Abstract
A simple and effective method was developed using phenol formaldehyde (PF) resins to immobilize hydrous ferric oxide (HFO) onto granular activated carbon (GAC). The resulting sorbent possesses advantages for both the ferric oxide and the GAC, such as a great As‐affinity of ferric oxide, large surface area of GAC, and enhanced physical strength. The studies showed that within one hour this sorbent was able to remove 85% of As(V) from water containing an initial As(V) concentration of 1.74 mg l−1. The As(V) adsorption onto the sorbent was found to follow a pseudo‐second order kinetics model. The adsorption isotherms were interpreted in terms of the Langmuir and Freundlich models. The equilibrium data fitted very well to both models. Column tests showed that this sorbent was able to achieve residual concentrations of As(V) in a range of 0.1–2.0 µg l−1 while continuously treating about 180 bed volume (BV, 130 ml‐BV) of arsenate water with an initial As(V) concentration of 1886 µg l−1 at a filtration rate of 13.5 ml min−1, i.e., an empty bed contact time (EBCT) of 9.6 min and a gram sorbent contact time (GSCT) of 0.15 min. After passing 635 BV of arsenate water, the exhausted sorbent was then tested by the Toxicity Characteristic Leaching Procedure (TCLP, US EPA Method 1311) test, and classified as non‐hazardous for disposal. Hence, this HFO‐PF‐coated GAC has the capability to remove As(V) from industrial wastewater containing As(V) levels of about 2 mg l−1.
INTRODUCTION
Arsenic contamination in water has become a worldwide environmental issue [Citation1]. It can be attributed to the mobility of arsenic and its compounds through a combination of natural processes [Citation2, Citation3], such as biological activity, volcanic emissions and erosion of arsenic‐containing rocks through geochemical reactions, in addition to anthropogenic activities. Arsenic‐containing waste streams are generated from mineral and chemical processes, arsenical pesticides and wood preservatives, and semiconductor devices using gallium arsenide (GaAs) [Citation4, Citation5]. Owing to its toxic and carcinogenic effects, arsenic contamination in water is creating potentially serious environmental problems for human beings and other living organisms [Citation6, Citation7]. The acceptable level of arsenic in drinking water was recently limited to 10 µg l−1 in the United States [Citation8]. This regulation has created challenges for the development of new, cost effective technologies capable of reducing arsenic to this level. Among the most commonly used technologies [Citation9–Citation11], adsorption is considered to be the most attractive treatment because it is economical and easy to implement.
Various types of sorbent have been used in the study of arsenic adsorption. The most common sorbents include activated alumina (AA) [Citation12, Citation13], activated carbon (AC) [Citation14], biomass [Citation15, Citation16], anion exchange resins [Citation17], metal‐loaded polymers [Citation18], and a variety of iron‐based adsorption media such as granular ferric hydroxide [Citation19], Fe(III)‐resins [Citation20, Citation21], and ferric modified materials [Citation22–Citation28]. Among these types, the most attractive sorbents are composed of aluminum and iron components, which show significant arsenic adsorption capability. This can be associated with the great metal‐affinity of Al and Fe [Citation29, Citation30] which have been utilized as conventional flocculating agents for metal removal in water. Activated alumina is the sorbent most commonly used to remove arsenic from drinking water. It has also been used in the treatment of arsenic in groundwater, but it was more expensive for both initial and ongoing operations as compared with other reported technologies.
Extensive studies show that hydrous ferric oxide (HFO) is a promising adsorption material for arsenic removal [Citation24, Citation31]. Moreover, it was reported that the HFO‐based sorbents had a preference for removal of As(V) over As(III) species [Citation32]. This could be associated with the high isoelectric point (pHiep = 7.9–8.2) of HFO [Citation30, Citation33]. Below the isoelectric pH, the surface of HFO has a slightly positive zeta potential (ζ > 0) which promotes interaction with arsenic anions. The strength of this ion‐dipole interaction depends on the charge and size of the ion and the magnitude of the dipole. Hence, the adsorption order for the HFO‐based sorbents is: AsO4 3− ≫ HAsO4 2− ≫ H2AsO4 − > H2AsO3 − ≫ H3AsO3. The dominant species of As(V) and As(III), existing in natural waters with a pH range of 6–9, are H2AsO4 −/HAsO4 2− and H3AsO3/H2AsO3 −, respectively [Citation32]. So, HFO‐based sorbents prefer negatively charged As(V) anions to As(III) species. Compared with other iron oxides, such as goethite and hematite, amorphous HFO has the greatest adsorption capability since it has the largest surface area [Citation33, Citation34]. However, HFO has two shortcomings similar to most iron oxides. Firstly, HFO is available only as fine powders, which are not suitable for column application because of their low hydraulic conductivity [Citation35]. Secondly, HFO tends to form crystalline iron oxides, which greatly reduce the surface area and thus capacity for arsenic removal [Citation31, Citation34]. To overcome these disadvantages, HFO granulation techniques have been developed for making granular ferric hydroxide (GFH) under high pressure [Citation19, Citation31, Citation33]. However, the physical integrity of the GFH beads was poor with extreme cracking and peeling at the surface, causing significant headloss pressure within a short operation time [Citation32].
