
Abstract
We investigated the regulation of hepatic ER stress in healthy liver and adult or perinatally programmed diet-induced non-alcoholic fatty liver disease (NAFLD). Female mice were fed either obesogenic or control diet before mating, during pregnancy and lactation. Post-weaning, offspring from each maternal group were divided into either obesogenic or control diet. At six months, offspring were sacrificed at 4-h intervals over 24 h. Offspring fed obesogenic diets developed NAFLD phenotype, and the combination of maternal and offspring obesogenic diets exacerbated this phenotype. UPR signalling pathways (IREα, PERK, ATF6) and their downstream regulators showed different basal rhythmicity, which was modified in offspring exposed to obesogenic diet and maternal programming. The double obesogenic hit increased liver apoptosis measured by TUNEL staining, active caspase-3 and phospho-JNK and GRP78 promoter methylation levels. This study demonstrates that hepatic UPR is rhythmically activated. The combination of maternal obesity (MO) and obesogenic diets in offspring triggered altered UPR rhythmicity, DNA methylation and cellular apoptosis.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome, which ranges from bland steatosis to steatohepatitis (NASH), cirrhosis and potentially hepatocellular carcinoma (HCC) (Farrell & Larter Citation2006). It is now the leading cause of chronic liver disease in the Western world with a prevalence rising in parallel to obesity. If current trends continue, the prevalence of NAFLD in the USA is projected to increase by 50% in 2030 (Younossi et al. Citation2011). Among children, the prevalence of NAFLD has more than doubled over the last two decades, now affecting approximately 11% of adolescents (Welsh et al. Citation2013). These dramatically rising rates could partially be explained by obesogenic dietary patterns, and there is emerging evidence that maternal obesity (MO) also predisposes offspring to NAFLD in animal models (Oben et al. Citation2010; Mouralidarane et al. Citation2013; Mouralidarane et al. Citation2015). However, the precise molecular mechanisms of programming in NAFLD pathogenesis remain unknown.
Endoplasmic reticulum (ER) is an organelle dedicated to the synthesis and folding of proteins. An ER stress situation is usually established when there is an accumulation of unfolded and misfolded proteins in the ER due to physiological and pathological insults, such as high-protein demand, viral infections, environmental toxins, inflammatory cytokines and mutant protein expression. These consequently trigger the unfolded protein response (UPR) as an adaptation mechanism (Ron & Walter Citation2007). There are three principal UPR sensors in mammals: inositol-requiring 1 alpha (IRE1α), protein kinase RNA (PKR)-like ER kinase (PERK) and activating transcription factor 6 (ATF6). Each sensor is bound by glucose-regulated protein 78 (GRP78), a chaperone protein and a master regulator of ER homeostasis, which inactivate those sensors and block the UPR. Excess misfolded proteins in ER force the dissociation of GRP78, thus activating the ER sensors to induce downstream UPR signalling. Consequently, the downstream UPR signalling increases the expression of ER chaperones to enhance protein-folding capacity, inhibit mRNA translation to reduce protein overload, and activate the degradation of the misfolded proteins. However, once the adaptation threshold is reached, UPR signalling induces apoptosis to eliminate the cell (Ron & Walter Citation2007).
Increasing evidence suggests that ER stress plays an important role in the pathogenesis of metabolic diseases including NAFLD (Dara et al. Citation2011; Hetz et al. Citation2013). Genetic ablation of ER stress-sensing pathways (PERK, IRE1α or ATF6) resulted in hepatic dyslipidaemia and NAFLD in response to chemical induction of ER stress (Basseri & Austin Citation2008; Fu et al. Citation2012). Furthermore, GRP78 overexpression modulated ER stress and reduced NAFLD in a murine model of obesity (Kammoun et al. Citation2009). UPR activation has also been reported in various animal and human models of NAFLD (Cao et al. Citation2012; Fu et al. Citation2012). It remains unclear, however, whether UPR activation plays a causative or adaptive role in the progression of NAFLD.
Accumulating evidence suggests that in addition to acute ER stress, UPR activation may also play a crucial role in normal physiological conditions to maintain proper cellular function, differentiation, development and survival (Ron & Walter Citation2007). Hepatic cells are exposed to nutrient fluctuation, and protein secretory pathways, lipogenesis and gluconeogenesis are activated in response to these fluctuations. It is plausible that ER regularly adjusts their function according to different stimuli. A recent study indicated that IRE1α is rhythmically activated in the liver, and it is synchronised by the circadian clock under normal physiological conditions (Cretenet et al. Citation2010). The absence of the circadian clock in mice perturbed this rhythmicity, leading to impaired lipid metabolism in the liver (Cretenet et al. Citation2010). In yeast, IRE1α is the only pathway responsible for UPR response; whereas in higher animals, UPR pathway also includes two other core pathways (PERK and ATF6) to cope with multiple signals and cell-specific reactions in a wide variety of cell types. Although some stressors activate all three pathways, it has been suggested that each pathway acts more selectively and in a manner dependent upon the cellular context physiological conditions (Wu et al. Citation2014). However, physiological UPR fluctuation in the liver and its relationship with liver pathophysiology are largely unknown. More comprehensive analyses of UPR pathways, including detailed time-course studies in intact organisms, and experiments under different physiological conditions are, therefore, needed.
