Abstract
Although the number of morphological characters provided by diatom frustules for species identification is unparalleled among unicellular eukaryotes, morphology alone has in several cases proven to be insufficient to detect species boundaries in this algal group. Our recent nuclear rDNA sequence survey indicated that Cyclotella meneghiniana, one of the most intensively studied freshwater diatoms, is another example of a species complex. We therefore sampled genetic variation in the D1/D2 regions of the nuclear 28S rDNA and in a segment of the plastid encoded psaA gene in a larger sample of C. meneghiniana strains to assess genetic diversity and genetic structure over a wider geographic range within this diatom. A strict congruence of nuclear rDNA and psaA variants was observed, indicative of the presence of eight genetically distinct, cryptic lineages, four of which were not represented in our previous study. To test whether the genetic structure is caused by the complete lack of allogamy, or by the presence of reproductive barriers between allogamous cryptic species, we generated AFLP fingerprints from two subsamples of sympatric strains. Phylogenetic incompatibility in AFLP banding patterns did not differ significantly from panmictic expectations. Therefore, we suggest that C. meneghiniana is a complex of several, reproductively isolated, sexual species, rather than a complex of either strictly self-fertilizing or clonal lineages.
Introduction
Diatoms are the most speciose, ecologically and practically important group of eukaryotic microalgae. They contribute significantly to global primary production (Field et al ., Citation1998) and are widely used in water quality monitoring and palaeo-environmental reconstructions (Whitton & Rott, Citation1996; Stoermer & Smol, Citation1999). Their most spectacular and unique feature is their finely ornamented silica cell wall (frustule), which is still the major information source for species identification and classification (Round et al ., Citation1990). Compared with other microorganisms, the frustules provide many potentially discriminative morphological characters and the existence of cryptic (morphologically indistinguishable, or at least indistinguishable using classical approaches) diatom species could be expected to be low (Knowlton, Citation1993, Citation2000). However, evidence from diverse taxonomic and ecological groups of diatoms indicates that morphospecies composed of cryptic species are not rare (Mann, Citation1989, Citation1999; Medlin et al ., Citation1991; Sarno et al ., Citation2005).
Cyclotella meneghiniana Kützing is one of the most extensively studied freshwater diatoms (Bourne et al ., Citation1992; Giri & Devi, Citation1992; Latala & Surosz, Citation1999; Lohr & Wilhelm, Citation2001; Tedrow et al ., Citation2002; Büchel, Citation2003; Lewandowska & Kosakowska, Citation2004), and also featured as the single most common diatom species in the review by Finlay et al . (Citation2002) of global diatom diversity. However, it is also one of the most problematic examples of a diatom morphospecies, the taxonomy of which cannot satisfactorily be clarified solely on the basis of frustule morphology. It shows large morphological variability (Schoeman & Archibald, Citation1980; Håkansson & Chepurnov, Citation1999) and occurs in a wide range of habitat types (Finlay et al ., Citation2002; Håkansson, Citation2002). Extreme morphological plasticity has been observed in the closely related Cyclotella cryptica Reimann, Lewin & Guillard: cultures of C. cryptica can display frustule morphologies considered characteristic of C. meneghiniana (Schultz, Citation1971). Cyclotella meneghiniana was also the first freshwater diatom in which molecular variation was investigated (Bourne, Citation1992), a study that revealed large (much greater than in closely related species) variation in its plastid genome restriction fragment length (RFLP) patterns and its paraphyly with respect to C. cryptica.
The idea that C. meneghiniana might not be a single, genetically homogeneous sexual species has been discussed previously in the literature and two alternative hypotheses have been suggested. Based on her observation of large genetic variation and paraphyly of plastid RFLPs, Bourne (Citation1992) considered that it might be a complex of several species. On the other hand, Drebes (Citation1977) suggested that C. meneghiniana had abandoned allogamy: although sexual reproduction had been studied intensively in this species (Iyengar & Subrahmanyan, Citation1944; Schultz & Trainor, Citation1968; Rao, Citation1970, Citation1971, Citation1996; Håkansson & Chepurnov, Citation1999), no observations of allogamous fertilization have ever been published.