Hence, there is a need to produce HFO‐based sorbents possessing great As‐adsorption capability, large surface area and satisfactory mechanical strength. The primary objective of this study was to develop a simple and effective method of using phenol formaldehyde (PF) resins for immobilizing HFO onto granular activated carbon (GAC). The cured PF resins can hinder the gradual crystallization of HFO. Accordingly, HFO‐PF‐coated GAC should have good mechanical strength without the problem of surface cracking and peeling, in addition to the favorable features of both HFO and GAC. The resulting sorbent was further assessed by examining its As(V) adsorption properties at equilibrium and kinetic conditions, and by testing its column performance with a view to providing an economically feasible solution for remediating industrial As‐contaminated waters.
MATERIALS AND METHODS
Materials
A sample of GAC (Type PCB, mesh 12×30) was provided by Calgon Carbon Corp. Phenol formaldehyde resins (55% solids, viscosity 169 cps) with low polymerization degree (n = 1–3) were obtained from Forintek Canada Corp. The FeCl3 solution (41% w/w, technical grade) was supplied by Brenntag Canada Inc. The solid chemicals of Na2HAsO4·7H2O and NaOH were reagent grade, and separately used to prepare the solutions of As(V) (1.5–1.9 mg l−1) and NaOH (10% w/w) with water. The arsenic content for each sample was determined at Vizon Scitec, using a Perkin Elmer Optima 3300DV Inductively Coupled Plasma Atomic Emission Spectrometer. Low levels of arsenic (< 20 µg l−1) were analyzed with a Thermo X Series II Inductively Coupled Plasma Mass Spectrometer.
Procedures for Preparing HFO‐PF‐Coated GAC
The sorbent was prepared by soaking 100 g of GAC in 60 g of FeCl3 (41% w/w), leading to the uniform coverage of the GAC surface with Fe(III). Then, FeCl3 was converted to HFO with NaOH (10% w/w) neutralization carried out under aeration using an air‐sparger. The HFO‐coated GAC was then filtered and washed with water (100 ml × 2) to remove soluble compounds. Finally, it was immersed in a diluted solution of PF resins (40 g, PF(55%)/water = 1/1). After thorough mixing, the treated GAC was collected by filtration, and baked in an oven with a temperature profile that started at room temperature and gradually increased to 150–175°C. It was held for two hours at this temperature, and then for an additional one hour under vacuum to remove volatile and thermal unstable compounds. Finally, the prepared sorbent (mesh 12×30, avg. 1.2 mm) contained about 6 g Fe(III) immobilized by PF thin film onto the surface (1150–1250 m2 g−1) of GAC with a pH value of 6.5–7.0 in 5% water solution. The chemical cost is about 1.5–2.0 USD kg−1 to prepare HFO‐PF‐coated GAC.
As‐adsorption Isotherm
Batch experiments were conducted at pH 6.9–7.0 to obtain As‐adsorption isotherms. About 400 mg of sorbent (HFO‐PF‐coated GAC), and 80 ml of arsenic solution with varying initial As(V) concentrations (0.67–29.0 mg l−1, Table ) were added to each of the six 250‐ml Erlenmeyer flasks. Then, the flasks were vigorously shaken with a wrist‐arm shaker at room temperature (21±2°C) for 24 hours and then left on the bench for 24 hours. Each solution was filtered through a 0.45 µm filter paper (Waterman®) for determining As concentration in water.
Table 1. As(V) adsorption isotherm for HFO‐PF‐coated GAC.
Kinetics of As‐adsorption
To study the effect of contact time on As(V) uptake, the experiments were performed at room temperature by using 10.1007 g of sorbent in 1050 ml of arsenic solution with an initial As(V) concentration of 1740 µg l−1. The pH of solution was maintained at pH 6.7–6.9. With vigorous mixing, samples were taken from the solution at 0, 3, 8, 15, 25, 40, 60, 90, 120 and 180 minutes, and filtered through a 0.45 µm filter paper for analyzing As concentration in water.