We recently reported that developmental programming models of NAFLD are an effective physiological animal model of human NAFLD (Oben et al. Citation2010; Mouralidarane et al. Citation2013). In these models, offspring of lean mothers fed an obesogenic diet developed mild NAFLD in adulthood, whereas those exposed to both MO and obesogenic diets developed more severe NASH. This model is a unique tool to investigate NAFLD progression, as well as understanding the developmental programming of NAFLD. Based on this model, we analysed the ER rhythmicity by measuring UPR fluctuation on a daily time course in the context of NAFLD, and the influence that maternal programming has on NAFLD pathogenesis and progression.
Materials and methods
Animal model
Female C57BL/6J mice (n = 20; Charles River Laboratories, Margate, UK), of 100 d old were randomly divided to be fed with a standard chow diet (RM1, Special Dietary Services, Essex, UK) or an obesogenic diet (824053, Special Dietary Services), supplemented with fortified sweetened condensed milk (Nestle, Vevey, Switzerland) for 6 weeks ad libitum (Oben et al. Citation2010; Mouralidarane et al. Citation2013) (dietary composition in ). Obesogenic fed mice (30% of increase in body weight) were mated with C57BL/6J males from the same litter. Conception was determined by vaginal plug formation. The female animals were maintained on their allocated diets throughout gestation and lactation. After birth, litters were standardised to six pups each with equal number of males and females when possible. At day 21 of life, offspring from both sexes were weaned onto either the standard chow or the obesogenic diet. Four experimental groups were generated: offspring of lean weaned onto standard chow diet (OffCon-SC), offspring of lean weaned onto obesogenic diet (OffCon-OD), offspring of obese weaned onto standard chow diet (OffOb-SC) and offspring of obese weaned onto obesogenic diet (OffOb-OD). At 6 months post-natal, offspring were euthanised by CO2 inhalation at 4 h intervals over a 24-h period (Zeitgeber Time (ZT) 0, 4, 8, 12, 16 and 20, where ZT0 is light and ZT12 is dark). Following the sacrifice, blood and liver samples were obtained and appropriately stored until analysis. All studies were approved by Local University College London Ethics Committee, and conducted under UK Home Office and Animal in Science Scientific Procedures Act 1986 guidelines. Animals were maintained on a 12 h light/12 h dark cycle and at 22 °C with food and water ad libitum during all the procedures.
Table 1. Composition of diets.
Liver phenotype analysis
Offspring livers at 6 months were formalin fixed and paraffin embedded before sectioning, and stained with haematoxylin and eosin (H&E) and Masson’s Trichrome to asses NAFLD activity score by an expert liver pathologist blinded to the group identities.
Alanine aminotransferase
Plasma ALT concentration was assayed by the Royal Free Hospital Clinical Biochemistry Department (London, UK).
Hepatic triglyceride content
Hepatic triglyceride content was determined by an adaptation of the Folch Method (Folch et al. Citation1957) for extraction followed by an enzymatic colorimetric assay for quantification (UNIMATE 5 TRIG, Roche Diagnostics, Sussex, UK).
Western blotting
Proteins were extracted and quantified as previously described (Soeda et al. Citation2012). We pooled together (n = 3–5) and analysed equal amounts for each group/time point. Proteins were loaded onto a polyacrylamide gel for electrophoresis and transferred to a polyvinylidene membrane by electroblotting. Membranes were blocked with 5% non-fat dried milk in TBS-T and incubated with the primary antibodies (). After washing in TBS-T, the membranes were incubated with the secondary antibody and developed by chemiluminescence (ECL, Amersham plc, Amersham, UK).
Table 2. Antibodies for western blotting.
Real time qPCR
Total RNA (n = 3–4 animals per experimental group/time point) was isolated from frozen hepatic tissue using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA was DNase treated and retrotranscripted with the Qiagen QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD). Quantitative real-time PCR was performed by triplicate using ABI PRISM 7500 HT Fast real-time PCR system (Applied Biosystems, Austin, TX) and SYBR Green PCR kit (Qiagen) and target genes normalised with GAPDH as an internal control. Fold change between groups was calculated using 2−ΔΔCt method. Gene-specific primer sequences are listed in .
Table 3. Rt-qPCR primers sequences.
TUNEL assay
TUNEL assay was carried out by using the TUNEL Apoptosis detection kit (Millipore, Temecula, CA). Paraffin embedded samples were cut, deparaffinised and incubated with proteinase K. The slides were incubated with TdT buffer and subsequently with a mix including TdT buffer, biotin-dUTP and TdT. After washing with TB and PBS, samples were incubated with a blocking solution and later with an Avidin-FITC solution. Finally, slides were counterstained and assessed by fluorescent microscopy.
