Abstract
Formation of harmful disinfection by‐products (DBPs), of which trihalomethanes (THMs) and haloacetic acids (HAAs) are the major groups, can be controlled by removal of natural organic matter (NOM) before disinfection. In the literature, removal of precursors is variable, even with the same treatment. The treatment of DBP precursors and NOM was examined with the intention of outlining precursor removal strategies for various water types. Freundlich adsorption parameters and hydroxyl rate constants were collated from the literature to link treatability by activated carbon and advanced oxidation processes (AOPs), respectively, to physico‐chemical properties. Whereas hydroxyl rate constants did not correlate meaningfully with any property, a moderate correlation was found between Freundlich parameters and log KOW, indicating activated carbon will preferentially adsorb hydrophobic NOM. Humic components of NOM are effectively removed by coagulation, and, where they are the principal precursor source, coagulation may be sufficient to control DBPs. Where humic species remaining post‐coagulation retain significant DBP formation potential (DBPFP), activated carbon is deemed a suitable process selection. Anion exchange is an effective treatment for transphilic species, known for high carboxylic acid functionality, and consequently is recommended for carboxylic acid precursors. Amino acids have been linked to HAA formation and are important constituents of algal organic matter. Amino acids are predicted to be effectively removed by biotreatment and nanofiltration. Carbohydrates have been found to reach 50% of NOM in river waters. If the carbohydrates were to pose a barrier to successful DBP control, additional treatment stages such as nanofiltration are likely to be required to reduce their occurrence.
Introduction
Although disinfection of drinking water is necessary to suppress microbial activity, a significant associated risk is the formation of disinfection by‐products (DBPs) through reactions of disinfectants with natural organic matter (NOM) [Citation1]. Many DBPs pose a health risk to humans [Citation1], and consequently two halogenated groups – the trihalomethanes (THMs) and haloacetic acids (HAAs) – are regulated in the USA, at 80 µg L−1 for total THMs (CHCl3, CHBrCl2, CHClBr2 and CHBr3) and 60 µg L−1 for five HAA species (monochloroacetic acid, dichloroacetic acid [DCAA], trichloroacetic acid [TCAA], monobromoacetic acid and dibromoacetic acid). Total THMs are also legislated in the UK, at 100 µg L−1. In chlorinated drinking water, the dominant THM species is typically chloroform (CHCl3), with DCAA and TCAA being the prevalent HAA species [Citation2]. Particularly at high bromide concentrations, which has been defined as over 200 µg L−1 [Citation3], the formation of mixed brominated and chlorinated DBPs is typical [Citation2]. In raw drinking waters bromide concentrations typically vary from a few micrograms per litre to several milligrams per litre [Citation3]. Note that, while typically only the brominated and chlorinated THMs and HAAs are considered, iodinated species can also form, and are currently thought of as emerging DBPs [Citation4]. There are several approaches to control disinfection by‐products (DBPs), including removal of precursor material before disinfection and altering disinfectant dose, type or dosing location [Citation5]. However, reducing disinfectant doses is limited by the need to provide sufficient residuals for distribution. A residual of 0.1–0.2 mg L−1 is typically viewed as suitable for this purpose [Citation6]. Moreover, switching disinfectant can result in the formation of alternative DBPs, as illustrated by links between chloramines and the formation of N‐nitrosodimethylamine (NDMA) and dichloroacetonitrile (DCAN) [Citation4,Citation7].
Therefore, in several ways, the removal of NOM, including precursor material, is more satisfactory [Citation5]. Not only is the production of alternative DBPs avoided, but precursor removal can often be attained through utilization of existing technology. Many studies have examined DBP precursor removal, particularly THM precursors, with limited research encompassing HAA precursor removal [Citation5]. There is wide geographical and seasonal variation in NOM composition [Citation8,Citation9], which is reflected in variable removal of NOM, even by the same treatment. Natural organic matter is typically characterized with adsorption chromatography into fractions of varying hydrophobicity [Citation8,Citation10]. Although nomenclature varies, in order of descending hydrophobicity, the following fractions can be isolated: hydrophobic, transphilic and hydrophilic. In addition, depending on the method, operationally defined acid, base and/or neutral subdivisions of the above three fractions can be separated [Citation8,Citation10]. Such procedures can be used to provide data about the relative importance of operationally defined fractions to DBP formation. Although hydrophobic fractions, which can constitute up to ∼75% of NOM [Citation11], and humic species, which account for much of hydrophobic NOM, are thought to be the major source of DBP precursors [Citation10], a range of species can be involved. This conclusion is supported by model compound work, where both hydrophobic and hydrophilic molecules, notably activated aromatic species, β‐dicarbonyl compounds and a small number of amino acids, have been identified as reactive DBP precursors [Citation12–Citation14]. Nitrogen‐enriched water sources, such as those impacted by wastewater and algae, have been highlighted as more likely to generate various nitrogen‐containing DBPs [Citation15]. Since the majority of water treatment research uses natural waters rather than model compounds, links between DBP formation and treatability are incompletely resolved. This is compounded by very limited knowledge about specific chemical identity of DBP precursors in drinking water and the unpredictability of DBP formation. Consequently, there is uncertainty about how to operate NOM removal processes for targeted precursor removal. The objectives of this review, therefore, were to analyze literature data regarding removal of NOM, THM precursors and HAA precursors and consequently highlight circumstances in which various treatment processes can be effectively deployed for precursor removal.
Precursor removal by coagulation
Coagulation followed by clarification and/or filtration is the standard process used to remove particulate matter and NOM from surface waters and is normally the first step of conventional water treatment [Citation16]. Coagulants are typically iron or aluminium salts which hydrolyse rapidly to form positively charged insoluble precipitates in water, removing NOM through a variety of principally electrostatic mechanisms (Table ). Doses of coagulants typically vary from 10 to 150 mg L−1 for aluminium sulphate, 10 to 250 mg L−1 for ferric sulphate and 5 to 150 mg L−1 for ferric chloride (Table [Citation17]) depending on turbidity and NOM concentration. For aluminium sulphate this range equates to 0.3 to 1.1 mgAl mgDOC−1 (Table ). There is a wide range of efficacy associated with coagulation, with total organic carbon (TOC) removal ranging from 7% to 76% and removal of THM and HAA precursors from 7% to 76% and 15% to 78%, respectively (Table [Citation18–Citation20]). The respective removals of TOC, ultraviolet absorption (UV), THM formation potential (THMFP), dihaloacetic acid formation potential (DXAAFP) and trihalacetic acid formation potential (TXAAFP) of 8%, 73%, 10%, 12% and 22% in a water with 1.1 mg L−1 dissolved organic carbon (DOC) illustrate the lower end of treatability (Table [Citation18]). Two key points arise from comparison of this data: the higher removal of UV‐absorbing material compared with other parameters, and higher removal of TXAAFP relative to DXAAFP. Both are features of the literature, as illustrated by a more treatable water, where removal of UV, TOC, THMFP, DXAAFP and TXAAFP were 52%, 74%, 62%, 65% and 75%, respectively (Table [Citation18]). The higher susceptibility of TCAA precursors to coagulation than DCAA precursors is linked to the former's more hydrophobic nature [Citation18]. High removal of high molecular weight (MW), hydrophobic organics is typical during coagulation of drinking water. This is shown by respective removals of 84%, 64%, 14% and 17% for the humic acid (HAF), fulvic acid (FAF) hydrophilic acid (HPIA), and hydrophilic non‐acid (HPINA) fractions of an upland water [Citation11]. Note that the hydrophobic acid fraction consists of the HAF and the FAF. In addition HPIA and HPINA are respectively equivalent to transphilic acid and hydrophilic fractions [Citation8,Citation10]. This data also correlates to charge, since high anionic charge is a feature associated with hydrophobic fractions of drinking water, as shown by charge densities of 6.8, 4.2 and 0.006 meq gDOC−1 for the HAF, FAF and HPIA fractions, respectively, of an upland water [Citation9]. A consequence of this selectivity for hydrophobic NOM is that the levels of HPINA in the raw water indicate the residual post‐coagulation [Citation11]. A similar reasoning explains the positive relationship between specific ultraviolet absorbance (SUVA) and treatability (Figure ), since high SUVA values indicate a high proportion of hydrophobic material in a water [Citation6]. High SUVA waters can be regarded as those with a SUVA above 4 L mg−1 m−1 (Figure ) [Citation6]. The charge‐driven nature of NOM coagulation means that electrophoretic monitoring is appropriate, for example it has been demonstrated how optimum removal can be achieved by operating within a zeta potential window of −10 to 3 mV [Citation11]. However, NOM removal is often indirectly controlled through coagulating between pH 4.5 and 6.0 or by using coagulant doses above those required for particle removal, termed enhanced coagulation [Citation16]. The effect of coagulating at acidic pH on downstream disinfection should also be considered, since chlorination at acidic pH has been reported to increase DCAA levels [Citation21]. However, as THMs have been found to increase and TCAA to decrease at higher pH, downstream consequences are complicated [Citation22] and would require empirical verification. The same applies to choice of coagulant. While it is thought that generally higher removals of precursors can be obtained with iron salts rather than aluminium salts, the latter may be more effective at low coagulant doses [Citation23]. Thus, coagulation can be expected to be successful for removal of DBP precursors which are anionic in character, which will typically also be hydrophobic and of high MW. However, in waters where reactive precursors are of low anionic charge or neutral, coagulation will have little impact upon their removal, and they will constitute part of the post‐coagulation NOM residual. Such waters are likely to have a high proportion on HPINA species and are also likely to be of low SUVA.