Pilot‐scale Column Tests
A glass column (2.54‐cm internal diameter and 30‐cm length) was carefully packed with sorbent (HFO‐PF‐coated GAC, 62 g), ensuring no air was trapped in the sorbent bed. Trapped air blocks As‐water from migrating to the adsorption sites and hinders the sorbent functionality. For de‐aerating (fully wetting) the sorbent, the column was pre‐filled with water and allowed to sit idle for 24 hours, then backwashed in an upflow direction to displace any air that has been trapped in the sorbent bed.
The sorbent bed volume (BV) was 130 ml, and the empty bed contact time (EBCT) was set at 9.6 minutes. That is, the contact time per gram of the sorbent (GSCT) was 0.15 minutes (= 9.6 min/62 g). Two batches of As‐water, containing As(V) 1886 µg l−1 and 1470 µg l−1, respectively, were prepared to simulate industrial As(V)‐contaminated wastewaters at pH 6.8. In the first batch, 23.4 l of As‐water (As(V) 1886 µg l−1) were pumped through the column at a filtration rate of 13.5 ml min−1. In the second batch, 59.2 l of As‐water (As(V) 1470 µg l−1) were pumped through the column at a slower rate (1.6–4.7 ml min−1) for the purpose of completely exhausting the sorbent. After finishing the column tests, the exhausted sorbent was left in the column with As‐water for two more months and then subjected to the Toxicity Characteristic Leaching Procedure (TCLP, US EPA Method 1311) [Citation36] test. The samples of filtrate were analyzed with the standard ICP method.
RESULTS AND DISCUSSION
HFO‐PF‐Coated GAC Preparation
GAC, made from coconut shell type PCB, was selected as a HFO support due to its high surface area (1150–1250 m2 g−1), macropore structure (>1000 Å in diameter), high density (0.85 g ml−1) and high pore volume activity (0.72 ml g−1) [Citation37]. Hydrous ferrous oxide is poorly crystalline, highly porous and unstable in hydrated (FeO(OH)·nH2O) or anhydrous (FeO(OH)) forms. Resol PF resins (Figure ), commonly used wood adhesives, were utilized to tightly combine both amorphous HFO and GAC through the formation of an Fe(III) complex with hydroxide groups of PF (Figure ) and the adhesion bonding between cured PF resins and GAC. It was reported that the technique to impregnate Fe‐GAC composites, resulting in mineralogical and structural changes, is an important means to increase the iron content [Citation24]. This paper presents the key techniques employing low polymerization degree (PD, n = 1–3) of PF resins with low viscosity to immobilize HFO onto the GAC surface and hinder HFO gradual crystallization. If using PF resins with a high concentration or with a high PD, PF resins could block the pores of the GAC leading to a poor As‐adsorption. The diluted PF solution can freely enter into the pores of the GAC to contact Fe(III) existing on the surface of the GAC. According to the procedures of preparation, soluble impurities (such as sodium salts) were removed with water washing, and volatile and unstable compounds (such as small species of PF) were eliminated under vacuum and high temperatures of 150–175°C. After curing the PF resins, the resulting sorbent possesses advantages for both ferric oxide and GAC, such as the great As‐affinity of ferric oxide, the large surface area of GAC, and enhanced physical strength. As a result, no extreme cracking or peeling at the surface was observed during the two‐month column test of this sorbent. With a view to its potential application in remediating As‐contaminated industrial wastewaters, the sorbent was assessed, using the As‐water containing ≈2 mg l−1 As(V), for As(V) adsorption properties under kinetic and equilibrium conditions, and its column performance was tested. Due to issues of cost and handling of hazardous As chemicals, the regeneration of the sorbent was not frequently practiced although it can be regenerated.
Figure 1 Proposed structure for: (A) Type resol PF resins, and (B) linked iron with PF.