Immunohistochemistry
Paraffin embedded tissue was cut, deparaffinised and treated with proteinase K and subsequently with a primary anti-rabbit Cleaved caspase-3 antibody (96645, Cell Signaling Technology, Danvers, MA). Then, samples were incubated with a diluted biotinylated secondary antibody (Vector Laboratories, Burlingame, CA). The slides were counterstained with haematoxylin and the images were captured using Nikon Eclipse e600 microscope and quantified by NIS-Elements Advance software (Nikon Corporation, Tokyo, Japan).
DNA methylation measurement
DNA from liver samples was isolated using the DNeasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany). gDNA methylation percentage was measured by the EpiTect Methyl II PCR primer assay for mouse Grp78 (CpG island 106178). After incubation with restriction enzymes, rt-qPCR was performed by using ABI PRISM 7500 HT Fast real-time PCR system (Applied Biosystems) and SYBR Green PCR kit (Qiagen). These results were analysed using Qiagen excel templates for the assay.
Statistical analyses
Data are expressed as mean ± standard error of the mean (SEM). The statistical unit used throughout the analysis is the number of dams. Means of each group were compared using a one-way ANOVA statistical test followed by Tukey’s post-hoc test when necessary (GraphPad Prism 5.0; GraphPad Software Inc., Cary, NC). Statistical significance was accepted as p < .05.
Results
Offspring exposed to maternal obesity and post-natal obesogenic diets developed profound NAFLD phenotype
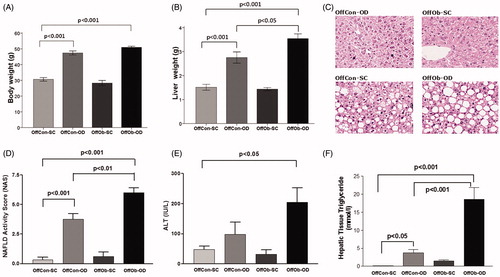
Consistent with our previous reports, offspring exposed to MO and an obesogenic diet (OffOb-OD) post-weaning developed a profound obesity-associated NAFLD phenotype, characterised by higher liver weight (), NAS score () and hepatic triglyceride content (), in comparison to offspring exposed to post-partum obesogenic diet only (OffCon-OD). Additionally, in comparison to the control group (OffCon-SC), there was also a significant increase in body weight () and plasma ALT levels (). Furthermore, the obesogenic potential of the diet was widely demonstrated when compare OffCon-SC and OffCon-OD groups, as OD diet intake after weaning induced increased body weight (), liver weight (), NAS score () and liver triglyceride content (). These results would indicate that MO programmes features of NAFLD in offspring, which is further exacerbated following exposure to an obesogenic diet post-weaning.
Figure 1. Phenotypical, histological and biochemical evidences of NAFLD phenotype in offspring at 6 months: (A) body weight, (B) liver weight, (C) H&E stains of representative liver sections, (D) NAFLD activity score, (E) plasma ALT concentrations and (F) hepatic triglyceride content. n = 5 per experimental group; values shown as mean ± SEM; OffCon-SC: offspring of lean weaned onto standard chow diet; OffCon-OD: offspring of lean weaned onto obesogenic diet; OffOb-SC: offspring of obese weaned onto standard chow diet; OffOb-OD: offspring of obese weaned onto obesogenic diet; one way ANOVA with Tukey’s post-hoc test for the main comparisons (OffCon-SC versus OffCon-OD; OffCon-SC versus OffOb-OD; OffCon-OD versus OffOb-OD).
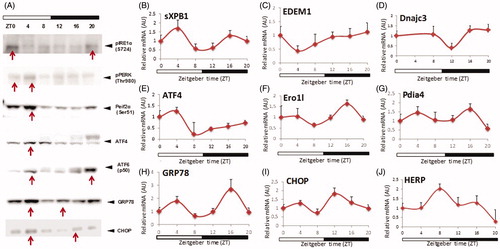
UPR pathways are rhythmically activated in normal physiological conditions
Initially, we investigated the activation patterns of the three major UPR pathways (IRE1α, PERK and ATF6) during a 24-h-time period under normal physiological conditions (OffCon-SC). Mice were maintained on strict 12:12 h light/dark cycles and sacrificed every 4 h over 24 h. IRE1α activation, as detected by their phosphorylation status, showed increased protein concentration at the end of the dark period and reached a maximum at the beginning of the light period (). IRE1α activation initiates the splicing of the transcriptional factor XBP1 to spliced XBP1 (sXBP1), an active transcriptional factor which induces ER metabolic components. sXBP1 mRNA expression showed weak biphasic peaks at ZT4 and ZT16 (). EDEM1 and DNajc3, sXBP1 specific downstream targets genes, reached their maximum mRNA expressions at the end of the dark period (). There was a long time delay between sXBP1 expression and downstream targets, consistent with the slow induction of EDEM1, which has been reported previously (Hampton Citation2003). Upon activation, PERK is autophosphorylated and promotes Eif2α phosphorylation (Ron & Walter Citation2007). pEif2α generally inhibits mRNA translation, but specifically induces ATF4 transcriptional factor expression (Ron & Walter Citation2007). Phospho-PERK and phospho-Eif2α showed peak levels at ZT4 (). ATF4 mRNA and protein expression also reached maximum at ZT4 (). Ero1l expression, which is thought to be relatively ATF4-dependent, showed a 24 h rhythm peak at ZT16 (). ATF6 activation, detected by active cleavage protein form (p50 ATF6), showed a clear bimodal pattern peaking at ZT4 and ZT20 (). Pdia4 mRNA expression, which is predominantly regulated by ATF6, also showed a similar bimodal pattern (). Other important components of UPR, GRP78 (predominantly ATF6 dependent) and CHOP (predominantly PERK-Eif2α-ATF4 dependent) also showed bimodal mRNA expression patterns () and protein expression (). HERP, which is induced by IRE1α-XBP1 and ATF6 showed a 24 h rhythmicity pattern (). From the results, all three UPR pathways and their downstream targets showed either 12 h or 24 h activation pattern in the liver under normal physiological conditions. The differences in the rhythmicity and time of peak activation among the three UPR sensors indicate that each arm of the sensors could be driven by different environmental cues. Furthermore, the different times of downstream gene upregulation suggests that the three UPR sensors are regulated by different output timeframes.
