to environmental stress. Cell Stem Cell. 24(6):958–973.e9.
- Doetsch F, García-Verdugo JM, Alvarez-Buylla A. 1999. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A. 96(20):11619–11624.
- Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. 2012. Defining the mode of tumour growth by clonal analysis. Nature. 488(7412):527–530.
- Durso DF, Bacalini MG, do Valle ÍF, Pirazzini C, Bonafé M, Castellani G, Faria AMC, Franceschi C, Garagnani P, Nardini C. 2017. Aberrant methylation patterns in colorectal cancer: a meta-analysis. Oncotarget. 8(8):12820–12830.
- Dwivedi VK, Pardo-Pastor C, Droste R, Kong JN, Tucker N, Denning DP, Rosenblatt J, Horvitz HR. 2021. Replication stress promotes cell elimination by extrusion. Nature. 593(7860):591–596.
- Ellis SJ, Gomez NC, Levorse J, Mertz AF, Ge Y, Fuchs E. 2019. Distinct modes of cell competition shape mammalian tissue morphogenesis. Nature. 569(7757):497–502.
- Fadul J, Rosenblatt J. 2018. The forces and fates of extruding cells. Curr Opin Cell Biol. 54:66–71.
- Fan Y, Bergmann A. 2008. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 14(3):399–410.
- Fearon ER, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell. 61(5):759–767.
- Feil R, Fraga MF. 2012. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 13(2):97–109.
- Feinberg AP, Ohlsson R, Henikoff S. 2006. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 7(1):21–33.
- Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, Jones PH. 2019. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell. 25(3):329–341.e6.
- Flanagan DJ, Pentinmikko N, Luopajärvi K, Willis NJ, Gilroy K, Raven AP, Mcgarry L, Englund JI, Webb AT, Scharaw S, et al. 2021. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature. 594(7863):430–435.
- Fratini E, Carbone C, Capece D, Esposito G, Simone G, Tabocchini MA, Tomasi M, Belli M, Satta L. 2015. Low-radiation environment affects the development of protection mechanisms in V79 cells. Radiat Environ Biophys. 54(2):183–194.
- Fujimichi Y, Otsuka K, Tomita M, Iwasaki T. 2019. An efficient intestinal organoid system of direct sorting to evaluate stem cell competition in vitro. Sci Rep. 9(1):20297.
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. 2004. A census of human cancer genes. Nat Rev Cancer. 4(3):177–183.
- Giroux V, Rustgi AK. 2017. Metaplasia: tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat Rev Cancer. 17(10):594–604.
- Goetz W, Morgan MNM, Baulch JE. 2011. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat Res. 175(5):575–587.
- Grant EJ, Brenner A, Sugiyama H, Sakata R, Sadakane A, Utada M, Cahoon EK, Milder CM, Soda M, Cullings HM, et al. 2017. Solid cancer incidence among the life span study of atomic bomb survivors: 1958-2009. Radiat Res. 187(5):513–537.
- Grant EJ, Yamamura M, Brenner AV, Preston DL, Utada M, Sugiyama H, Sakata R, Mabuchi K, Ozasa K. 2021. Radiation risks for the incidence of kidney, bladder and other urinary tract cancers: 1958-2009. Radiat Res. 195(2):140–148.
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. 2015. Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature. 526(7575):715–718.
- Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, Leedham SJ, Deheragoda M, Sasieni P, Novelli MR, et al. 2006. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci USA. 103(3):714–719.
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 491(7423):254–258.
- Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 11(3):291–302.
- Guiu J, Hannezo E, Yui S, Demharter S, Ulyanchenko S, Maimets M, Jørgensen A, Perlman S, Lundvall L, Mamsen LS, et al. 2019. Tracing the origin of adult intestinal stem cells. Nature. 570(7759):107–111.
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, et al. 2008. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci USA. 105(48):18913–18918.
- Hageman JH, Heinz MC, Kretzschmar K, van der Vaart J, Clevers H, Snippert HJG. 2020. Intestinal regeneration: regulation by the microenvironment. Dev Cell. 54(4):435–446.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell. 144(5):646–674.
- Harrison JD, Balonov M, Bochud F, Martin C, Menzel HG, Ortiz-Lopez P, Smith-Bindman R, Simmonds JR, Wakeford R. 2021. ICRP Publication 147: use of dose quantities in radiological protection. Ann ICRP. 50(1):9–82.