Figure 1 Relationships between raw water SUVA and removal of bulk NOM, THM precursors and HAA precursors by coagulation.
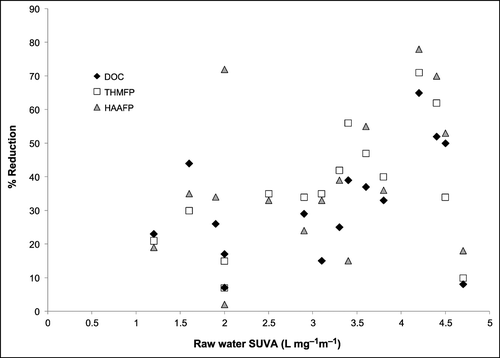
Table 1. Selectivity of NOM removal processes.
Table 2. NOM and DBP precursor removal by coagulation and MIEX®.
Precursor removal by ion exchange
Ion‐exchange removal mechanisms relate to the exchange of an ion in the aqueous phase for one in the solid phase attached to the ion exchanger (Table ). For example, the MIEX® anion exchange resin developed for NOM removal works by exchanging anionic NOM for a chloride ion attached to the cationic resin surface [Citation24]. MIEX® is a relatively novel process used as an alternative to coagulation or as an adjunct to reduce coagulant doses. Reports on its use have shown improved removal of NOM and THM precursors relative to coagulation [Citation25]. For instance, coagulation of one water with alum at 60 mg L−1 attained respective removals of DOC, UV, THMFP and HAAFP of 33%, 38%, 40% and 36% (Table [Citation19]). Equivalent values with 5 mL L−1 MIEX® were 75%, 84%, 69% and 71%. A combined treatment, with 5 mL L−1 MIEX® then 16 mg L−1 alum, was even more effective, with equivalent values of 76%, 85%, 76% and 73% (Table ), indicating synergistic benefits of a combined treatment and that MIEX® pretreatment can reduce coagulant doses. Whilst there is still debate regarding the type of NOM that can be treated more effectively by MIEX® than by coagulation, evidence suggests transphilic NOM is involved. Lee and co‐workers [Citation24] found MIEX® removed 63–75%, 70–89% and 2–67% of the hydrophobic, transphilic and hydrophilic fractions respectively, in three waters. The transphilic acid fraction was also found to have a higher affinity for MIEX® than did the other fractions [Citation26], this being explained by its higher charge density. While the exact chemical identity of transphilic acids is unknown, they are assumed to be more hydrophilic than the hydrophobic acids and with a high proportion of carboxylic acid functionality [Citation27]. In a recent study, uptake of a water of hydrophobic character deteriorated from 65% to 25% with consecutive MIEX® use designed to simulate full‐scale operation, whereas removal of two waters of hydrophilic and algogenic character were more consistent at ∼60% and ∼30%, respectively [Citation25]. These differences were explained by the hydrophobic‐character water containing more high MW species capable of blocking resin exchange sites and the algogenic water containing a higher proportion of neutral species. Thus, as with coagulation, NOM composition has a strong influence on treatability, although it appears the amount of hydrophobic species is not the determining factor for anion exchange. An added benefit of MIEX® is that it offers some removal of bromide, although this decreases with increasing alkalinity and bromide concentration [Citation28]. This is significant as bromine, formed by the oxidation of bromide in the presence of chlorine, is a more effective substitution agent than chlorine and has been found to increase DBP levels [Citation2]. However, removal is inconsistent: in one water with alkalinity of 20 mg L−1 as CaCO3, MIEX® effected a reduction in bromide from 163 to <10 µg L−1, whereas with another water with alkalinity of 155 mg L−1 as CaCO3 and bromide of 38 µg L−1, no bromide reduction was observed [Citation20]. This variability was rationalized by an increasing competition for ion exchange sites by bicarbonate ions in the higher alkalinity water. Residual NOM remaining after ion exchange is thought mainly to comprise neutral compounds [Citation29]. Thus ion exchange is likely to be most effective for treatment of hydrophobic and especially transphilic DBP precursors, but also offers removal of low‐MW anionic material. As with coagulation, residual NOM remaining post‐treatment will be neutral and low‐charge species, properties associated with the hydrophilic components of NOM [Citation9].
Analysis methods for activated carbon and advanced oxidation processes
To elucidate relationships between compound properties and removal by activated carbon (AC) and advanced oxidation processes (AOPs), Freundlich adsorption parameters (log KF [capacity parameter] and 1/n [intensity parameter]) and aqueous hydroxyl rate constants (k·OH) were collated [Citation17,Citation28–Citation30] for 158 compounds (Appendix 1). The following compound physico‐chemical properties were also assembled: MW, octanol–water partition coefficient (log KOW), molar volume (MV), surface tension (γ), polar surface area (PSA), polarizability (α), density (ρ) and the soil–water partition coefficient (log KOC) (Appendix 1). Properties were taken from [Citation33–Citation36], and experimental values were used wherever available. Log KOC values were estimated with the Estimation Programs Interface Suite [Citation33], using two different models. Literature THM formation data was included where available [Citation37,Citation38]. The Pearson product‐moment correlation coefficient (r), calculated with Minitab 15, was used to assess relationships between adsorption parameters and compound physico‐chemical properties (Tables and ). This coefficient is used to measure the degree of linear relationship between two variables and can assume a value from −1 to +1, these values respectively indicating a negative and positive linear relationship, while a value of 0 indicates no relationship.
Table 3. Correlations between compound properties and log KF for all compounds.
Table 4. Correlations between compound properties and log KF for non‐halogenated compounds.