Figure 2. Protein and mRNA concentration of UPR pathways in normal physiological condition (OffCon-SC experimental group; offspring of lean weaned onto standard chow diet) during 24 h. n = 3–4 per experimental time.
Offspring exposure to maternal obesity and post-natal obesogenic diet-disrupted UPR homeostasis
We next investigated how MO and obesogenic diets post-weaning can influence the rhythmic activation of UPR. Our preliminary studies showed very similar UPR activation pattern between OffCon-SC and OffOb-SC (). Given this, the OffOb-SC cohort was excluded from further studies.
Western blotting of phosopho-IRE1α showed different activation patterns among the three experimental groups (). In OffCon-OD, IRE1α activation occurred more broadly at the beginning of dark and beginning of light compared to OffCon-SC, showing an activation peak at ZT12, whereas OffOb-OD showed a stronger constant activation of IRE1α. sXBP1 mRNA expression presented a single peak at ZT4 in OffCon-OD and it was constantly downregulated in OffOb-OD (). DNajc3, a target gene of sXBP1 showed slight early peak at ZT16 in OffCon-SC but constantly lower expression in OffOb-OD parallel to sXBP1 mRNA levels (). PERK was strongly activated at ZT4-ZT12 in OffCon-OD, although it was more constantly activated in OffOb-OD (). As a result, phosopho-Eif2α was constantly overexpressed in OffOb-OD (). Conversely, ATF4 protein concentration and mRNA expression was constantly suppressed in this group (). In contrast to other two pathways, ATF6 activation showed the same bimodal pattern of activation among all three groups whereas it had much broader strong activation in OffCon-OD and OffOb-OD (). Pdia4 also presented a similar bimodal expression pattern among the three groups, but their magnitudes were not statistically different between them (). GRP78 and HERP expression, which is thought to be a sensitive marker of UPR activation, showed OffCon-OD single peak at ZT0, whereas this was constantly downregulated in OffOb-OD (). GRP78 protein expression was also decreased in OffOb-OD compared to OffCon-SC (). The pro-apoptotic transcription factor C/EBP homologous protein (CHOP) was consistently expressed in OffOb-OD at all time-points. However, in OffCon-OD, peak CHOP expression time was shifted from the end of the dark period to the beginning of light period (). CHOP protein expression levels were significantly high at ZT0 in OffOb-OD compared to OffCon-SC ().
Figure 3. Protein and mRNA hepatic concentrations of UPR pathways depending on maternal feeding and offspring post-weaning diet during 24 h. n = 3–4 per experimental group/time point; values shown as mean ± SEM; OffCon-SC: offspring of lean weaned onto standard chow diet; OffCon-OD: offspring of lean weaned onto obesogenic diet; OffOb-OD: offspring of obese weaned onto obesogenic diet; *statistical differences (p < .05) between OffCon-SC and OffCon-Ob. #Statistical differences (p < .05) between OffCon-SC and OffOb-Ob.
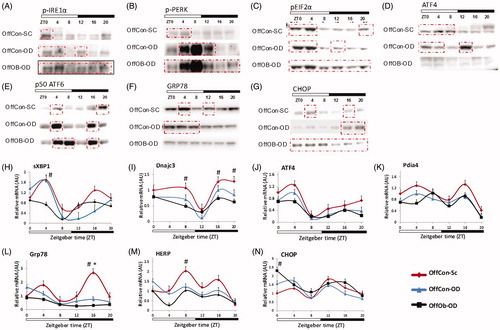
These results indicate that post-weaning, an obesogenic diet changes the timing of IRE1α, PERK and ATF6 pathway activation, and the combination of MO and post-weaning obesogenic diet induces constant activation of these UPR sensors. Furthermore, downstream signalling of these pathways was unresponsive to proximal UPR sensor activation mainly in OffOb-OD, whereas in OffCon-OD it could still be responsive.