- Hazelton W, Moolgavkar S, Curtis S, Zielinski J, Patrick Ashmore J, Krewski D. 2006. Biologically based analysis of lung cancer incidence in a large Canadian occupational cohort with low-dose ionizing radiation exposure, and comparison with Japanese atomic bomb survivors. J Toxicol Environ Health A: Curr Iss. 69(11):1013–1038.
- Heidenreich WF, Paretzke HG. 2001. The two-stage clonal expansion model as an example of a biologically based model of radiation-induced cancer. Radiat Res. 156(5 Pt 2):678–681.
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. 2003. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 100(25):15178–15183.
- Hendry JH, Otsuka K. 2016. The role of gene mutations and gene products in intestinal tissue reactions from ionising radiation. Mutat Res Rev Mutat Res. 770(Pt B):328–339.
- Hendry JH, Moore J. v, Potten CS. 1984. The proliferative status of microcolony‐forming cells in mouse small intestine. Cell Tissue Kinet. 17(1):41–47.
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. 2007. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 1(3):313–323.
- Ho MM, Ng A. v, Lam S, Hung JY. 2007. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 67(10):4827–4833.
- Hoffmann MJ, Schulz WA. 2005. Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol. 83(3):296–321.
- Hogan C, Dupré-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling E, Piddini E, Baena-López LA, Vincent J, Itoh Y, et al. 2009. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 11(4):460–467.
- Horemans N, Spurgeon DJ, Lecomte-Pradines C, Saenen E, Bradshaw C, Oughton D, Rasnaca I, Kamstra JH, Adam-Guillermin C. 2019. Current evidence for a role of epigenetic mechanisms in response to ionizing radiation in an ecotoxicological context. Environ Poll. 251:469–483.
- Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis D, et al. 2018. SEER cancer statistics review, 1975-2018. National Cancer Institute (US); [accessed 2020 Aug 5]. https://seer.cancer.gov/archive/csr/1975_2018/.
- Hua G, Wang C, Pan Y, Zeng Z, Lee SG, Martin ML, Haimovitz-Friedman A, Fuks Z, Paty PB, Kolesnick R. 2017. Distinct levels of radioresistance in Lgr5+ colonic epithelial stem cells versus Lgr5+ small intestinal stem cells. Cancer Res. 77(8):2124–2133.
- Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O'Connor W, Jr., Murphy AJ, et al. 2012. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 491(7423):259–263.
- Huels DJ, Sansom OJ. 2015. Stem vs non-stem cell origin of colorectal cancer. Br J Cancer. 113(1):1–5.
- Igarashi M, Guarente L. 2016. mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell. 166(2):436–450.
- Iwasaki T, Takashima Y, Suzuki T, Yoshida MA, Hayata I. 2011. The dose response of chromosome aberrations in human lymphocytes induced in vitro by very low-dose γ rays. Radiat Res. 175(2):208–213.
- Jayalekshmi PA, Nair RA, Nair RRK, Hoel DG, Akiba S, Nakamura S, Endo K. 2021. Background radiation and cancer excluding leukemia in Kerala, India – Karunagappally cohort study. Radiat Environ Med. 10:74–81.
- Jones PA, Baylin SB. 2002. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 3(6):415–428.
- Jones PA, Gonzalgo ML. 1997. Altered DNA methylation and genome instability: a new pathway to cancer? Proc Natl Acad Sci U S A. 94(6):2103–2105.
- Jordan CT, Guzman ML, Noble M. 2006. Cancer stem cells. N Engl J Med. 355(12):1253–1261.
- Kai M, Luebeck EG, Moolgavkar SH. 1997. Analysis of the incidence of solid cancer among atomic bomb survivors using a two-stage model of carcinogenesis. Radiat Res. 148(4):348–358.
- Kaiser JC, Blettner M, Stathopoulos GT. 2021. Biologically based models of cancer risk in radiation research. Int J Radiat Biol. 97(1):2–11.
- Kajita M, Hogan C, Harris AR, Dupré-Crochet S, Itasaki N, Kawakami K, Charras G, Tada M, Fujita Y. 2010. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J Cell Sci. 123(2):171–180.
- Kamiya K, Yasukawa-Barnes J, Mitchen JM, Gould MN, Clifton KH. 1995. Evidence that carcinogenesis involves an imbalance between epigenetic high-frequency initiation and suppression of promotion. Proc Natl Acad Sci U S A. 92(5):1332–1336.
- Kanwal R, Gupta S. 2010. Epigenetics and cancer. J Appl Physiol. 109(2):598–605.
- Kelsey G, Stegle O, Reik W. 2017. Single-cell epigenomics: recording the past and predicting the future. Science. 358(6359):69–75.