As‐adsorption Isotherm
The Langmuir equation was used to describe the equilibrium distribution of As(V) between the water and the sorbent. The adsorbate used in the isotherm equation is expressed as its molar concentration. However, in the literature, there is a common misapplication using the volumetric concentration of adsorbate with no theoretical consideration. The volumetric concentration of adsorbate can be used in the Langmuir isotherm equation only when the molecular weight of the adsorbate is much smaller than that of the sorbent [Citation38]. In this experiment, since HFO was immobilized onto the surface of the GAC by cured PF resins through chemical complexing and bonding, the HFO‐PF‐coated GAC was then considered as an integral sorbent having macro‐molecular weight. The effects of adsorbed As(V) on the weight of the much heavier sorbent could be neglected. Hence, the volumetric concentration of As(V) can be used in the Langmuir isotherm equation instead of the original molar concentration.
where Qe is the amount of As(V) adsorbed per unite weight of sorbent (µg g−1), Qm is the saturated monolayer adsorption capacity (µg g−1), Ce is the As(V) equilibrium concentration (mg l−1), and b is the Langmuir constant expressed in l mg−1, representing the balance relationships among As(V), sorbent, and the binding complex of As(V)‐sorbent. The value of b is related to the strength or affinity of As(V) for the sorbent at specific conditions. Equation (Equationi) is rearranged to the form:
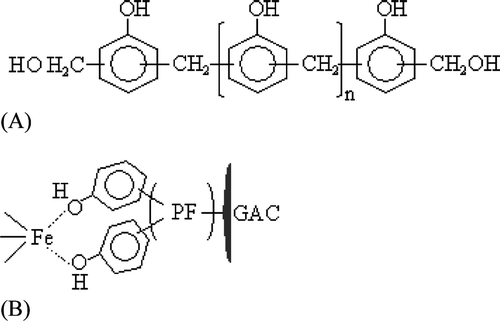
The experimental data (Table ) was plotted using the 1/Qe vs. 1/Ce version of the Langmuir isotherm Equation (Equationii). The least‐squares fit showed a good linear quality (R2 = 0.968, Figure ). Although this suggests the Langmuir isotherm equation is suitable to describe the As(V) adsorption process, the Qm value (1000 µg g−1) calculated from the intercept of Equation (Equationiii) is not reliable. It is too far below Qe (1880.5 and 2330.7 µg g−1) obtained for the initial As(V) concentrations (Ci ) of 19.3 and 29.0 mgl−1 (Table ).
Figure 2 As‐adsorption isotherm for adsorbent of HFO‐PF‐coated GAC at different initial As(V) concentrations: (A) the plot for the 1/Qe vs. 1/Ce version of Langmuir isotherm, and (B) the plot for the Qe vs. Ce version of Langmuir isotherm.
Correspondingly, the plot of Qe vs. Ce for the Langmuir isotherm Equation (Equationi) in Figure shows the data are far from total site saturation which is commonly represented by a flat line, i.e. Qe approaches a constant (Qm ) as Ce continually increases. This shows that the active sites were still present at equilibrium with an initial As(V) concentration of 19.3 or 29 mg l−1. Therefore, the assumption of surface homogeneity for deriving the Langmuir equation could not explicitly express the As(V) adsorption onto HFO‐PF‐coated GAC.
The Langmuir equation assumes that the sorbent surface has a homogeneous distribution of adsorption sites. Each site is capable of adsorbing one molecule of the adsorbate equally. Once the surface is covered with a monolayer of adsorbed molecules, it is saturated and has no further influence on the system. However, real systems are likely to deviate from the theoretical model. The Langmuir equation quite often yields only an empirical isotherm, which could be caused : (i) complex interactions among solvent‐solute, solvent‐sorbent, and solute‐sorbent; (ii) a heterogeneous surface composed of adsorption sites with various adsorption potentials; and (iii) multi‐layers of adsorbed molecules.
Thus, the Fruendlich isotherm equation, which assumes each adsorption site has different adsorption potentials, was used to express the As(V) adsorption onto HFO‐PF‐coated GAC.
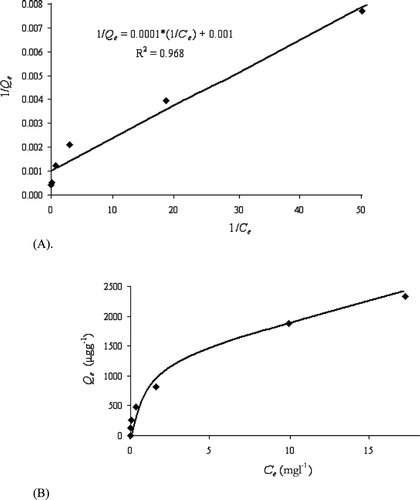
where K and n are Fruendlich constants related to the adsorption capability and the adsorption intensity, respectively. Equation (Equationiv) gives the mathematical relationships between K and 1/n. For a given value of K, a smaller value of 1/n means stronger adsorption strength. For a fixed value of 1/n, a larger value of K means a greater adsorption capacity. The resulting K value was 0.724 mg g−1 and 1/n value was 0.4. Similar magnitudes of K, 0.734 and 0.689 mg g−1, were reported for As(III) removal by the sorbents of iron oxide‐impregnated activated alumina and iron oxide‐coated cement respectively [Citation39]. The K values for As(V) adsorption onto different impregnated Fe‐GAC composites were reported to be within the range 0.01–0.29 mg g−1, and the 1/n values, within a range of 0.49–1.19 [Citation24]. Obviously, HFO‐PF‐coated GAC compares well with most of the related sorbents, hence, it can be considered to be a viable sorbent for As(V) removal from industrial wastewater containing ≈2 mg l−1 of As(V).