Precursor removal by activated carbon adsorption
The primary adsorbent used for water treatment is activated carbon, which can be applied as powdered activated carbon (PAC) or granular activated carbon (GAC). While PAC can be applied at various stages of water treatment, GAC is typically utilized after coagulation–filtration/sedimentation but before post‐disinfection [Citation6]. Typical PAC doses are between 5 and 25 mg L−1, with average doses towards the lower end of this range [Citation17]. Activated carbon is variously employed for removal of specific contaminants, such as pesticides, as well as taste‐ and odour‐causing compounds and bulk NOM. Granular activated carbon can offer preferential removal of DBP precursors over bulk NOM (Table [Citation39–Citation41]). After 50 days operation, removal of DOC, THM precursors and HAA precursors was high at 80%, 95% and 89%, respectively (Table [Citation41]). After 250 days, the respective removals were 42%, 40% and 71%. This data is from a full‐scale trial with an empty bed contact time (EBCT) of 21 min and illustrates how initially high removal levels decline over the bed life of the GAC. A minimum EBCT of 10–15 min is generally recommended for DBP precursor removal [Citation41]. Reversible physical adsorption caused by non‐specific mechanisms, such as van der Waals forces dipole interactions and hydrophobic interactions, are considered the commonest means of sorption (Table [Citation42]). In the presence of oxygen, it is thought that AC can act as a catalyst for oxidative coupling reactions between phenolic compounds, which can affect the degree of sorption [Citation43].
Table 5. NOM and DBP precursor removal by AOPs and activated carbon.
In the literature, many Freundlich parameters are for toxic and halogenated compounds; thus, to more accurately reflect the nature of NOM, correlations for non‐halogenated compounds are also presented (Table ). For both sets of compounds, log KF shows moderate correlations with log KOW, MW and MV. For the complete set of compounds, these relationships have correlations of 0.568 (Figure ), 0.557 and 0.609 (157, 157 and 151 data pairs), respectively (Table ). For the non‐halogenated compounds, the equivalent correlations are 0.473, 0.4 and 0.483 (81, 81 and 77 data pairs), respectively (Table ). In addition, for all compounds, log KF exhibits positive correlations with log KOC and polarizability, at 0.546 and 0.556 (134 and 150 data pairs), respectively. These trends indicate that adsorbability increases with compound size and hydrophobicity but, while MW and log KOW provide an indication of adsorption performance, these relationships are not strong enough to be used as accurate predictors. Correlations with MW and α are in accordance with Traube's rule, which states adsorbability increases with size for a series of homologous organic compounds, corresponding to increasing polarizability. Part of the reason for the weakness of the correlations is the very wide range of values exhibited for log KF: from −4.17 for N‐dimethylnitrosamine to 4.14 for PCB, which equate to KF values ranging from 6.8 × 10–5 to 1.41 × 104 (Figure ). In contrast, the other properties examined do not vary by such a magnitude. The formation of THM does not correlate with any physico‐chemical property (Tables and ), in accordance with a more extensive study of the formation of a model compound DBP [Citation15].
Figure 2 Relationship between log KF and log KOW for all compounds.
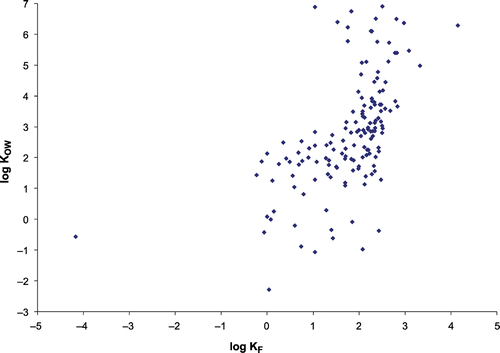
However, the literature suggests that for NOM adsorption these correlations are complicated by size exclusion and electrostatic effects. It has been reported that smaller humic acids were preferentially removed by an activated carbon [Citation44], this being explained by size exclusion, with smaller GAC pores being less accessible to high‐MW components of NOM molecules. While average NOM size is thought to be in the range 4–40 Å, the mean pore radius of F400 carbon, commonly used in water treatment, is 12 Å [Citation44]. This is the likely explanation for carbons with larger pore size having been found to perform more effectively in regard to NOM uptake [Citation45], while a more recent study recommended selection of a carbon with pores >1 nm [Citation46]. Increased NOM uptake, particularly of HAA precursors, has been observed for a steam‐treated carbon with increased mesopores, relative to non‐modified carbon, although, for a more hydrophilic water, differences were negligible [Citation46]. However, since there is limited knowledge about DBP precursor size, assessing the benefits of using a carbon with increased mesopores for precursor uptake requires empirical investigation and is site specific. Electrostatics also affect adsorption, with coulombic repulsion between anionic solutes and acidic groups on the carbon surface being the most relevant interactions [Citation45]. The same study recommended selection of a carbon with a basic point of zero charge (pHpzc), to facilitate coulombic attraction between NOM and AC. As most molecules listed in Appendix 1 are neutral, pKa values were not included as a compound property. Overall, while the correlation of 0.557 between log KF and MW (Table ) is influenced by the carbon pore size distribution and electrostatic interactions are affected by the charge of the carbon surface, hydrophobic molecules are more treatable than hydrophilic ones (Figure ). Activated carbon will be most successful when reactive precursors belong to this category.
Precursor removal by advanced oxidation processes
Advanced oxidation processes are characterized by the in situ generation of hydroxyl radicals (·OH), and are currently considered an advanced water treatment. There are various ways of generating AOPs, among them ozone/UV, ozone/H2O2, UV/H2O2, vacuum UV and Fenton's reactions [Citation47]. Although the means of ·OH production varies, all these processes share the same method of degrading NOM (Table ), through fast and non‐selective reactions with organic compounds [Citation17]. The average second‐order rate constants for reactions between NOM and ·OH in 17 waters was 3.9 × 108 M−1 s−1 [Citation48], with values determined by competition kinetics. More recently, rate constants were directly measured as 1 × 108 M−1 s−1 to 5 × 108 M−1 s−1 for ·OH and NOM reactions [Citation49]. These values are typical for organics and are three to four orders of magnitude higher than for other oxidants used in water treatment [Citation17].
For the complete set of compounds, r = 0.309 for the relationship between kOH and log KOC (51 data pairs) (Table ). All others correlations were between −0.168 and 0.187 (Table ), which is in accordance with the non‐selective nature of ·OH reactions. For the non‐halogenated compounds, the correlations of 0.396 for the relationship between kOH and log KF (30 data pairs) and 0.412 for the relationship between kOH and log KF (29 data pairs) were the highest recorded (Table ).