- Keshav S. 2006. Paneth cells: Leukocyte-like mediators of innate immunity in the intestine. J Leukoc Biol. 80(3):500–508.
- Kim JG, Park MT, Heo K, Yang KM, Yi JM. 2013. Epigenetics meets radiation biology as a new approach in cancer treatment. Int J Mol Sci. 14(7):15059–15073.
- Kizil C, Kyritsis N, Brand M. 2015. Effects of inflammation on stem cells: together they strive? EMBO Rep. 16(4):416–426.
- Klose RJ, Bird AP. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 31(2):89–97.
- Kon S, Fujita Y. 2021. Cell competition-induced apical elimination of transformed cells, EDAC, orchestrates the cellular homeostasis. Dev Biol. 476:112–116.
- Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, Kajita M, Ishikawa S, Yamauchi H, Yako Y, et al. 2017. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 19(5):530–541.
- Koturbash I, Pogribny I, Kovalchuk O. 2005. Stable loss of global DNA methylation in the radiation-target tissue - a possible mechanism contributing to radiation carcinogenesis? Biochem Biophys Res Commun. 337(2):526–533.
- Kovalchuk O, Burke P, Besplug J, Slovack M, Filkowski J, Pogribny I. 2004. Methylation changes in muscle and liver tissues of male and female mice exposed to acute and chronic low-dose X-ray-irradiation. Mutat Res. 548(1-2):75–84.
- Kozar S, Morrissey E, Nicholson AMM, van der Heijden M, Zecchini HII, Kemp R, Tavaré S, Vermeulen L, Winton DJJ, van der Heijden M, et al. 2013. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. 13(5):626–633.
- Kreuzer M, Auvinen A, Cardis E, Durante M, Harms-Ringdahl M, Jourdain JR, Madas BG, Ottolenghi A, Pazzaglia S, Prise KM, et al. 2018. Multidisciplinary European Low Dose Initiative (MELODI): strategic research agenda for low dose radiation risk research. Radiat Environ Biophys. 57(1):5–15.
- Krndija D, Marjou F e, Guirao B, Richon S, Leroy O, Bellaiche Y, Hannezo E, Vignjevic DM. 2019. Active cell migration is critical for steady-state epithelial turnover in the gut. Science. 365(6454):705–710.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. 1994. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 367(6464):645–648.
- Laurier D, Richardson DB, Cardis E, Daniels RD, Gillies M, O'Hagan J, Hamra GB, Haylock R, Leuraud K, Moissonnier M, et al. 2017. The international nuclear workers study (INWORKS): a collaborative epidemiological study to improve knowledge about health effects of protracted low-dose exposure. Radiat Protect Dosimetry. 173(1–3):21–25.
- Lawson DA, Witte ON. 2007. Stem cells in prostate cancer initiation and progression. J Clin Invest. 117(8):2044–2050.
- Leung CT, Brugge JS. 2012. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 482(7385):410–413.
- Levayer R, Dupont C, Moreno E. 2016. Tissue crowding induces caspase-dependent competition for space. Curr Biol. 26(5):670–677.
- Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, et al. 2015. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 528(7583):560–564.
- Little JB. 2000. Radiation carcinogenesis. Carcinogenesis. 21(3):397–404.
- Little MP, Hawkins MM, Charles MW, Hildreth NG. 1992. Fitting the Armitage-Doll model to radiation-exposed cohorts and implications for population cancer risks. Radiat Res. 132(2):207–221.
- Lochhead P, Chan AT, Nishihara R, Fuchs CS, Beck AH, Giovannucci E, Ogino S. 2015. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol. 28(1):14–29.
- Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. 2010. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 330(6005):822–825.
- López-Otín C, Kroemer G. 2021. Hallmarks of health. Cell. 184(1):33–63.
- Marnef A, Cohen S, Legube G. 2017. Transcription-coupled DNA double-strand break repair: active genes need special care. J Mol Biol. 429(9):1277–1288.
- Martin ML, Adileh M, Hsu KS, Hua G, Lee SG, Li C, Fuller JD, Rotolo JA, Bodo S, Klingler S, et al. 2020. Organoids reveal that inherent radiosensitivity of small and large intestinal stem cells determines organ sensitivity. Cancer Res. 80(5):1219–1227.
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. 2018. Somatic mutant clones colonize the human esophagus with age. Science. 362(6417):911–917.
- Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, Lasitschka F, Mastitsky SE, Brors B, Hielscher T, et al. 2014. Cell competition is a tumour suppressor mechanism in the thymus. Nature. 509(7501):465–470.