Figure shows an improved linear quality (R2 = 0.9937) for the logarithmic version of the Fruendlich isotherm Equation (Equationv). Hence, the Fruendlich equation should more closely approximate to the real As(V) adsorption process. That is, the surface for HFO‐PF‐coated GAC was distributed with heterogeneous adsorption sites, such as various sizes of HFO micro‐particles. Langmuir adsorption would occur at these sites, each having different adsorption potentials.
Figure 3 Freundlich plot at different initial As(V) concentrations.
As‐adsorption Kinetics
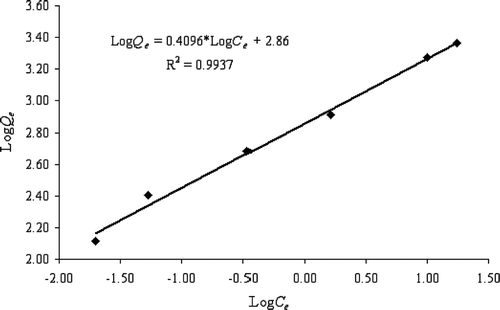
The kinetics data are listed in Table and plotted in Figure showing that the sorbent was able to remove 92% of As(V) in approximately 90 minutes. The As(V) level was reduced from an initial 1.74 mg l−1 to 0.04 mg l−1. For modeling the rate of As(V) adsorption onto HFO‐PF‐coated GAC in a well‐agitated system, the pseudo‐second order equation [Citation40] was applied to the As(V) adsorption kinetics data. Assuming the As(V) adsorption capacity of the sorbent is proportional to the number of active sites on its surface, then the rate for the pseudo‐second order equation is given by Equation (Equationvi).
Table 2. Kinetics data for As(V) adsorption onto HFO‐PF‐coated GAC.
Figure 4 Kinetics of As(V) removal by HFO‐PF‐coated GAC.
where k2 is the rate constant of adsorption (g mg−1min), qt is the amount of adsorbed As(V) at time t (mg g−1), and qe is the equilibrium uptake (mg g−1). At t = 0, qt = 0, Equation (Equationvi) is converted to Equation (Equationvii) expressing the initial adsorption rate, ri .
Equation (Equationvi) can be integrated and rearranged to the form:
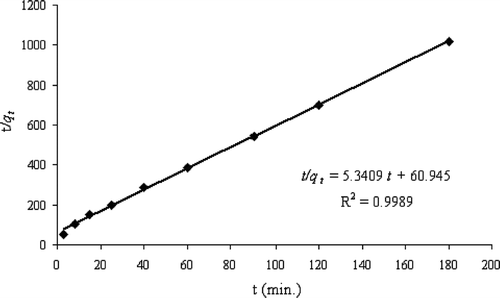
Figure shows a straight line of t/qt vs. t for the As(V) adsorption onto the sorbent. The correlation coefficient is close to one (R2 = 0.9989) indicating that the experimental data are in good agreement with the pseudo‐second order equation. Therefore, it is believed that the As(V) adsorption onto HFO‐PF‐coated GAC in this study followed the pseudo‐second order kinetics model. The values for qe and ri are calculated from the slope and intercept, respectively, of Equation (Equationix).
and ri = 0.0164 mg g−1min
Thus, k2 = ri / qe 2 = 0.468 g mg−1min
Figure 5 The plot for the t/qt vs. t version of the pseudo‐second order Equation (Equationviii) at initial As(V) concentration of 1.74 mgl−1 and sorbent dose of 10.1 gl−1.