Ultraviolet radiation doses of 0.5–3.0 J cm−2 and H2O2 doses of 10–20 mg L−1 for UV/H2O2 treatment are typical of those employed for NOM oxidation [Citation39]. At 0.5 J cm−2, reductions in DOC, UV, THMFP, DCAAFP and TCAAFP of −11%, 24%, 8%, −35% and 8% were recorded [Citation39]. With 3.0 J cm−2, equivalent values were 20%, 59%, 73%, −74% and 69%, thus AOPs can increase formation of DBPs, and DCAA in particular, across a range of UV fluence values. Note the similarity in behaviour between TCAA and THM precursors and the disparate nature of DCAA precursors, as noted elsewhere [Citation50]. Such increases occur through formation of reactive DCAA precursors on oxidation. The specific identity of these precursors is uncertain but a rise in DCAA has been linked to formation of diketones and then aldehydes [Citation51]. Higher respective removals of DOC, UV, THMFP and HAAFP of 56%, 91%, 89% and 83%, with a similar water source and an ozone–UV AOP (UV: 1.61 W s cm−2 ozone 0.062 mg mL−1), shows what can be achieved with higher energy and chemical input [Citation40]. However, while AOPs can completely mineralize NOM to carbon dioxide, the high costs involved mean that partial oxidation is the more feasible means of operation. Since AOP products, including aldehydes and carboxylic acids, tend to be biodegradable [Citation47], there has been interest in applying AOPs in synergy with biodegradation. Thus, in contrast to UV/H2O2 alone (UV: 3 J cm−2; H2O2: 10–20 mg L−1), the same AOP dose combined with biological activated carbon (BAC) achieved reductions in DOC, UV, THMFP, DCAAFP and TCAAFP of 80%, 81%, 85%, 63% and 85%, respectively (Table [Citation39]), and thus effected significant DCAAFP removal. Finally, the presence of carbonate and bicarbonate ions can scavenge ·OH and suppress the success of AOPs [Citation17]. This would be most likely in high alkalinity waters, such as the one with alkalinity above 200 mg L−1 as CaCO3 (Table ). Thus, while AOPs are an effective technology for removing a variety of NOM, careful assessment of downstream DBP formation is advised before they are utilized for DBP control.
Table 6. NOM and DBP precursor removal by ozone and biotreatment.
Precursor removal by ozone
Compared with AOPs, ozone is an established part of water treatment, with well over 1000 ozone water treatment plants worldwide [Citation31]. Ozone is typically employed for disinfection, taste and odour control, and degradation of target organic contaminants, rather than bulk DOC removal. This is partly because rate constants for reactions of ozone with organics are much lower than with ·OH. For example apparent rate constants range from 3 × 10–5 M−1 s−1 for acetic acid to 20 × 103 M−1 s−1 for dimethylamine to 18 × 106 M−1 s−1 for phenol [Citation17]. Ozone can be operated as an AOP by adding UV or H2O2 to generate ·OH. In fact, ·OH is also produced naturally through reactions between NOM and ozone, and this is thought to be the major degradation route for target compounds [Citation48]. There is a consensus that ozone alone, under typical water treatment conditions, which involve doses of ∼1 mgO3 mgDOC−1 and a contact time of ∼15 min, is relatively ineffective for DBP precursor removal, though higher doses may enable improved performance [Citation52]. To illustrate, in one reservoir water at an ozone dose of 0.85 mg mgDOC−1 removals of DOC, UV and THM precursors were 5%, 47% and 6%, respectively (Table [Citation53–Citation56]). At an ozone dose of 3 mg mgDOC−1, removal of these parameters had increased to 16%, 72% and 43%, respectively, which also illustrates the selectivity for UV‐absorbing species typical of the process. Particularly at high doses, ozone has the potential to increase HAA and THM levels, though these effects are unpredictable. For example, in one study, with a high ozone dose of 5 mg mgDOC−1 an increase in HAA formation of 50% was observed, contrasting with a 12% decrease at 0.5 mg mgDOC−1 and a 12% increase at 1.0 mg mgDOC−1. Elsewhere, 5% and 4% increases in THMFP and HAAFP, respectively, were observed with combined ozone (2.0 mg mgDOC−1) and biotreatment (Table [Citation56]). Increased levels of bicarbonate concentration during ozonation have been reported to decrease subsequent HAA formation [Citation57]. Since bicarbonate reduces the formation of hydroxyl radicals through scavenging reactions, this indicates that ozone may be more effective than hydroxyl radicals at reacting with precursor sites. In one water with a SUVA of 2.5 L mg−1 m−1, mean removals of THMFP and HAAFP for ozone–coagulation were, respectively, 57% and 66% and consequently similar to coagulation–ozone, where equivalent values of 54% and 71% were observed [Citation53]. As intimated by the rate constants listed, ozone reacts preferentially with activated aromatic compounds. Although nucleophilic reactions are possible, they are slow and electrophilic addition to unsaturated bonds is the main reaction route [Citation58]. Hence, as shown by the generally higher reduction in UV‐absorbing species than other parameters (Table ), ozone primarily reacts with humic species, with the main product groups being aldehydes, ketones and carboxylic acids [Citation58]. As such compounds are typically biodegradable, using ozone upstream of biodegradation can improve precursor removal. Values of −5% to 54% and −4% to 70% for removal of THM and HAA precursors, respectively, have been reported for combined ozone–biofiltration (Table [Citation52,Citation55,Citation56]). Given the propensity for ozone to react selectively with aromatics, the effectiveness even of combined ozone–biofiltration for precursor removal may be limited by the amount of aromatic/humic material. This idea is supported by Wricke and co‐workers [Citation59], who found the maximum production of biodegradable DOC was only around 30% of the total DOC. Ozone can also form bromate, a suspected human carcinogen and inorganic DBP, through reactions with bromide in drinking water [Citation60]. Bromate is currently regulated in the USA at 10 µg L−1 and, while its formation is most acute in high bromide waters, its presence can be mitigated by ozonation at below pH 7, though this is not straightforward in waters with a pH above this value [Citation3,Citation60–Citation61].
Precursor removal by biotreatment
Biological processes in water treatment typically entail development of a biofilm on a sand or activated carbon filter and are more widely employed in Europe than in the USA [Citation62]. Biotreatment can remove NOM through enzyme‐controlled microbial degradation as well as adsorption (Table ). The rate of biodegradation is controlled by substrate mass transport and biodegradation kinetics [Citation63]. Natural organic matter can be divided into easily biodegradable and recalcitrant material [Citation64]. Typical EBCTs for NOM removal are 19–36 min and 5–10 min for activated carbon and sand filters, respectively [Citation63,Citation64]. Small non‐UV‐absorbing molecules tend to be biodegradable [Citation65]. In general, small compounds are more biodegradable because of increased ease of transport across the cell membrane [Citation66]. For example, aldehydes were found to be readily biodegradable [Citation55], with average removal of 70% found for various amino acids [Citation67]. Lower biodegradation of amino acids has been reported elsewhere, such as 46% removal reported by Prévost et al. [Citation68]. These values were interpreted as indicating either aggregation to humic structures or the lower biodegradability of specific amino acids. The amount of biodegradable material in a water body is linked to characteristics of the catchment. Waters with a higher proportion of biologically derived NOM are likely to be low in aromaticity, with high nitrogen content and relatively biodegradable [Citation69]. Amounts of biodegradable NOM in rivers in Europe and the USA were found to vary from a few per cent to around 40% [Citation67]. Despite this, higher removal of HAA precursors, in particular, are found in the literature, with reductions of 37% and 62% for THM and HAA precursors, respectively, by bioactive sand (Table [Citation55]). These values indicate that at least in certain waters, reactive HAA precursors can belong to a readily biodegradable group, possibly aldehydes or amino acids. A more moderate reduction of precursor concentration is found in other studies, and DBP levels can even increase slightly post‐treatment. This is demonstrated by one study using a 3 d contact with BAC, where increases in THMFP and DCAAFP of up to 9% and 11%, respectively, were measured (Table [Citation39]). Interestingly, TCAA precursors proved to be more biodegradable than DCAA precursors, with respective maximum removals of 29% and 46%, respectively (Table [Citation39]). This is the opposite of what would be predicted based on the more hydrophilic character of DCAA precursors compared with TCAA precursors [Citation18] and high biodegradability of low MW, aliphatic molecules [Citation65], and highlights the uncertain identity of aquatic precursors. To summarize, biotreatment will only have a significant impact on precursor removal where reactive precursors are readily biodegradable. Such situations are more probable in waters with high amounts of biologically derived NOM, and are likely to involve HAA precursors. As discussed above, oxidative pretreatment can also be used to increase the amount of biodegradable material.