- Matsumura H, Liu N, Nanba D, Ichinose S, Takada A, Kurata S, Morinaga H, Mohri Y, de Arcangelis A, Ohno S, et al. 2021. Distinct types of stem cell divisions determine organ regeneration and aging in hair follicles. Nat Aging. 1(2):190–204.
- McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ, Taylor RW, Lee CY, Preston SL, Lovell M, et al. 2008. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology. 134(2):500–510.
- Merino MM, Levayer R, Moreno E. 2016. Survival of the fittest: essential roles of cell competition in development, aging, and cancer. Trends Cell Biol. 26(10):776–788.
- Miousse IR, Kutanzi KR, Koturbash I. 2017. Effects of ionizing radiation on DNA methylation: from experimental biology to clinical applications. Int J Radiat Biol. 93(5):457–469.
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. 2012. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science. 338(6103):108–113.
- Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk MEG, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S, et al. 2011. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 108(1):179–184.
- Moolgavkar SH, Knudson AG. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66(6):1037–1052.
- Moolgavkar SH, Day NE, Stevens RG. 1980. Two-stage model for carcinogenesis: epidemiology of breast cancer in females. J Natl Cancer Inst. 65(3):559–569.
- Morgan WF. 2003. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 159(5):581–596.
- Morton LM, Karyadi DM, Stewart C, Bogdanova TI, Dawson ET, Steinberg MK, Dai J, Hartley SW, Schonfeld SJ, Sampson JN, et al. 2021. Radiation-related genomic profile of papillary thyroid cancer after the Chernobyl accident. Science. 372(6543):eabg2538.
- Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. 1995. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. 31(7-8):1061–1064.
- Moya IM, Halder G. 2019. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 20(4):211–226.
- Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, Nagy T, Cagan A, Goulding D, Klein AM, et al. 2018. Epidermal tissue adapts to restrain progenitors carrying clonal p53 mutations. Cell Stem Cell. 23(5):687–699.e8.
- Nair RRK, Rajan B, Akiba S, Jayalekshmi P, Nair MK, Gangadharan P, Koga T, Morishima H, Nakamura S, Sugahara T. 2009. Background radiation and cancer incidence in Kerala, India-Karunagappally cohort study. Health Phys. 96(1):55–66.
- Nam AS, Chaligne R, Landau DA. 2021. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 22(1):3–18.
- Naito Y, Takagi T, Handa O, Ishikawa T, Nakagawa S, Yamaguchi T, Yoshida N, Minami M, Kita M, Imanishi J, et al. 2003. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J Gastroenterol Hepatol. 18(5):560–569.
- Nanki K, Fujii M, Shimokawa M, Matano M, Nishikori S, Date S, Takano A, Toshimitsu K, Ohta Y, Takahashi S, et al. 2020. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature. 577(7789):254–259.
- Negrini S, Gorgoulis VG, Halazonetis TD. 2010. Genomic instability an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 11(3):220–228.
- Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, Drenth JP, Van der Meer JW. 2004. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol. 34(7):2052–2059.
- Niwa O, Barcellos-Hoff MH, Globus RK, Harrison JD, Hendry JH, Jacob P, Martin MT, Seed TM, Shay JW, Story MD, et al. 2015. ICRP Publication 131: stem cell biology with respect to carcinogenesis aspects of radiological protection. Ann ICRP. 44(3–4):7–357.
- Norman M, Wisniewska KA, Lawrenson K, Pablo GM, Tada M, Kajita M, Mano H, Ishikawa S, Ikegawa M, Shimada T, et al. 2012. Loss of scribble causes cell competition in mammalian cells. J Cell Sci. 125(Pt 1):59–66.
- Nusse YM, Savage AK, Marangoni P, Rosendahl-Huber AKM, Landman TA, de Sauvage FJ, Locksley RM, Klein OD. 2018. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature. 559(7712):109–113.
- Ohsawa S, Vaughen J, Igaki T. 2018. Cell extrusion: a stress-responsive force for good or evil in epithelial homeostasis. Dev Cell. 44(3):284–296.
- Otsuka K, Iwasaki T. 2015. Effects of dose rates on radiation-induced replenishment of intestinal stem cells determined by Lgr5 lineage tracing. J Radiat Res. 56(4):615–622.
- Otsuka K, Suzuki K. 2016. Differences in radiation dose response between small and large intestinal crypts. Radiat Res. 186(3):302–314.
- Otsuka K, Tomita M. 2018. Concurrent live imaging of DNA double-strand break repair and cell-cycle progression by CRISPR/Cas9-mediated knock-in of a tricistronic vector. Sci Rep. 8(1):17309.