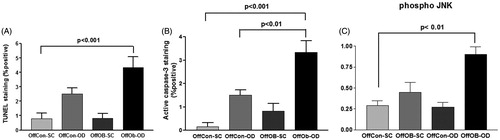
Offspring exposure to maternal obesity and post-natal obesogenic diet-induced increased apoptosis and hypermethylation of GRP78
The previous results suggested an unresolved UPR-induced apoptosis in the mice liver. The TUNEL assay was used to detect cell nuclei in apoptotic cells. As predicted, OffOb-OD showed significantly higher degrees of apoptosis according to the TUNEL index score as well as to the caspase-3 probe (). The apoptotic effector protein caspase-3 was also activated in OffOb-OD as an effect of obesogenic feeding. Finally, the addition of both obesogenic stimuli (maternal and offspring) significantly increased the activation of the proapoptotic protein JNK ().
Figure 4. Inflammation, apoptosis and autophagy assessment by (A) TUNEL staining assay, (B) active caspase-3 staining and (C) phospho-JNK protein concentration measurement. n = 5 per experimental group; values shown as mean ± SEM; OffCon-SC: offspring of lean weaned onto standard chow diet; OffCon-OD: offspring of lean weaned onto obesogenic diet; OffOb-SC: offspring of obese weaned onto standard chow diet; OffOb-OD: offspring of obese weaned onto obesogenic diet; one way ANOVA with Tukey’s post-hoc test for the main comparisons (OffCon-SC versus OffCon-OD; OffCon-SC versus OffOb-OD; OffCon-OD versus OffOb-OD).
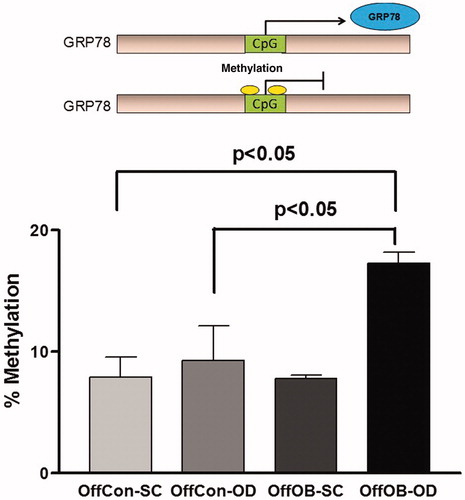
GRP78 is considered as a master regulator of UPR, and ATF6 is primarily responsible for the transcriptional induction of this chaperone protein. The constantly low GRP78 expression despite the elevated ATF6 activation in OffOb-OD prompted the investigation of the possible mechanism of downregulation of this gene. As other previous perinatal studies have suggested (Martinez et al. Citation2012), epigenetic changes play a significant role in pathophysiological phenotypes derived from developmental programming (Cordero et al. Citation2015). Thus, we hypothesised that alterations in DNA methylation might play a role in regulating the expression of this gene. Methyl specific quantitative PCR assay revealed that GRP78 was significantly hypermethylated in OffOb-OD compared with OffCon-SC and OffCon-OD (), which suggests that these changes require additional stimulation by maternal programming and offspring obesogenic feeding. However, as downregulation of GRP78 expression was partly opposed to gene methylation patterns, and higher methylation is usually associated with lower expression, GRP78 mRNA levels could partially be explained by epigenetic alterations.
Figure 5. Specific DNA methylation measurement in GRP78 promoter region. n = 5 per experimental group; values shown as mean ± SEM; OffCon-SC: offspring of lean weaned onto standard chow diet; OffCon-OD: offspring of lean weaned onto obesogenic diet; OffOb-SC: offspring of obese weaned onto standard chow diet; OffOb-OD: offspring of obese weaned onto obesogenic diet; one way ANOVA with Tukey’s post-hoc test for the main comparisons (OffCon-SC versus OffCon-OD; OffCon-SC versus OffOb-OD; OffCon-OD versus OffOb-OD).
Discussion
ER stress is increasingly thought to play an important role in the pathogenesis and progression of NAFLD. As such, it is now considered a potential therapeutic target (Dara et al. Citation2011; Fu et al. Citation2012; Hetz et al. Citation2013), however, further understanding of the complex physiology of the UPR pathways, and their relation to NAFLD is required. We have shown here that even in healthy conditions, three major UPR pathways are dynamically activated in the liver. Obesogenic feeding appears to change their rhythmicity, and the combination of MO and post-weaning obesogenic diets can significantly alter UPR rhythmicity and homeostasis in parallel with the development of a NAFLD phenotype.