Precursor removal by membranes
Membrane processes are an increasingly common feature of water treatment [Citation6]. Four types are utilized: microfiltration, ultrafiltration (UF), nanofiltration (NF) and reverse osmosis, listed in order of decreasing pore size and size of molecules rejected, though there is overlap between these classifications [Citation70]. Meanwhile, the majority of NOM has an apparent MW between a few hundred and 10,000 Da, with the mean value towards the lower end of this range [Citation71,Citation72]. Rejection of molecules occurs through size exclusion and electrostatic repulsion for charged membranes, whereas for tighter membranes differing diffusion rates of various solutes across the membrane also participate [Citation70]. Thus, the properties of the membrane surface affect the type of molecules removed. In general, owing to the small size of DBP precursors, NF membranes are required for successful precursor removal. Ultrafiltration has been found to have only limited efficacy for DBP control, with removals up to 44%, 50% and 32% found for bulk DOC, THM precursors and HAA precursors, respectively, by a membrane with molecular weight cut‐off (MWCO) of 60,000 Da (Table [Citation73–Citation75]). These values are higher than those of other work using UF membranes, where respective retentions as low as 17% [Citation73], 10% [Citation76] and 13% have been recorded. It is feasible that the highest value [Citation73] relates to increased high MW species in the particular water, which had a high SUVA of 6.2 L mg−1 m−1. Elsewhere SUVA has been found to positively correlate with MW [Citation77,Citation78]. In contrast, while requiring higher operating pressures, NF has proved extremely effective for precursor removal (Table [Citation73–Citation75]). The maximum retention achievable with NF is represented by a study using four different membranes with MWCO of 100–300 Da where, for one water with 3.8 mg L−1 DOC, removals of 93%, 98% and 99%, respectively, were recorded for DOC, THM and HAA precursors (Table [Citation73]). The minimum retention from a study using five waters, a thin film composite (TFC) and a negative membrane of MWCO 200 Da were 67%, 66% and 67%, respectively (Table [Citation75]), values presumed to correspond to a large proportion of low MW NOM. Several studies have suggested that optimum precursor removal is obtained with a membrane with a MWCO of around 200 Da [Citation79], at which pore size rejection of THMs and HAAs themselves can also be expected.
Table 7. NOM and DBP precursor removal by membrane processes.
In addition to studies using natural waters, model compounds have also been used to assess membrane performance, with removal found to be affected by properties other than size. Hydrophilic model compounds were found to be preferentially removed, compared with hydrophobic compounds, for three different NF membranes [Citation80,Citation81]. The latter study found that, for a group of neutral molecules of MW 146–154 Da, retention varied from 0% to 91% and 0% to 82% for two NF membranes with MWCO of 180 and 150–300 Da, respectively [Citation81]. There was found to be a linear relationship between log KOW and retention. The preferential rejection of acids by a negatively charged membrane [Citation82] can be explained by coulombic repulsion between solute acids and membrane surface. One potential problem with NF is the low removal of bromide, which can cause a shift towards brominated DBPs upon chlorination of the permeate stream [Citation74]. Despite this, NF is still highly effective for DBP precursor removal and may perhaps be used to best effect for removal of low MW, hydrophilic precursors recalcitrant to other treatment processes.
Physical properties of NOM groups
The analysis of removal mechanisms shows treatability of NOM is largely determined by physical properties, especially size, charge and hydrophobicity (Table ). Thus, to assess the treatability of NOM groups by different treatment processes it was necessary to assign these properties. Several assumptions were made while compiling Table [Citation83–Citation87], because of the uncertainty about the precise characteristics of NOM. This ambiguity is complicated by aggregation and overlap in functionality between the listed groups. For example, amino acids and humic species can have carboxylic acid functionality, whereas amino acids may be associated with humic substances in natural waters [Citation88]. It is proposed that humic species are the largest, most hydrophobic and highly charged of the NOM groups. This is because charge is primarily a feature of hydrophobic fractions [Citation9], while MW and aromaticity have been reported to be directly proportional to SUVA254 [Citation77,Citation78]. Although fragmentation of large humic species may occur naturally, it is assumed fragments will retain the character of the whole. Carboxylic acids in NOM are assumed to be smaller and more hydrophilic than humic species, properties consistent with the NOM transphilic fraction known for high carboxylic acid functionality [Citation26,Citation27]. One specific example would be citric acid [Citation69]; other examples include mixed keto‐acid compounds [Citation13] or, more simply, still fatty acids. Thurman considered glutamic acid, glycine, serine and aspartic acid to be the most abundant aqueous amino acids [Citation87]. These species are relatively small (MW 75–147 g mol−1) and hydrophilic (log KOW −3.21 to −3.89), while only glutamic and aspartic acid have a single negative charge based on pKa values. However, combined amino acids are considered to be four to five times more common than free species [Citation83], hence amino acids are considered to be of intermediate MW (Table ). Proteins in water often originate from algae or phytoplankton and, based on pyrolysis data, can include phenol, pyridine, toluene and styrene groups [Citation10]. Glucose is considered the most common sugar in drinking water [Citation87], though arabinose and mannose are also thought to be widespread [Citation89]. These three carbohydrates are neutral, relatively hydrophilic (log KOW −2.39 to −3.24) and relatively small (MW 150–180 g mol−1) and are taken as representative of species found in aquatic environments.
Table 8. Proposed DBP formation, physical properties and treatability of NOM groups.
Discussion: implications for DBP control
Because of the difficulty of identifying precursor material, characterizing waters to predict DBP formation is more complicated than predicting treatability. Formation of DBPs is not straightforward to predict from bulk characters, except where a majority of precursors belong to a group which correlates to a bulk property, as has been observed for humic species and UV absorbance [Citation50]. Since the treatability of NOM groups can be predicted, assuming their physical characteristics are understood, guidance can be provided for their targeted removal. The high DBPFP of humic species is well known (Table ), and they can represent up to 75% of NOM in temperate upland catchments (Table ). Fortunately, because of their charge and size, humic substances are the NOM group most treatable by coagulation (Table ). Therefore well‐optimized coagulation may be sufficient for DBP control where humic species contain the bulk of precursor material. There are precedents for high precursor removal by coagulation in hydrophobic‐rich waters, for example the maximum removals of 71% and 78% for THM and HAA precursors, respectively, reported by Singer and Bilyk [Citation20]. There is also evidence that TCAA precursors are more treatable than DCAA and THM precursors by coagulation [Citation18]. Residual humic substances remaining after coagulation are perhaps most likely fragments of lower size and charge. Owing to their hydrophobicity, in situations where they retain significant DBPFP, activated carbon adsorption is recommended for their removal. Anion exchange or oxidation by ozone/AOP followed by biofiltration may also be successful. Such situations will be indicated by hydrophobic fractions of a post‐coagulation holding relatively high DBPFP.
The variety, identity and amount of carboxylic acids present in NOM have not been fully elucidated (Table ). Assuming carboxylic acids are generally smaller and consequently have fewer charged groups than do humic substances, their removal by coagulation is also presumed to be lower (Table ). The transphilic fraction of NOM, having high DBPFP, is hypothesized to coincide with carboxylic acids being an important source of precursors. Ion exchange is proposed to be an effective choice for their treatment, given its efficiency in treating the transphilic fraction of NOM. Otherwise activated carbon, biotreatment, membranes and AOPs can all be expected to have some success, depending on the nature of the acids present. Owing to electrostatic repulsion, lower removal by activated carbon and charged membranes can be expected than by neutral analogues.