- Otsuka K, Koana T, Tauchi H, Sakai K. 2006. Activation of antioxidative enzymes induced by low-dose-rate whole-body γ irradiation: adaptive response in terms of initial DNA damage. Radiat Res. 166(3):474–478.
- Otsuka K, Hirabayashi Y, Tsuboi I, Inoue T. 2010. Regeneration capability of Lin-/c-Kit+/Sca-1+ cells with or without radiation exposure for repopulation of peripheral blood in lethally irradiated mice monitored using Ly5.1 isotype on days 35, 90, and 270 after transplantation. Exp Hematol. 38(5):417–425.
- Otsuka K, Hamada N, Magae J, Matsumoto H, Hoshi Y, Iwasaki T. 2013. Ionizing radiation leads to the replacement and de novo production of colonic Lgr5(+) stem cells. Radiat Res. 179(6):637–646.
- Otsuka K, Suzuki K, Fujimichi Y, Tomita M, Iwasaki T. 2018. Cellular responses and gene expression profiles of colonic Lgr5 + stem cells after low-dose/low-dose-rate radiation exposure. J Radiat Res. 59(suppl_2):ii18–ii22.
- Otsuka K, Fujimichi Y. 2021. Challenges to radiation biology using organoids and live-cell imaging. Radiat Biol Res Commun. 56:224–235.
- Ozasa K, Shimizu Y, Suyama A, Kasagi F, Soda M, Grant EJ, Sakata R, Sugiyama H, Kodama K. 2012. Studies of the mortality of atomic bomb survivors, Report 14, 1950 – 2003: an overview of cancer and noncancer diseases. Radiat Res. 177(3):229–243.
- Pacchierotti F, Spanò M. 2015. Environmental impact on DNA methylation in the germline: state of the art and gaps of knowledge. Biomed Res Int. 2015:123484.
- Paunesku T, Stevanović A, Popović J, Woloschak GE. 2021. Effects of low dose and low dose rate low linear energy transfer radiation on animals – review of recent studies relevant for carcinogenesis. Int J Radiat Biol. 97(6):757–768.
- Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB, et al. 2020. Mutational signature in colorectal cancer caused by genotoxic pks + E. coli. Nature. 580(7802):269–273.,.
- Pogribny I, Raiche J, Slovack M, Kovalchuk O. 2004. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun. 320(4):1253–1261.
- Potten CS, Clarke RB, Wilson J, Renehan AG. 2006. Tissue stem cells. Boca Raton (FL): CRC Press.
- Potten CS, Gandara R, Mahida YR, Loeffler M, Wright NA. 2009. The stem cells of small intestinal crypts: where are they? Cell Prolif. 42(6):731–750.
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. 2012. The pan-ErbB negative regulator lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 149(1):146–158.
- Preston SL, Wong WM, Chan AOO, Poulsom R, Jeffery R, Goodlad RA, Mandir N, Elia G, Novelli M, Bodmer WF, et al. 2003. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 63(13):3819–3825.
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. 2010. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12(6):662–667.
- Raiche J, Rodriguez-Juarez R, Pogribny I, Kovalchuk O. 2004. Sex- and tissue-specific expression of maintenance and de novo DNA methyltransferases upon low dose X-irradiation in mice. Biochem Biophys Res Commun. 325(1):39–47.
- Ramasundara M, Leach ST, Lemberg DA, Day AS. 2009. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol. 24(2):202–208.
- Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature. 414(6859):105–111.
- Richardson DB, Cardis E, Daniels RD, Gillies M, Haylock R, Leuraud K, Laurier D, Moissonnier M, Schubauer-Berigan MK, Thierry-Chef I, et al. 2018. Site-specific solid cancer mortality after exposure to ionizing radiation: a cohort study of workers (INWORKS). Epidemiology. 29(1):31–40.
- Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. 2007. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 26(33):4833–4841.
- Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capellà G, Ribas M, et al. 2006. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 66(17):8462–9468.
- Roskams T. 2006. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 25(27):3818–3822.
- Rossi DJ, Jamieson CHM, Weissman IL. 2008. Stems cells and the pathways to aging and cancer. Cell. 132(4):681–696.
- Rothkamm K, Löbrich M. 2003. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 100(9):5057–5062.
- Rühm W, Azizova TV, Bouffler SD, Little MP, Shore RE, Walsh L, Woloschak GE. 2016. Dose-rate effects in radiation biology and radiation protection. Ann ICRP. 45(1_suppl):262–279.