Under normal physiological conditions, IRE1α and PERK activation reached maximum peaks at the end of dark periods up to the beginning of light periods. ATF6 showed a clear peak in the middle of the light period, and at the end of the dark period. The expression levels of the downstream targets of these pathways followed a similar rhythmicity, although induction of each gene seems to have a different time lag from the time of proximal sensor activation. These results further expand on previous findings which showed a 12 h rhythmic activation of the IRE1α pathway (Cretenet et al. Citation2010). The discrepancy between our results showing a 24 h cycle of IRE1α activation and these previous results might be related to the age of mice (24 versus 8 weeks), as there is an aged-linked decrease in the expression and activity of key ER-related mediators (Brown & Naidoo Citation2012). An additional possibility for this discrepancy could rely on the sampling interval, longer in this study (4 h versus 2 h). The findings of a bimodal pattern of ATF6 downstream mRNA expression being dependent upon the weight and feeding of offspring is consistent with previous studies (Vollmers et al. Citation2009). Multiple, critical functions of the liver are precisely timed processes (Liu et al. Citation2013; Oishi & Itoh Citation2013).
In addition to hepatic functions being timed, UPR rhythmicity appears to be broadly modified by exposure to dietary modifications. Post-weaning obesogenic diets resulted in an alteration of the time of activation for both IRE1α and PERK, while the combination of MO and post-weaning obesogenic diets induced constant activation of these two sensors. IRE1α and PERK share considerable sequence homology in their luminal domains, while ATF6 does not, suggesting that substrate specific activation exists for each sensor (Wu et al. Citation2014). ATF6 has been shown to be preferentially activated by tissue ischaemia (Doroudgar et al. Citation2009), while other UPR pathways may be associated more with protein overloading, and abnormalities in liver calcium and lipid metabolism (Fu et al. Citation2011). Furthermore, both classical ER stress due to misfolded protein and other environmental cues, such as feeding (lipid and glucose fluctuations) can trigger UPR (Kanda et al. Citation2015). Bimodal ATF6 target gene expression in the liver, which reflects bimodal feeding patterns (i.e. consolidate feeding event in the middle of the night and several events during the day), seems to be closely coupled with a daily rhythm in feeding rather than the circadian clock (Pendergast et al. Citation2013). Our control group data supports this feeding-induced ATF6 activation. It was found that a high-fat-diet-induced changes in feeding behaviour, in which the main feeding was during the night, with extra calories consumed by the mice on a high-fat-diet mostly during the daytime (Kohsaka et al. Citation2007). Thus, obesogenic diets enhanced the ATF6 activation during the day. Contrary to ATF6, IRE1α activation appeared to be governed instead by clock regulated lipid metabolism. Kitai et al. recently demonstrated that IRE1α and PERK can sense lipid saturation by their transluminal domains without misfolded protein, while ATF6 was unaffected (Kitai et al. Citation2013). ATF6 was also reported to be activated by the expression of physiological levels of ER membrane bound protein, possibly by sensing their transluminal domains (Maiuolo et al. Citation2011). These mechanisms might explain anticipatory UPR, where environmental stimuli or developmental programming induce UPR activation, rather than misfolded proteins. Indeed, our results showed that IRE1α and PERK were more constantly activated by an obesogenic diet, possibly secondary to increased saturated fatty acid content (Wang et al. Citation2006), supporting specific membrane lipid-saturation sensing mechanisms with IRE1α and PERK. More sensitive assays, shorter interval sampling and metabolomics or proteomic approaches might further illustrate the precise rhythmicity and link between liver metabolic rhythm and UPR activation.
The continuous activation of three UPR sensors was a hallmark of UPR status in OffOb-OD, while downstream pathways were not upregulated. In OffCon-OD, although there was the time shift and difference of magnitude of proximal sensor activation, downstream signalling was partially maintained. Thus, dysregulation of UPR homeostasis might be a key factor in the progression of an obesity-associated NAFLD phenotype. This UPR dysregulation has also been reported in human NAFLD (Puri et al. Citation2008). A possible explanation for the decreased expression of sXBP1 observed in OffOb-OD mice together with higher IRE1α activation could be the regulation by IRE1-dependent decay (RIDD), which induces selective post-transcriptional mRNA degradation including XBP1 and cellular apoptosis (Tam et al. Citation2014). Sustained stress attenuates IRE1α-XBP1 while activating RIDD, which in turn degrades UPR target genes and upregulates caspase-2-induced cellular apoptosis (Lin et al. Citation2007).
Failure of UPR adaptation induces apoptotic cell death (Rutkowski et al. Citation2006). Therefore, significant increase in apoptotic cell death in OffOb-OD could be explained by UPR-induced cell death, as IRE1α activates the JNK pathway, which is related to inflammation and apoptosis (Van Rooyen et al. Citation2013). Furthermore, ATF4 regulates amino acid biosynthesis, glutathione and oxidative stress defence (Harding et al. Citation2003). In our model, the downregulation of ATF4 could lead to a susceptibility to cellular stress. Moreover, CHOP is a transcriptional factor that plays a role in UPR-induced apoptosis, and its upregulation induces reactive oxygen species (ROS) production. All these effects induced by JNK, ATF4 and CHOP are key mechanisms of NAFLD pathogenesis. It is not clear how these downstream pathways were downregulated, but ATF4 translation has been reported to be inhibited by Toll-like receptor 4 (TLR4) activation in macrophages (Woo et al. Citation2009), and TLR4 on hepatocytes activated by increasing circulating LPS in NAFLD (Li et al. Citation2011). CHOP expression is predominantly regulated by PERK-ATF4 axis, as well as affected by ubiquitin-proteasome dependent degradation (Hattori et al. Citation2003). The continuous expression of CHOP in OffOb-OD with ATF4 downregulation, and transient expression in OffCon-SC and OffCon-OD might be adaptive responses to UPR activation, and the continuous expression could induce cellular apoptosis, as demonstrated by the TUNEL assay and caspase-3 results.