Amino acids and proteins are particularly important constituents of NOM in waters with high algal activity, wastewater influence, or generally higher amounts of biologically derived NOM. Where amino acids and proteins are reactive precursors, it is feasible that the concentration of nitrogen‐containing NOM will correlate to DBP formation, in particular non‐regulated nitrogen‐containing DBPs [Citation7]. Further, l‐aspartic acid and l‐asparagine are known to be reactive HAA precursors [Citation14], and are probably significant DBP precursors in such waters. Coagulation and ion exchange may provide uptake of the charged amino acids, but, because of the low charge of the commonest aquatic species, high removal is not expected for these two processes (Table ). Since amino acids are known to be readily biodegradable, biotreatment is a recommended process option, and nanofiltration is also likely to be effective. The efficacy of adsorption would depend on other NOM present, since, owing to their low hydrophobicity, they will be less adsorbable than similar hydrophobic NOM components. Because of their relatively non‐selective nature, AOPs are proposed to be a suitable process across the range of NOM, including amino acids (Table ). It is predicted that since proteins are larger than amino acids and may incorporate hydrophobic and/or changed side groups, this will make them relatively more responsive than amino acids to all treatments except biodegradation (Table ). This observation is in accordance with the successful removal of algae by coagulation previously reported [Citation90]. Finally, carbohydrates have been found to constitute 50% of NOM in river waters and to form significant THM levels at pH 8 (Table ). On current knowledge the predominant carbohydrates in water are small, neutral and relatively hydrophilic. Thus, they are not expected to be treatable by either coagulation or ion exchange. Instead additional treatment may be necessary in waters where there are important sources of precursors. Nanofiltration may perhaps be most effective, while activated carbon, biotreatment and AOPs may also meet with success. In summary, more effective process selection criteria for precursor removal would come with increased knowledge of precursor identity in an individual water. This would facilitate choice of appropriate technologies for precursor treatment. Depending on the nature of reactive precursors present, optimized coagulation treatment may be proved sufficient for precursor control in hydrophobic waters. Where the post‐coagulation residual remains reactive regarding DBP formation, the deployment of MIEX® for carboxylic acid precursors, and/or GAC for hydrophobic precursors, and/or NF for hydrophilic precursors is recommended.
Acknowledgements
Anglian Water, Northumbrian Water, Severn Trent Water, United Utilities and Yorkshire Water are gratefully acknowledged for their financial support.
References
- Muellner , M.G. , Wagner , E.D. , McCalla , K. , Richardson , S.D. , Woo , Y.‐T. and Plewa , M.J. 2007 . Haloacetonitriles vs. regulated haloacetic acids: Are nitrogen‐containing DBPs more toxic? . Environ. Sci. Technol. , 41 : 645 – 651 .
- Krasner , S.W. 1999 . “ Chemistry of disinfection by‐product formation ” . In Formation and Control of Disinfection By‐Products in Drinking Water , Edited by: Singer , P.C. 27 – 52 . Denver, CO : American Water Works Association .
- von Gunten , U. and Hoigné , J. 1994 . Bromate formation during ozonation of bromide‐containing waters: Interaction of ozone and hydroxyl radical reactions . Environ. Sci. Technol. , 28 : 1234 – 1242 .
- Krasner , S.W. , Weinberg , H.S. , Richardson , S.D. , Pastor , S.J. , Chinn , R. , Sclimenti , M.J. , Onstad , G.D. and Thruston , A.D. Jr . 2006 . Occurrence of a new generation of disinfection byproducts . Environ. Sci. Technol. , 40 : 7175 – 7185 .
- Singer , P.C. , ed. 1999 . “ Formation and Control of Disinfection By‐Products in Drinking Water ” . Denver, CO : American Water Works Association .
- Parsons , S.A. and Jefferson , B. 2006 . “ Introduction to Potable Water Treatment Processes ” . Oxford : Blackwell .
- Lee , W. , Westerhoff , P. and Croué , J.‐P. 2007 . Dissolved organic nitrogen as a precursor for chloroform, dichloroacetonitrile, N‐nitrosodimethylamine and trichloronitromethane . Environ. Sci. Technol. , 41 : 5485 – 5490 .
- Croué , J.‐P. , Debroux , J.‐C. , Amy , G.L. , Aiken , G.R. and Leenheer , J.A. 1999 . “ Natural organic matter: Structural characteristics and reactive properties ” . In Formation and Control of Disinfection By‐Products in Drinking Water , Edited by: Singer , P.C. 65 – 93 . Denver, CO : American Water Works Association .
- Sharp , E.L. , Parsons , S.A. and Jefferson , B. 2006 . Seasonal variations in natural organic matter and its impact on coagulation in water treatment . Sci. Total Environ. , 363 : 183 – 194 .
- Croué , J.‐P. , Korshin , G.V. and Benjamin , M. 2006 . “ Characterization of Natural Organic Matter in Drinking Water ” . Denver, CO : American Water Works Association Research Foundation . report number 90780
- Sharp , E.L. , Jarvis , P. , Parsons , S.A. and Jefferson , B. 2006 . Impact of fractional character on the coagulation of NOM . Colloids Surf. A , 286 : 104 – 111 .
- Rook , J.J. 1977 . Chlorination reactions of fulvic acids in natural waters . Environ. Sci. Technol. , 11 : 478 – 482 .
- Dickenson , E.R. , Summers , R.S. , Croué , J.‐P. and Gallard , H. 2008 . Haloacetic acid and trihalomethane formation from the chlorination and bromination of aliphatic β‐dicarbonyl acid model compounds . Environ. Sci. Technol. , 42 : 3226 – 3233 .
- Reckhow , D.A. and Kim , J. Role of proteins and amino acids in DBP formation . IWA conference on NOM: From Source to Tap . Bath.
- Mitch , W.A. , Krasner , S.W. , Westerhoff , P. and Dotson , A. 2009 . Occurrence and Formation of Nitrogenous Disinfection By‐Products, report number 91250 , Denver, CO : Water Research Foundation .
- Randtke , S.J. 1999 . “ Disinfection by‐product precursor removal by coagulation and precipitate softening ” . In Formation and Control of Disinfection By‐Products in Drinking Water , Edited by: Singer , P.C. 237 – 258 . Denver, CO : American Water Works Association .
- MWH , Crittenden , J.C. , Rhodes Trussell , R. , Hand , D.W. , Howe , K.J. and Tchobanoglous , G. 2006 . Water Treatment: Principles and Design, , 2nd ed. , New York : Wiley .
- Liang , L. and Singer , P.C. 2003 . Factors influencing the formation and relative distribution of haloacetic acids and trihalomethanes in drinking water . Environ. Sci. Technol. , 37 : 2920 – 2928 .
- Boyer , T.H. and Singer , P.C. 2005 . Bench‐scale testing of a magnetic ion exchange resin for removal of disinfection by‐product precursors . Water Res. , 39 : 1265 – 1276 .
- Singer , P.C. and Bilyk , K. 2002 . Enhanced coagulation using a magnetic ion exchange resin . Water Res. , 36 : 4009 – 4022 .
- Selcuk , H. , Rizzo , L. , Nikolaou , A.N. , Meric , S. , Belgiorno , V. and Bekbolet , M. 2007 . DBPs formation and toxicity monitoring in different origin water treated by ozone and alum/PAC coagulation . Desalination , 210 : 31 – 43 .
- Stevens , A.A. , Moore , L.A. and Miltner , R.J. 1989 . Formation and control of non‐trihalomethane disinfection by‐products . J. Amer. Water Works Assoc. , 81 : 54 – 60 .