Column Studies
The column tests were performed to determine the As(V) removal efficiency of HFO‐PF‐coated GAC with a bulk density of 0.48 g ml−1. The results (Table ) showed that this sorbent was able to achieve residual concentrations of As(V) in a range of 0.1–2.0 µg l−1 while continuously treating about 180 BV (130 ml‐BV) of arsenate water with an initial As(V) concentration of 1886 µg l−1. Furthermore, As(V) removal was close to 100% during a contact time of 29 hours at a flow rate of 13.5 ml min−1. That is, the EBCT was set at 9.6 minutes, and the GSCT was 0.15 minutes. The EBCT is not directly related to the kinetic adsorption properties of the sorbent, as EBCT varies depending on the column size and flow rate. The required GSCT for As(V) removal is closely linked with its kinetic adsorption properties. A 0.15‐min. of GSCT is practically acceptable. Based on the GSCT value, the required column volume, EBCT and flow rate can be calculated, and then the acceptability of the GSCT can be determined. For example, it was reported that a small‐scale column, packed with 1.5 g sorbent, was tested at an EBCT of 3.3 minutes [Citation28]. Although it demonstrated positive results, its GSCT of 2.2 minutes is not acceptable for the design of column size and flow rate.
Table 3. The column test data for As(V) adsorption onto HFO‐PF‐coated GAC.
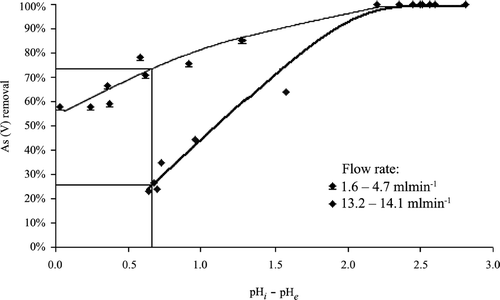
It was found that a slow flow rate, at around the breakthrough point of 225 BV, could improve the efficiency of arsenic adsorption, owing to the fact that the film diffusion became a control factor for the adsorption rate as As(V) migrated from solution to sorbent [Citation41]. When BV passed 635, the amount of adsorbed As(V) on the exhausted sorbent was about 1.52 mg g−1, which was far below the observed maximum adsorption capacity Qm (2.33 mg g−1, Table ) suggesting the exhausted sorbent retained some As‐adsorption capability. Consistently, the leachable As and Fe were < 0.1 mg l−1 and < 0.5 mg l−1, respectively, when the exhausted sorbent was extracted for 18 hours at a final pH of 5.5 according to the TCLP test. Thus, the exhausted sorbent was classified as non‐hazardous for disposal with no stabilization of adsorbed As(V) required.
It is noticeable that the pH value of treated effluent tended to decrease during the filtration at low (1.6–4.7 ml min−1) or high (13.2–14.1 ml min−1) flow rates. It was found that this pH decrease was closely related to the efficiency of As(V) adsorption. The greater As(V) adsorption corresponded to a greater pH drop (Figure ). This observation could be associated with proton dissociation from the slightly positively charged surface of sorbent and also from the formed Fe‐As(V) complexes (I) and (II), Equations (Equationxi) and (Equationxii) [Citation30]:
Figure 6 The plot for the As(V) removal vs. ΛpH at different flow rates.
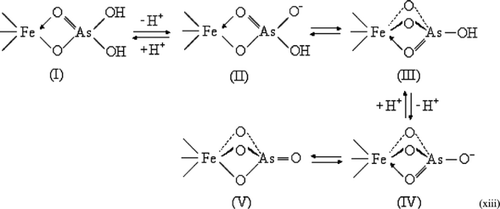
Below the isoelectric point (pHiep = 7.9–8.2) of HFO [Citation30], the sorbent surface is slightly positively charged, Equation (Equationx). In natural waters, As(V) occurs as H3AsO4 with three pK a values of 2.19, 6.94 and 11.5 [Citation33]. In the typical pH range of 6–7, the dissolved As(V) ions are predominantly present as H2AsO4 − and HAsO4 2−. While As(V) is adsorbed onto the surface of HFO‐PF‐coated GAC, Fe‐As(V) complexes are formed as illustrated by the proposed structures (I) – (V) in Equation (xiii). The unfilled d‐orbital of Fe(III) favors the distribution of electron density in the hydroxide group through an electron‐withdrawing inductive effect, resonance structures, and O atoms as exchangeable ligands which bind to Fe(III). Accordingly, the hydroxide protons in the complexes (I) and (II) become more dissociable leading to a lower pH value in comparison with H2AsO4 − and HAsO4 2−.