The presence of cryptic species and strict clonality or autogamy leads to similar deviations from patterns of genetic variation expected in a panmictic population, i.e. a relative lack of heterozygosity and linkage between physically unlinked loci (Tibayrenc & Ayala, Citation2002). However, the implications for variation patterns within genetically distinct groups are different. In the case of cryptic species, relative geno- and phenotypic homogeneity within (because of sexual reproduction) and larger differentiation among (because of reproductive barriers) cryptic species are expected. On the other hand, within-group homogenization due to sexual reproduction is absent in a complex lacking allogamy. Instead, a general pattern of correlations between unrelated features can be expected (Tibayrenc & Ayala, Citation2002). Whereas within an allogamous sexual species different parts of an individual's genome have different genealogies (because each locus might come from either parent in each generation), loci within genomes of autogamous or agamous (strictly clonal) organisms share a common genealogy and thus, similar patterns of divergence. In spite of these differences, the alternatives of strict clonality versus the presence of cryptic species are rarely tested in molecular studies to clarify species boundaries in diatoms. The importance of testing these possibilities explicitly is further underlined by the fact that effective clonality has been observed in protists that were previously considered as obligate sexual species (Ajzenberg et al ., Citation2002). Thus, despite the long history of study, it remains unclear whether C. meneghiniana is, (1) a single allogamous sexual species, (2) a complex of several allogamous species, or (3) a strictly self-fertilizing (autogamous) complex
In a previous study (Beszteri et al ., Citation2005a ), we surveyed rDNA (D1/D2 regions of the 28S rDNA and the internal transcribed spacer, ITS) sequence variation in a limited collection of strains of what was originally believed to be a single population of this species. Numerous copies of the ribosomal cistron are present in typical eukaryotic genomes and polymorphisms can occur among copies. Thus, even clonal cultures may show within-strain polymorphisms for this cistron, especially in the more variable regions. To test for the presence of genetic substructure accounting for intra-genomic rDNA polymorphisms we compared sequence variation within single strains (clones) with variation among strains. Our results revealed large between-strain rDNA sequence variation, accompanied by much smaller within-strain variation, the first strong evidence for the presence of genetic structure within C. meneghiniana. The data suggested that four, genetically distinct (presumably reproductively isolated), but morphologically (apparently) indistinguishable groups were present at a single locality. Patterns of variation in the ITS regions (no significant within-group between-strain variance component) suggested that the genetic structure was more probably explained by the presence of multiple allogamous species than by the complete lack of allogamy. However, our confidence in this conclusion was constrained by the small ITS sequence variation present in each subgroup. It has also remained unclear whether the four groups found in our population study represent the global diversity within C. meneghiniana, or whether additional genetically distinct groups exist.
A larger survey of molecular variation in C. meneghiniana was undertaken to test whether this morphospecies includes additional candidate cryptic species. We therefore sampled a large collection of strains, including most cultures of this species available from public culture collections and strains isolated from a number of geographically diverse localities (mainly in Europe). We also included strains of closely related species, including C. cryptica, which is capable of producing C. meneghiniana-like morphologies (Schultz, Citation1971), Cyclotella quillensis Bailey, Cyclotella choctawatcheeana Prasad, and Cyclotella cf. scaldensis to determine whether, and to what extent, molecular markers can distinguish these morphospecies. One Stephanodiscus Ehrenberg and three Discostella Houk & Klee strains were also analysed to serve as outgroups in phylogenetic analyses. To increase confidence in the results but avoid the time-consuming cloning steps necessary to evaluate intragenomic polymorphism in rDNA sequences, we complemented screening of the variable D1/D2 regions of the nuclear 28S rDNA by sequencing a portion of the single-copy, plastid-encoded psaA gene. In the case of psaA, within-strain polymorphism is not an issue. Furthermore, apparent linkage (non-random association) between two physically unlinked loci is a stronger diagnostic of genetic structure than a single molecular marker.
For testing the hypothesis that allogamy is absent in natural populations of C. meneghiniana, we also generated amplified fragment length polymorphism (AFLP) fingerprints from a subset of strains, isolated from two, closely located populations, belonging to the same genotypic group (based on their nuclear rDNA and psaA sequences). The hypothesis of strict clonality was tested using this data set, by exploring phylogenetic incompatibility among loci (Mes, Citation1998).
Materials and methods
Cultures
lists the cultures used in this study and their origins. Cultures established in the course of our study were isolated and grown as described previously (Beszteri et al ., Citation2005b ). Because Discostella has only recently been separated from the genus Cyclotella (Houk & Klee, Citation2004), its strains are generally still referred to as Cyclotella spp. in culture collections. The group of cultures previously called ‘ambiguous’ C. scaldensis morph (Beszteri et al ., Citation2005a , Citation b ) will be designated C. cf. scaldensis in this paper and the three morphospecies, C. meneghiniana, C. quillensis and C. cryptica, will be referred to as the C. meneghiniana complex. Cyclotella cf. scaldensis and C. choctawatcheeana can be distinguished from them by the possession of closed alveolar chambers. Morphological data and images of the cultures used are available under www.algaterra.org and from the corresponding author upon request.
Table 1. List of strains used, their origins, GenBank accession numbers and abbreviations of LSU rDNA and psaA types. The grouping of the strains reflects their genotypes at the two loci investigated. This corresponds to morphospecies with the exception of the doubtfully identified strain CCMP 336 (see below and in text), and C. meneghiniana, which contains several genotypic groups. In the case of the 28S rDNA types, the presence of one variant is indicated when no ambiguous positions were observed. In the presence of ambiguous positions, rDNA types were reconstructed assuming the presence of two different variants, these are listed separated by commas
DNA extraction, PCR and sequencing
Cultures were harvested by filtration and DNA was extracted using a modified CTAB protocol (Doyle & Doyle, Citation1990) or the PAN Plant Kit (PAN Biotech, Aidenach, Germany). The D1/D2 regions of the nuclear 28S rDNA were amplified and sequenced using primers from Scholin et al . (Citation1994). A ∼700 basepair (bp) fragment of the psaA gene was chosen as the chloroplast genetic marker and amplified using PCR primers from Yoon (2002). PCR products were purified with the QIAQuick PCR Product Purification Kit (QIAGEN, Germany) and directly sequenced on both strands using Big Dye Terminator v3.1 sequencing chemistry (Applied Biosystems, CA, USA). Sequencing products were electrophoresed on an ABI 3100 Avant sequencer (Applied Biosystems, CA, USA).