- Rühm W, Eidemüller M, Kaiser J. 2017. Biologically-based mechanistic models of radiation-related carcinogenesis applied to epidemiological data. Int J Radiat Biol. 93(10):1093–1117.
- Sada A, Jacob F, Leung E, Wang S, White BS, Shalloway D, Tumbar T. 2016. Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat Cell Biol. 18(6):619–631.
- Sadakane A, French B, Brenner A. v, Preston DL, Sugiyama H, Grant EJ, Sakata R, Utada M, Cahoon EK, Mabuchi K, et al. 2019. Radiation and risk of liver, biliary tract, and pancreatic cancers among atomic bomb survivors in Hiroshima and Nagasaki: 1958-2009. Radiat Res. 192(3):299–310.
- Sage E, Shikazono N. 2017. Radiation-induced clustered DNA lesions: repair and mutagenesis. Free Radic Biol Med. 107:125–135.
- Sakai K, Okada S. 1981. An alkaline separation method for detection of small amount of DNA damage. J Radiat Res. 22(4):415–424.
- Sakamaki A, Katsuragi Y, Otsuka K, Tomita M, Obata M, Iwasaki T, Abe M, Sato T, Ochiai M, Sakuraba Y, et al. 2015. Bcl11b SWI/SNF-complex subunit modulates intestinal adenoma and regeneration after γ-irradiation through Wnt/ß-catenin pathway. Carcinogenesis. 36(6):622–631.
- Sakata R, Preston DL, Brenner A. v, Sugiyama H, Grant EJ, Rajaraman P, Sadakane A, Utada M, French B, Cahoon EK, et al. 2019. Radiation-related risk of cancers of the upper digestive tract among Japanese atomic bomb survivors. Radiat Res. 192(3):331–344.
- Saha S, Aranda E, Hayakawa Y, Bhanja P, Atay S, Brodin NP, Li J, Asfaha S, Liu L, Tailor Y, et al. 2016. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun. 7(1):13096.
- Sancho M, Di-Gregorio A, George N, Pozzi S, Sánchez JM, Pernaute B, Rodríguez TA. 2013. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell. 26(1):19–30.
- Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, Nakatani T, Kunisawa J, Fujita Y. 2018. Obesity suppresses cell-competition-mediated apical elimination of RasV12-transformed cells from epithelial tissues. Cell Rep. 23(4):974–982.
- Satoh Y, Asakawa J i, Nishimura M, Kuo T, Shinkai N, Cullings HM, Minakuchi Y, Sese J, Toyoda A, Shimada Y, et al. 2020. Characteristics of induced mutations in offspring derived from irradiated mouse spermatogonia and mature oocytes. Sci Rep. 10(1):1–4.
- Scaramozza A, Park D, Kollu S, Beerman I, Sun X, Rossi DJ, Lin CP, Scadden DT, Crist C, Brack AS. 2019. Lineage tracing reveals a subset of reserve muscle stem cells capable of clonal expansion under stress. Cell Stem Cell. 24(6):944–957.e5.
- Searle AG. 1974. Mutation induction in mice. Adv Radiat Biol. 4:131–207.
- Seibold P, Auvinen A, Averbeck D, Bourguignon M, Hartikainen JM, Hoeschen C, Laurent O, Noël G, Sabatier L, Salomaa S, et al. 2020. Clinical and epidemiological observations on individual radiation sensitivity and susceptibility. Int J Radiat Biol. 96(3):324–339.
- Shaker A, Swietlicki EA, Wang L, Jiang S, Onal B, Bala S, DeSchryver K, Newberry R, Levin MS, Rubin DC. 2010. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J Clin Invest. 120(6):2081–2093.
- Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis. 31(1):27–36.
- Shibata A, Jeggo PA. 2020. Canonical DNA non-homologous end-joining; capacity versus fidelity. Br J Radiol. 93(1115):20190966.
- Shimura T. 2021. The role of mitochondrial oxidative stress and the tumor microenvironment in radiation-related cancer. J Radiat Res. 62(Supplement_1):i36–i43.
- Singh SK, Clarke ID, Hide T, Dirks PB. 2004. Cancer stem cells in nervous system tumors. Oncogene. 23(43):7267–7273.
- Slaughter DP, Southwick HW, Smejkal W. 1953. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 6(5):963–968.
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. 2010. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 143(1):134–144.
- Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. 2014. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 15(1):62–69.
- Stewart-Ornstein J, Cheng HWJ, Lahav G. 2017. Conservation and divergence of p53 oscillation dynamics across species. Cell Syst. 5(4):410–417.e4.