- Edwards , M. 1997 . Predicting DOC removal during enhanced coagulation . J. Amer. Water Works Assoc. , 89 : 78 – 89 .
- Lee , N. , Sinha , S. , Amy , G. and Bourke , M. Evaluation of magnetic ion exchange resin treatment for preferential removal of NOM fractions . American Water Works Association – Water Quality Technology Conference . Seattle. November .
- Mergen , M.R.D. , Jefferson , B. , Parsons , S.A. and Jarvis , P. 2008 . Magnetic ion‐exchange resin treatment: Impact of water type and resin use . Water Res. , 42 : 1977 – 1988 .
- Boyer , T.H. , Singer , P.C. and Aiken , G.R. 2008 . Removal of dissolved organic matter by anion exchange: Effect of dissolved organic matter properties . Environ. Sci. Technol. , 42 : 7431 – 7437 .
- Aiken , G.R. , McKnight , D.M. , Thorn , K.A. and Thurman , E.M. 1992 . Isolation of hydrophilic organic acids from water using nonionic macroporous resins . Org. Geochem. , 18 : 567 – 573 .
- Johnson , C.J. and Singer , P.C. 2003 . Impact of a magnetic ion exchange resin on ozone demand and bromate formation during drinking water treatment . Water Res. , 38 : 3738 – 3750 .
- Bolto , B. , Dixon , D. , Eldridge , R. , King , S. and Linge , K. 2002 . Removal of natural organic matter by ion exchange . Water Res. , 36 : 5057 – 5065 .
- American Water Works Association and Pontius , F.W. , eds. 2006 . Water Quality and Treatment, , 4th ed. , New York : McGraw‐Hill .
- Metcalf and Eddy Inc. , Tchobanoglous , G. , Burton , F.L and Stensel , H.D. 2006 . Wastewater Engineering Treatment and Reuse, , 4th ed. , New York : McGraw‐Hill .
- Buxton , G.V. , Greenstock , C.L. , Helman , W.P. and Ross , A.B. 1988 . Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in aqueous solution . J. Phys. Chem. Ref. Data , 17 : 513 – 886 . available and updated at http://www.rcdc.nd.edu/compilations/INTRO.HTM
- US EPA . December 2006 . “ Estimation Programs Interface Suite™ for Microsoft® Windows, v 3.2 ” . December , Washington, D.C., , USA : United States Environmental Protection Agency .
- Chemspider, Chemical data base search. Available at http://www.chemspider.com/Search.aspx (accessed December 2008)
- US National Library of Medicine . “ Chem ID Plus advanced database search ” . Available at http://www.chem.sis.nlm.nih.gov/chemidplus/ (accessed December 2008)
- National Center for Biotechnology Information (NCBI), PubChem database search. Available at: http://pubchem.ncbi.nlm.nih.gov/ (accessed December 2008)
- Chaidou , C.I. , Georgakilas , V.I. , Stalikas , C. , Saraçi , M. and Lahaniatis , E.S. 1999 . Formation of chloroform by aqueous chlorination of organic compounds . Chemosphere , 39 : 587 – 594 .
- De Laat , J. , Merlet , N. and Dore , M. 1982 . Chloration de composés organiques: Demande en chlore et réactivite vis‐a‐vis de la formation des trihalométhanes. Incidence de l'azote ammoniacal . Water Res. , 16 : 1437 – 1450 . (In French)
- Toor , R. and Mohseni , M. 2007 . UV‐H2O2 based AOP and its integration with biological activated carbon treatment for DBP reduction in drinking water . Chemosphere , 66 : 2087 – 2095 .
- Chin , A. and Bérubé , P.R. 2005 . Removal of disinfection by‐product precursors with ozone‐UV advanced oxidation process . Water Res. , 39 : 2136 – 2144 .
- Jacangelo , J.G. , DeMarco , J. , Owen , D.M. and Randtke , S.J. 1995 . Selected processes for removing NOM: An overview . J. Amer. Water Works Assoc. , 87 : 64 – 77 .
- Weber , W.J. , McGinley , P.M. and Katz , L.E. 1991 . Sorption phenomena in subsurface systems: Concepts, models and effects on contaminant fate and transport . Water Res. , 25 : 499 – 528 .
- Karanfil , T. , Schlautman , M.A. , Kilduff , J.E. and Weber , W.J. 1996 . Adsorption of organic macromolecules by granular activated carbon. 2. Influence of dissolved oxygen . Environ. Sci. Technol. , 30 : 2195 – 2201 .
- Kilduff , J.E. , Karanfil , T. , Chin , Y.‐U. and Weber , W.J. 1996 . Adsorption of natural organic electrolytes by activated carbon: A size exclusion chromatography study . Environ. Sci. Technol. , 30 : 1336 – 1343 .
- Karanfil , T. , Kitis , M. , Kilduff , J.E. and Wigton , A. 1999 . Role of granular activated carbon surface chemistry on the adsorption of organic compounds. 2. Natural organic matter . Environ. Sci. Technol. , 33 : 3325 – 3333 .
- Karanfil , T. , Cheng , W. , Guo , Y. , Dastgheib , S.A. and Song , H. 2006 . “ DBP formation control by modified activated carbons ” . Denver, CO : American Water Works Association Research Foundation . Report number 91181
- Parsons , S.A. , ed. 2006 . “ Advanced Oxidation Processes for Water and Wastewater Treatment ” . London : IWA .
- Westerhoff , P. , Aiken , G. , Amy , G. and Debroux , J. 1999 . Relationships between the structure of natural organic matter and its reactivity towards molecular ozone and hydroxyl radicals . Water Res. , 33 : 2265 – 2276 .
- Westerhoff , P. , Mezyk , S.P. , Cooper , W.J. and Minakata , D. 2007 . Electron pulse radiolysis determination of hydroxyl radical rate constants with Suwannee River fulvic acid and other dissolved organic matter isolates . Environ. Sci. Technol. , 41 : 4640 – 4646 .
- Reckhow , D.A. , Singer , P.C. and Malcolm , R.L. 1990 . Chlorination of humic materials: Byproduct formation and chemical interpretations . Environ. Sci. Technol. , 24 : 1655 – 1664 .
- Reckhow , D.A. and Singer , P.C. 1985 . “ Mechanisms of organic halide formation during fulvic acid chlorination and implications with respect to preozonation ” . In Water Chlorination: Environmental Impact and Health Effects , Edited by: Jolley , R.L. , Bull , R.J. and Davis , W.P. Vol. 5 , 1229 – 1257 . Chelsea, MI : Lewis Publishers .
- Speitel , G.E. , Symons , J.M. , Diehl , A.C. , Sorenson , H.W. and Cipparone , L.A. 1993 . Effect of ozone dosage and subsequent biodegradation on removal of DBP precursors . J. Amer. Water Works Assoc. , 85 : 89 – 95 .
- Chaiket , T. , Singer , P.C. , Miles , A. , Moran , M. and Pallotta , C. 2002 . Effectiveness of coagulation, ozonation and biofitration in controlling DBPs . J. Amer. Water Works Assoc. , 92 : 81 – 95 .
- Galapate , R.P. , Baes , A.U. and Okada , M. 2001 . Transformation of dissolved organic matter during ozonation: Effects on trihalmethane formation potential . Water Res. , 35 : 2201 – 2206 .
- Miltner , R.J. , Shukairy , H.M. and Summers , R.S. 1992 . Disinfection by‐product formation and control by ozonation and biotreatment . J. Amer. Water Works Assoc. , 84 : 53 – 62 .
- Siddiqui , M.S. , Amy , G.L. and Murphy , B.D. 1997 . Ozone enhanced removal of natural organic matter from drinking water sources . Water Res. , 31 : 3098 – 3106 .