CONCLUSIONS
This paper presents a simple and effective method for immobilizing HFO onto GAC with PF resins. The resulting sorbent was used for As(V) adsorption studies. The kinetics of As(V) adsorption by this sorbent was rapid with 85% of the total adsorption occurring within the first hour while treating water with an initial As(V) concentration of 1.74 mg l−1. The As(V) adsorption onto the sorbent was found to follow a pseudo‐second order kinetics model. The As(V) adsorption equilibrium data fit the Langmuir and Freundlich adsorption models very well. With reference to the Fruendlich model, it is believed that the surface of HFO‐PF‐coated GAC was distributed with heterogeneous adsorption sites, such as various sizes of HFO micro‐particles. Each of these sites could have a different adsorption potential where Langmuir adsorption would occur. The column results showed that this sorbent was able to reduce the As(V) concentration from 1886 µg l−1 to 0.1–2.0 µg l−1 while treating about 180 BV of arsenate water at a filtration rate of 13.5 ml min−1. The exhausted sorbent was classified as non‐hazardous for disposal according to the TCLP test. Accordingly, this HFO‐PF‐coated GAC can be considered to be a viable sorbent for As(V) removal from industrial wastewater.
REFERENCES
- Mandal , B.K. and Suzuki , K.T. 2002 . Arsenic round the world: a review . Talanta , 58 : 201 – 235 .
- Matschullat , J. 2000 . Arsenic in the geosphere — a review . Sci. Total Environ. , 249 : 297 – 312 .
- Bissen , M. and Frimmel , F.H. 2003 . Arsenic — a review. Part I: Occurrence, toxicity, speciation, mobility . Acta Hydrochim. Hydrobiol. , 31 : 9 – 18 .
- Smedley , P.L. and Kinniburgh , D.G. 2002 . A review of the source, behavior and distribution of arsenic in natural waters . Appl. Geochem. , 17 : 517 – 569 .
- Lebow , S. , Williams , R.S. and Lebow , P. 2003 . Effect of simulated rainfall and weathering on release of preservative elements from CCA treated wood . Environ. Sci. Technol. , 37 : 4077 – 4082 .
- Karim , M. M. 2000 . Arsenic in groundwater and health problems in Bangladesh . Water Res. , 34 : 304 – 310 .
- Tisler , T. and Zagorc–Koncan , J. 2002 . Acute and chronic toxicity of As to some aquatic organisms . Bull. Environ. Contamin. Toxicol. , 69 : 421 – 429 .
- US EPA Office of Water . Fact Sheet: EPA to Implement 10 ppb Standard for Arsenic in Drinking Water http://www.epa.gov/safewater/ars/implement.html (Accessed January 2006)
- Shih , M. 2005 . An overview of arsenic removal by pressure‐driven membrane processes . Desalination , 172 : 85 – 97 .
- Thirunavukkarasu , O.S. , Subramanian , K.S. , Chaalal , O. and Islam , M. R. 2005 . Arsenic removal in drinking water. Impacts and novel removal technologies . Energy Sources , 27 : 209 – 219 .
- Wickramasinghe , S.R. , Han , B. , Zimbron , J. , Shen , Z. and Karim , M.N. 2004 . Arsenic removal by coagulation and filtration: comparison of groundwaters from the United States and Bangladesh . Desalination , 169 : 231 – 244 .
- Wang , L. , Chen , A. and Fields , K. 2000 . “ Arsenic removal from drinking water by ion exchange and activated alumina plants ” . Washington, D.C. : US Environmental Protection Agency . Report EPA‐600/R00‐/088
- Lin , T.‐F. and Wu , J.‐K. 2001 . Adsorption of arsenite and arsenate within activated alumina grains: equilibrium and kinetics . Water Res. , 35 : 2049 – 2057 .
- Pattanayak , J. , Mondal , K. , Mathew , S. and Lalvani , S.B. 2000 . A parametric evaluation of the removal of As(V) and As(III) by carbon‐based adsorbents . Carbon , 38 : 589 – 596 .
- Loukidou , M.X. , Matis , K.A. , Zouboulis , A.I. and Liakopoulou‐Kyriakidou , M. 2003 . Removal of As(V) from wastewaters by chemically modified fungal biomass . Water Res. , 37 : 4544 – 4552 .
- Ghimire , K.N. , Inoue , K. , Yamaguchi , H. , Makino , K. and Miyajima , T. 2003 . Adsorptive separation of arsenate and arsenite anions from aqueous medium by using orange waste . Water Res. , 37 : 4945 – 4953 .
- Korngold , E. , Belayev , N. and Aronov , L. 2002 . Removal of arsenic from drinking water by anion exchangers . Desalination , 141 : 81 – 84 .
- Zhu , X. and Jyo , A. 2001 . Removal of arsenic(V) by zirconium(IV)‐loaded phosphoric acid chelating resin . Sep. Sci. Technol. , 36 : 3175 – 3189 .