- Strong LC. 1949. A new theory of mutation and the origin of cancer. Yale J Biol Med. 21(4):293–299.
- Sugiyama H, Misumi M, Brenner A, Grant EJ, Sakata R, Sadakane A, Utada M, Preston DL, Mabuchi K, Ozasa K. 2020. Radiation risk of incident colorectal cancer by anatomical site among atomic bomb survivors: 1958–2009. Int J Cancer. 146(3):635–645.
- Taki K, Wang B, Nakajima T, Wu J, Ono T, Uehara Y, Matsumoto T, Oghiso Y, Tanaka K, Ichinohe K, et al. 2009. Microarray analysis of differentially expressed genes in the kidneys and testes of mice after long-term irradiation with low-dose-rate γ-rays. J Radiat Res. 50(3):241–252.
- Tamori Y, Bialucha CU, Tian AG, Kajita M, Huang YC, Norman M, Harrison N, Poulton J, Ivanovitch K, Disch L, et al. 2010. Involvement of Lgl and mahjong/VprBP in cell competition. PLoS Biol. 8(7):e1000422.
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. 2015. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 519(7541):57–62.
- Tao Z, Akiba S, Zha Y, Sun Q, Zou J, Li J, Liu Y, Yuan Y, Tokonami S, Morishoma H, et al. 2012. Cancer and non-cancer mortality among Inhabitants in the high background radiation area of Yangjiang, China (1979-1998). Health Phys. 102(2):173–181.
- Tauchi H, Kobayashi J, Morishima KI, van Gent DC, Shiraishi T, Verkaik NS, VanHeems D, Ito E, Nakamura A, Sonoda E, et al. 2002. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 420(6911):93–98.
- Tawa R, Kimura Y, Komura JI, Miyamura Y, Kurishita A, Sasaki MS, Sakurai H, Ono T. 1998. Effects of X-ray irradiation on genomic DNA methylation levels in mouse tissues. J Radiat Res. 39(4):271–278.
- Thirlwell C, Will OCC, Domingo E, Graham TA, McDonald SAC, Oukrif D, Jeffrey R, Gorman M, Rodriguez-Justo M, Chin-Aleong J, et al. 2010. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology. 138(4):1441–1454.e7.
- Thliveris AT, Halberg RB, Clipson L, Dove WF, Sullivan R, Washington MK, Stanhope S, Newton MA. 2005. Polyclonality of familial murine adenomas: analyses of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc Natl Acad Sci U S A. 102(19):6960–6965.
- Thompson DE, Mabuchi K, Ron E, Soda M, Tokunaga M, Ochikubo S, Sugimoto S, Ikeda T, Terasaki M, Izumi S, et al. 1994. Cancer incidence in atomic bomb survivors. Part II: Solid tumors, 1958-1987. Radiat Res. 137(2):S17–S67.
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. 2011. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 478(7368):255–259.
- Timp W, Feinberg AP. 2013. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 13(7):497–510.
- Tomasetti C, Vogelstein B. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 347(6217):78–81.
- Tomasetti C, Li L, Vogelstein B. 2017. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 355(6331):1330–1334.
- Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, Palles C, Broderick P, Jaeger EE, Farrington S, et al. 2011. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 7(6):e1002105.
- Totafurno J, Bjerknes M, Cheng H. 1987. The crypt cycle. Crypt and villus production in the adult intestinal epithelium. Biophys J. 52(2):279–294.
- Totsuka Y, Watanabe M, Lin Y. 2021. New horizons of DNA adductome for exploring environmental causes of cancer. Cancer Sci. 112(1):7–15.
- Tümpel S, Rudolph KL. 2012. The role of telomere shortening in somatic stem cells and tissue aging: Lessons from telomerase model systems. Ann N Y Acad Sci. 1266:28–39.
- Uchinomiya K, Yoshida K, Kondo M, Tomita M, Iwasaki T. 2020. A mathematical model for stem cell competition to maintain a cell pool injured by radiation. Radiat Res. 194(4):379–389.
- UNSCEAR. 2017. UNSCEAR 2017 report: “sources, effects and risks of ionizing radiation” Annex B – epidemiological studies of cancer risk due to low-dose-rate radiation from environmental sources.
- UNSCEAR. 2020. UNSCEAR 2020/2021 report vol. II: “sources, effects and risks of ionizing radiation” Scientific Annex B – levels and effects of radiation exposure due to the accident at the Fukushima Daiichi Nuclear Power Station: implications of information published since the UNSCEAR 2013 Rep.