- Reckhow , D.A. , Legube , B. and Singer , P.C. 1986 . The ozonation of organic halide precursors: Effect of bicarbonate . Water Res. , 20 : 987 – 998 .
- Goel , S. , Hozalski , R.M. and Bouwer , E.J. 1995 . Biodegradation of NOM: Effect of NOM source and ozone dose . J. Amer. Water Works Assoc. , 87 : 90 – 105 .
- Wricke , B. , Petzoldt , H. , Heiser , H. and Bornmann , K. 2006 . “ NOM – removal by biofiltration after ozonation – results of a pilot plant test ” . In Advances in Slow Sand and Alternative Biological Filtration , Edited by: Nigel , G. and Robin , C. New York : Wiley .
- von Gunten , U. and Hoigne , J. 1992 . Factors controlling the formation of bromate during ozonation of bromide‐containing waters . AQUA , 41 : 299 – 304 .
- Weinberg , H.S. , Delcomyn , C.A. and Unnam , V. 2003 . Bromate in chlorinated drinking waters: Occurrence and implications for future regulation . Environ. Sci. Technol. , 37 : 3104 – 3110 .
- Shukairy , H.M. and Summers , R.S. 1992 . The impact of preozonation and biodegradation on disinfection by‐product formation . Water Res. , 26 : 1217 – 1227 .
- Huck , P.M. , Zhang , S. and Price , M.L. 1994 . BOM removal through biological treatment: A first order model . J. Amer. Water Works Assoc. , 86 : 61 – 71 .
- Yavich , A.A. and Masten , S.J. 2003 . Use of ozonation and FBT to control THM precursors . J. Amer. Water Works Assoc. , 94 : 159 – 171 .
- Hwang , C.J. , Krasner , S.W. , Schlimenti , M.J. , Amy , G.L. , Dickenson , E. , Bruchet , A. , Prompsy , C. , Filippi , G. , Croué , J.‐P. and Violleau , D. 2006 . “ Polar NOM: Characterization, DBPs, treatment ” . Denver, CO : American Water Works Association Research Foundation . report number 98077
- Leisinger , T. , ed. 2006 . “ Microbial Degradation of Xenobiotics and Recalcitrant Compounds ” . London : Academic Press .
- Jadas‐Hécart , A. 1989 . “ Contribution à l'étude de la demande en chlore à long terme d'une eau potable: modélisation et identification des précursors organiques, Ph.D. thesis ” . Université de Potiers . (In French)
- Prévost , M. , Gauthier , C. , Hureïki , L. , Desjardins , R. and Servais , P. 1998 . Removal of amino acids, biodegradable organic carbon and chlorine demand by biological filtration in cold water . Environ. Technol. , 19 : 903 – 911 .
- Leenheer , J.A. and Croué , J.‐P. 2003 . Characterizing aquatic dissolved organic matter . Environ. Sci. Technol. , 37 : 18A – 26A .
- Judd , S. and Judd , C. , eds. 2006 . “ The MBR Book ” . Amsterdam : Elsevier .
- Pelekani , C. , Newcombe , G. , Snoeyink , V.L. , Hepplewhite , C. , Assemi , S. and Beckett , R. 1999 . Characterization of natural organic matter using high performance size exclusion chromatography . Environ. Sci. Technol. , 33 : 2807 – 2813 .
- Tadanier , C.J. , Berry , D.F. and Knocke , W.R. 2000 . Dissolved organic matter apparent molecular weight distribution and number‐average apparent molecular weight by batch ultrafiltration . Environ. Sci. Technol. , 34 : 2348 – 2353 .
- Siddiqui , M.S. , Amy , G. , Ryan , J. and Odem , W. 2000 . Membranes for the control of natural organic matter from surface waters . Water Res. , 34 : 3355 – 3370 .
- Chellam , S. 2000 . Effects of nanofiltration on trihalomethane and haloacetic acid precursor removal and speciation in waters containing low concentrations of bromide ion . Environ. Sci. Technol. , 34 : 1813 – 1820 .
- Allgeier , S.C. and Summers , R.S. 1995 . Evaluating NF for DBP control with the RBSMT . J. Amer. Water Works Assoc. , 87 : 87 – 99 .
- Agbekodo , M.K. , Legube , B. and Cote , P. 1996 . Organics in NF permeate . J. Amer. Water Works Assoc. , 88 : 67 – 74 .
- Chin , Y.P. , Aiken , G. and Oloughlin , E. 1994 . Molecular‐weight, polydispersity, and spectroscopic properties of aquatic humic substances . Environ. Sci. Technol. , 28 : 1853 – 1858 .
- Weishaar , J.L. , Aiken , G.R. , Bergamanschi , B.A. , Fram , M.S. , Fujii , R. and Mopper , K. 2003 . Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon . Environ. Sci. Technol. , 37 : 4702 – 4708 .
- Van der Bruggen , B. and Vandecasteele , C. 2003 . Removal of pollutants from surface water and groundwater by nanofiltration: Overview of possible applications in the drinking water industry . Environ. Pollut. , 122 : 435 – 445 .
- Amy , G. , Kim , T.‐U. , Yoon , J. , Bellona , C. , Drewes , J. , Pellegrino , J. and Heberer , T. 2005 . Removal of micropollutants by NF/RO membranes . Water Sci. Technol. Water Supply , 5 : 25 – 33 .
- Braeken , L. , Ramaekers , R. , Zhang , Y. , Maes , G. , Van der Bruggen , B. and Vandecasteele , C. 2005 . Influence of hydrophobicity on retention in nanofiltration of aqueous solutions containing organic compounds . J. Membr. Sci. , 252 : 195 – 203 .
- Cho , J. , Amy , G.L. and Pellegrino , J. 2000 . Membrane filtration of natural organic matter: Factors and mechanisms affecting rejection and flux decline with charged ultrafiltration (UF) membrane . J. Membr. Sci. , 164 : 89 – 110 .
- Hureïki , L. , Croué , J.‐P. and Legube , B. 1994 . Chlorination studies of free and combined amino acids . Water Res. , 28 : 2521 – 2531 .
- Navalon , S. , Alvaro , M. and Garcia , H. 2008 . Carbohydrates as trihalomethanes precursors. Influence of pH and the presence of Cl− and Br− on trihalomethane formation potential . Water Res. , 42 : 3990 – 4000 .
- Navalon , S. , Alvaro , M. and Garcia , H. 2008 . Characterization of dissolved organic matter from Turia river. Prevalence of low‐molecular mass oligosaccharides . Chemosphere , (submitted)
- Scully , F.E.J. , Howell , G.D. , Kravitz , R. and Jewell , J.T. 1988 . Proteins in natural waters and their relation to the formation of chlorinated organics during water disinfection . Environ. Sci. Technol. , 22 : 537 – 542 .
- Thurman , E.M. 2006 . “ Organic Geochemistry of Natural Waters ” . Dordrecht : Nijhoff/Junk Publishers .
- Volk , C.J. , Volk , C.B. and Kaplan , L.A. 1997 . Chemical composition of biodegradable dissolved organic matter in streamwater . Limnol. Oceanogr. , 42 : 39 – 44 .
- McKnight , D.M. , Thurman , E.M. and Wershaw , R.L. 1985 . Biogeochemistry of aquatic humic substances in Thoreau's bog, Concord, Massachusetts . Ecology , 66 : 1339 – 1352 .
- Henderson , R.K. , Parsons , S.A. and Jefferson , B. 2008 . Successful removal of algae through the control of zeta potential . Sep. Sci. Technol. , 43 : 1653 – 1666 .