- Thirunavukkarasu , O.S. , Viraraghavan , T. and Subramanian , K. S. 2003 . Arsenic removal from drinking water using granular ferric hydroxide . Water SA , 29 : 161 – 170 .
- Kim , J. and Benjamin , M.M. 2004 . Modeling a novel ion exchange process for arsenic and nitrate removal . Water Res. , 38 : 2053 – 2062 .
- Bissen , M. and Frimmel , F.H. 2003 . Arsenic‐ a review. Part II: Oxidation of arsenic and its removal in water treatment . Acta Hydrochim. Hydrobiol. , 31 : 7 – 107 .
- Gu , Z. and Deng , B. 2007 . Use of iron‐containing mesoporous carbon (IMC) for arsenic removal from drink water . Environ. Eng. Sci. , 24 : 113 – 121 .
- Gupta , V.K. , Saini , V.K. and Jain , N. 2005 . Adsorption of As(III) from aqueous solutions by iron oxide‐coated sand . J. Colloid Interface Sci. , 288 : 55 – 60 .
- Chen , W. , Parette , R. , Zou , J. and Cannon , F. S. 2007 . Arsenic removal by iron‐modified activated carbon . Water Res. , 41 : 1851 – 1858 .
- Min , J.H. and Hering , J. G. 1998 . Arsenate sorption by Fe(III)‐doped alginate gels . Water Res. , 32 : 1544 – 1552 .
- Lenoble , V. , Laclautre , C. , Deluchat , V. , Serpaud , B. and Bollinger , J.‐C. 2004 . Arsenic removal by adsorption on iron(III) phosphate . J. Hazard. Mater. , B113 : 157 – 164 .
- Zhang , F.‐S. and Itoh , H. 2005 . Iron oxide‐loaded slag for arsenic removal from aqueous system . Chemosphere , 60 : 319 – 325 .
- Jang , M. , Min , S.H. , Kim , T.‐H. and Park , J.K. 2006 . Removal of arsenite and arsenate using hydrous ferric oxide incorporated into naturally occurring porous diatomite . Environ. Sci. Technol. , 40 : 1636 – 1643 .
- Drever , J.I. 1997 . The Geochemistry of Natural Waters , 3rd ed. , Upper Saddle River, NJ, , USA : Prentice‐Hall Inc. .
- Dzombak , D.A. and Morel , F.M.M. 1990 . “ Surface complexation modeling — hydrous ferric oxide ” . 89 – 191 . New York, , USA : John Wiley & Sons .
- Dixit , S. and Hering , J.G. 2003 . Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility . Environ. Sci. Technol. , 37 : 4182 – 4189 .
- Gu , Z. , Fang , J. and Deng , B. 2005 . Preparation and evaluation of GAC‐based iron‐containing adsorbents for arsenic removal . Environ. Sci. Technol. , 39 : 3833 – 3843 .
- Dambies , L. 2004 . Existing and prospective sorption technologies for the removal of arsenic in water . Sep. Sci. Technol. , 39 : 603 – 627 .
- Roberts , L.C. , Hug , S.J. , Ruettimann , T. , Khan , A.W. and Rahman , M.T. 2004 . Arsenic removal with iron(II) and iron(III) in waters with high silicate and phosphate concentrations . Environ. Sci. Technol. , 38 : 307 – 315 .
- Zeng , L. 2003 . A method for preparing silica‐containing iron(III) oxide adsorbents for arsenic removal . Water Res. , 37 : 4351 – 4358 .
- Toxicity Characteristic Leacing Procedure . 1992 . “ US EPA Method 1311 ” . Washington, D.C. www.epa.gov/epaswer/hazwaste/test/pdfs/1311.pdf
- 2003 . “ Calgon Carbon (product data, Type PCB Granular Carbon) ” . Pittsburgh, PA, , USA : Calgon Carbon Corporation .
- Liu , Y. 2006 . Some consideration on the Langmuir isotherm equation . Colloid. Surf. A , 274 : 34 – 36 .
- Kundu , S. and Gupta , A. K. 2006 . Adsorptive removal of As(III) from aqueous solution using iron oxide coated cement (IOCC): Evaluation of kinetic, equilibrium and thermodynamic models . Separa. and Purif. Technol. , 51 : 165 – 172 .
- Ho , Y. S. and Mckay , G. 1999 . Pseudo‐second order model for sorption process . Process Biochem. , 34 : 451 – 465 .
- Badruzzaman , M. , Westerhoff , P. and Knappe , D.R.U. 2004 . Intraparticle diffusion and adsorption of arsenate onto granular ferric hydroxide (GFH) . Water Res. , 38 : 4002 – 4012 .