The downregulation of GRP78, a master regulator for ER stress, might play an important role. It functions as a major ER chaperone and a controller of UPR signalling activation (Ron & Walter Citation2007). GRP78 knockout showed lethality in embryos day 3.5 with reduced proliferation rate of embryonic cells and massive apoptotic death (Luo et al. Citation2006). Furthermore, a decrease in GRP78 has been reported in the liver of diabetic db/db mice (Yamagishi et al. Citation2012); however, proteomic analysis has revealed increased hepatic GRP78 in human NASH, but no changes in a NAFLD phenotype (Gonzalez-Rodriguez et al. Citation2014). These findings suggest that GRP78 might be a potential therapeutic target for NAFLD. In our model, downregulation of GRP78 in OffOb-OD might be due to DNA methylation of its promoter, while degradation of GRP78 by RIDD may be related other mechanisms. According to epigenetic regulation, it has been described in rodents that maternal feeding during perinatal stages could affect global and specific DNA methylation status in the liver of offspring (Cordero et al. Citation2013; Chen et al. Citation2015). Further studies based on nutritional epigenetic programming of ER stress will be important in better delineating UPR activation mechanisms.
Interestingly, observational studies in high-fat diet fed mice across three generations (F0, F1 and F2) showed that transgenerational accumulation of epigenetic modifications may lead to up-regulation of lipogenesis and ER stress pathways in the liver (Li et al. Citation2012).
Our previous report on the same mouse model of maternal transmission of obesity demonstrated that obese dams had raised breast milk leptin levels compared to lean dams (Oben et al. Citation2010). Leptin deficient mice are one of the most widely used animal model of obesity (ob/ob) (Li et al. Citation2004). Leptin protein has been consistently shown to inhibit PERK-mediated ER stress and apoptosis in vitro in hepatocytes (Xiong et al. Citation2014). Mice with leptin deficiency displayed increased hepatic ER stress response and liver injury (Kim et al. Citation2015), and there is a strong association between hypothalamic ER stress and leptin resistance (Mardones et al. Citation2014; Ramirez & Claret Citation2015). An intriguing mechanistic hypothesis to explain our findings would be that the increase in leptin observed in the setting of obesity might be a compensatory mechanism to reduce ER stress in NAFLD.
Conclusions
Our findings demonstrate that major UPR pathways are rhythmically activated in the liver under normal physiological conditions, and this rhythm can be profoundly affected by maternal obesogenic feeding and post-natal intake. In each of the UPR pathways, an obesogenic diet caused phase shifts in the time of the peak activation. Furthermore, the combination of MO and an obesogenic diet post-weaning induced continuous activation of IRE1α and PERK pathways, perturbation of downstream gene expressions, and disruption of UPR pathways associated with several apoptotic-related factors, leading to greater cell death and changes on DNA methylation patterns.
Disclosure statement
No competing financial interest exists.
Additional information
Funding
References
- Basseri S, Austin RC. 2008. ER stress and lipogenesis: a slippery slope toward hepatic steatosis. Dev Cell. 15:795–796.
- Brown MK, Naidoo N. 2012. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 3:263.
- Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang Q, Chen J, Cheng WH, Shen W, Yang ZX. 2012. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. 364:115–129.
- Cordero P, Li J, Oben JA. 2015. Epigenetics of obesity: beyond the genome sequence. Curr Opin Clin Nutr Metab Care. 18:361–366.
- Cordero P, Milagro FI, Campion J, Martinez JA. 2013. Maternal methyl donors supplementation during lactation prevents the hyperhomocysteinemia induced by a high-fat-sucrose intake by dams. Int J Mol Sci. 14:24422–24437.
- Cretenet G, Le Clech M, Gachon F. 2010. Circadian clock-coordinated 12 Hr period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 11:47–57.
- Chen G, Broseus J, Hergalant S, Donnart A, Chevalier C, Bolanos-Jimenez F, Gueant JL, Houlgatte R. 2015. Identification of master genes involved in liver key functions through transcriptomics and epigenomics of methyl donor deficiency in rat: relevance to nonalcoholic liver disease. Mol Nutr Food Res. 59:293–302.
- Dara L, Ji C, Kaplowitz N. 2011. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 53:1752–1763.
- Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. 2009. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 284:29735–29745.
- Farrell GC, Larter CZ. 2006. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 43:S99–S112.
- Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 226:497–509.
- Fu S, Watkins SM, Hotamisligil GS. 2012. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15:623–634.
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. 2011. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 473:528–531.
- Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillon J, Lo Iacono O, Corazzari M, Fimia GM, et al. 2014. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 5:e1179.