- UNSCEAR. 2021. UNSCEAR 2020/2021 report vol. III: “sources, effects and risks of ionizing radiation” Scientific Annex C – biological mechanisms relevant for the inference of cancer risks from low-dose and low-dose-rate radiation.
- Urbán N, Cheung TH. 2021. Stem cell quiescence: the challenging path to activation. Development. 148(3):dev165084.
- van der Flier LG, Clevers H. 2009. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 71:241–260.
- van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, Warmerdam DO, Kakkar V, Moreno LF, Vieira Braga FA, et al. 2021. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature. 594(7863):436–441.
- Vaiserman AM. 2011. Hormesis and epigenetics: Is there a link? Ageing Res Rev. 10:413–421.
- van Velthoven CTJ, Rando TA. 2019. Stem cell quiescence: dynamism, restraint, and cellular idling. Cell Stem Cell. 24(2):213–225.
- Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavaré S, Winton DJ. 2013. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 342(6161):995–998.
- Vilenchik MM, Knudson AG. 2006. Radiation dose-rate effects, endogenous DNA damage, and signaling resonance. Proc Natl Acad Sci U S A. 103(47):17874–17879.
- Vishwakarma M, Piddini E. 2020. Outcompeting cancer. Nat Rev Cancer. 20(3):187–198.
- Visvader JE. 2011. Cells of origin in cancer. Nature. 469(7330):314–322.
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. 1988. Genetic alterations during colorectal-tumor development. N Engl J Med. 319(9):525–532.
- Wagner DE, Klein AM. 2020. Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet. 21(7):410–427.
- Wakabayashi T, Kato H, Ikeda T, Schull WJ. 1983. Studies of the mortality of A-bomb survivors, report 7. Part III. Incidence of cancer in 1959-1978, based on the tumor registry, Nagasaki. Radiat Res. 93(1):112–146.
- Weber M, Schübeler D. 2007. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 19(3):273–280.
- Waghmare I, Roebke A, Minata M, Kango-Singh M, Nakano I. 2014. Intercellular cooperation and competition in brain cancers: lessons from Drosophila and human studies. Stem Cells Transl Med. 3(11):1262–1268.
- WHO Report on Cancer. 2020. https://www.who.int/publications/i/item/who-report-on-cancer-setting-priorities-investing-wisely-and-providing-care-for-all.
- Wong WM, Mandir N, Goodlad RA, Wong BCY, Garcia SB, Lam SK, Wright NA. 2002. Histogenesis of human colorectal adenomas and hyperplastic polyps: the role of cell proliferation and crypt fission. Gut. 50(2):212–217.
- Wong WY, Allie S, Limesand KH. 2019. PKCζ and JNK signaling regulate radiation-induced compensatory proliferation in parotid salivary glands. PLoS One. 14(7):e0219572.
- Wu S, Powers S, Zhu W, Hannun YA. 2016. Substantial contribution of extrinsic risk factors to cancer development. Nature. 529(7584):43–47.
- Yamazaki M, Fujii E, Watanabe T, Kato A, Suzuki M. 2020. Histopathological evaluation of crypt fission during intestinal development in neonatal mice. J Toxicol Pathol. 33(1):39–46.
- Ye S, Yuan D, Xie Y, Pan Y, Shao C. 2013. Role of DNA methylation in long-term low-dose γ-rays induced adaptive response in human B lymphoblast cells. Int J Radiat Biol. 89(11):898–906.
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. 2019. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 565(7739):312–317.
- Yum MK, Han S, Fink J, Wu SHS, Dabrowska C, Trendafilova T, Mustata R, Chatzeli L, Azzarelli R, Pshenichnaya I, et al. 2021. Tracing oncogene-driven remodelling of the intestinal stem cell niche. Nature. 594(7863):442–447.
- Zhang G, Xie Y, Zhou Y, Xiang C, Chen L, Zhang C, Hou X, Chen J, Zong H, Liu G. 2017. P53 pathway is involved in cell competition during mouse embryogenesis. Proc Natl Acad Sci USA. 114(3):498–503.
- Zhao Y, Epstein RJ. 2008. Programmed genetic instability: a tumor-permissive mechanism for maintaining the evolvability of higher species through methylation-dependent mutation of DNA repair genes in the male germ line. Mol Biol Evol. 25(8):1737–1749.
- Zhu L, Finkelstein D, Gao C, Shi L, Wang Y, López-Terrada D, Wang K, Utley S, Pounds S, Neale G, et al. 2016. Multi-organ mapping of cancer risk. Cell. 166(5):1132–1146.e7.
- Zingg JM, Jones PA. 1997. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 18(5):869–882.