- Hampton RY. 2003. IRE1: a role in UPREgulation of ER degradation. Dev Cell. 4:144–146.
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 11:619–633.
- Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K. 2003. C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene. 22:1273–1280.
- Hetz C, Chevet E, Harding HP. 2013. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 12:703–719.
- Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. 2009. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 119:1201–1215.
- Kanda Y, Hashiramoto M, Shimoda M, Hamamoto S, Tawaramoto K, Kimura T, Hirukawa H, Nakashima K, Kaku K. 2015. Dietary restriction preserves the mass and function of pancreatic β cells via cell kinetic regulation and suppression of oxidative/ER stress in diabetic mice. J Nutr Biochem. 26:219–226.
- Kim SH, Kim KH, Kim HK, Kim MJ, Back SH, Konishi M, Itoh N, Lee MS. 2015. Fibroblast growth factor 21 participates in adaptation to endoplasmic reticulum stress and attenuates obesity-induced hepatic metabolic stress. Diabetologia. 58:809–818.
- Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. 2013. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells. 18:798–809.
- Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, Bass J. 2007. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 6:414–421.
- Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. 2012. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 56:900–907.
- Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, Hou YJ, Chang YX, Tu QQ, Feng GS, et al. 2011. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 54:1620–1630.
- Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, Thomas SA, Diehl AM. 2004. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 40:434–441.
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science. 318:944–949.
- Liu S, Brown JD, Stanya KJ, Homan E, Leidl M, Inouye K, Bhargava P, Gangl MR, Dai L, Hatano B, et al. 2013. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature. 502:550–554.
- Luo S, Mao C, Lee B, Lee AS. 2006. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 26:5688–5697.
- Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N. 2011. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci USA. 108:7832–7837.
- Mardones P, Dillin A, Hetz C. 2014. Cell-nonautonomous control of the UPR: mastering energy homeostasis. Cell Metab. 20:385–387.
- Martinez JA, Cordero P, Campion J, Milagro FI. 2012. Interplay of early life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc Nutr Soc. 71:276–283.
- Mouralidarane A, Soeda J, Sugden D, Bocianowska A, Carter R, Ray S, Saraswati R, Cordero P, Novelli M, Fusai G, et al. 2015. Maternal obesity programs offspring non-alcoholic fatty liver disease through disruption of 24-h rhythms in mice. Int J Obes (Lond). 39:1339–1348.
- Mouralidarane A, Soeda J, Visconti-Pugmire C, Samuelsson AM, Pombo J, Maragkoudaki X, Butt A, Saraswati R, Novelli M, Fusai G, et al. 2013. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology. 58:128–138.
- Oben JA, Mouralidarane A, Samuelsson AM, Matthews PJ, Morgan ML, McKee C, Soeda J, Fernandez-Twinn DS, Martin-Gronert MS, Ozanne SE, et al. 2010. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol. 52:913–920.
- Oishi K, Itoh N. 2013. Disrupted daily light-dark cycle induces the expression of hepatic gluconeogenic regulatory genes and hyperglycemia with glucose intolerance in mice. Biochem Biophys Res Commun. 432:111–115.
- Pendergast JS, Branecky KL, Yang W, Ellacott KL, Niswender KD, Yamazaki S. 2013. High-fat diet acutely affects circadian organisation and eating behavior. Eur J Neurosci. 37:1350–1356.
- Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. 2008. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 134:568–576.
- Ramirez S, Claret M. 2015. Hypothalamic ER stress: a bridge between leptin resistance and obesity. FEBS Lett. 589:1678–1687.
- Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 8:519–529.
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. 2006. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4:e374.
- Soeda J, Morgan M, McKee C, Mouralidarane A, Lin C, Roskams T, Oben JA. 2012. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun. 417:17–22.
- Tam AB, Koong AC, Niwa M. 2014. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 9:850–858.
- Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, Teoh NC, Farrell GC. 2013. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol. 59:144–152.
- Vollmers C, Gill S, DiTacchio L, Pulivarthy SR, Le HD, Panda S. 2009. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci USA. 106:21453–21458.
- Wang D, Wei Y, Pagliassotti MJ. 2006. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 147:943–951.
- Welsh JA, Karpen S, Vos MB. 2013. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr. 162:496–500. e491.
- Woo CW, Cui D, Arellano J, Dorweiler B, Harding H, Fitzgerald KA, Ron D, Tabas I. 2009. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol. 11:1473–1480.
- Wu H, Ng BS, Thibault G. 2014. Endoplasmic reticulum stress response in yeast and humans. Biosci Rep. 34:e00118.
- Xiong Y, Zhang J, Liu M, An M, Lei L, Guo W. 2014. Human leptin protein activates the growth of HepG2 cells by inhibiting PERK-mediated ER stress and apoptosis. Mol Med Rep. 10:1649–1655.
- Yamagishi N, Ueda T, Mori A, Saito Y, Hatayama T. 2012. Decreased expression of endoplasmic reticulum chaperone GRP78 in liver of diabetic mice. Biochem Biophys Res Commun. 417:364–370.
- Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. 2011. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 9:524–530.