Sequence assembly and analyses
Sequences were assembled using SeqScape 2.1 (Applied Biosystems, CA, USA) as described previously (Beszteri et al ., Citation2005a ). In the nuclear rDNA electropherograms, ‘high confidence ambiguities’ were identified based on the relative heights of two peaks (0.3 or more) occurring in the same position without sequencing noise around that position in both sequencing reactions. Sequences containing such ambiguities were considered to represent within-strain polymorphisms. Individual 28S rDNA variants producing the observed ambiguities were reconstructed from these sequences following Clark (Citation1990), described in more detail by Beszteri et al . (Citation2005a ).
Phylogenetic analyses and calculations of evolutionary distances were performed using PAUP* 4.0b10 (Swofford, Citation1998). For maximum likelihood (ML) and distance-based tree calculations, likelihood scores of different nucleotide substitution models were compared on a neighbour joining (NJ) tree using Modeltest 3.0 (Posada, Citation1998). The best-fit model according to the Akaike Information Criterion (AIC) was used for phylogenetic analyses using ML and NJ tree inference with ML distances. The chosen models had the following parameters: for the 28S rDNA data set, base frequencies were: A: 0.262, C: 0.204, G: 0.289 and T: 0.245; substitution rates were: A–C: 0.487, A–G: 2.255, A–T: 1.491, C–G: 0.379 and G–T: 7.666; the proportion of invariant sites was 0.414, and among-site rate heterogeneity was described by a gamma distribution with a shape parameter of 0.59. For the psaA data set, base frequencies were: A: 0.310, C: 0.146, G: 0.196 and T: 0.348; substitution rates were: A–C 1.000, A–G: 2.488, A–T: 0.372, C–G: 0.372 and G–T: 6.939; the proportion of invariant sites was 0.6425, and the gamma shape parameter was 0.87. Maximum parsimony (MP) and ML trees were obtained in heuristic searches with 10 random taxa addition sequences. Bootstrapping (BS) of MP and NJ analyses was performed with 1,000 replicates.
AFLP
AFLPs were used to test the hypothesis of strict clonality within one of the apparently genetically distinct lineages within C. meneghiniana, which were revealed by the DNA sequence data. Twelve strains (with prefixes AT-D and AT-N, see ) belonging to group A based on their partial 28S rDNA sequences were used for the AFLP genotyping. These strains were isolated from samples taken at two localities from the River Weser (Northern Germany) on the same day (21 July 2003). The two sampling sites are situated ∼40 km from each other (near Nienburg, 52°38,54′N; 9°12,41′E, and Daverden, 52°57,96′N; 9°9,38′E). DNA extraction was performed using the PAN Plant Kit (PAN Biotech, Aidenach, Germany). DNA quality was verified by agarose gel electrophoresis; concentration was measured spectrophotometrically.
Digestion of genomic DNA, ligation, and pre-amplification were performed as described in John et al , (Citation2004) beginning with 250 ng genomic DNA. Selective amplifications using 2–3 additional bases were also performed as described in John et al , (Citation2004), but with the following modifications: three primer combinations were used (EcoRI + AC − MseI + CG; EcoRI + AC − MseI + CCT; EcoRI + AAG − MseI + CCT) and EcoRI-specific primers were marked with the fluorescent dye 6-FAM. 1 µl of the product with 0.5 µl of GeneScan-500 ROX Size Standard (Applied Biosystems) internal DNA size marker were diluted to 10 µl with Hi-Di Formamide and run on an ABI 3100 Avant sequencer (all Applied Biosystems, CA, USA). AFLP bands were sized and manually scored using GeneMapper v3.5 (Applied Biosystems, CA, USA).
AFLP analyses
The hypothesis that allogamous reproduction was absent was tested by compatibility analyses. Two loci are called incompatible when the evolution of the two-locus genotypes can be explained on a phylogenetic tree without homoplasy. If this is not the case and either homoplasious mutations or genetic exchange between lineages is required to explain observed two-locus genotypes, the two loci are deemed incompatible. In the case of binary genotypes (i.e. only presence-absence alleles are distinguished), as with AFLP, two loci are incompatible when all four possible two-locus genotypes are observed. Using multilocus AFLP genotypes of the two population samples, the numbers of incompatible pairs of loci are compared with expectations for panmixia (random mating) estimated by a randomization procedure. Compatibility analyses were performed using programs from the package PICA 95 (Wilkinson, Citation1995). The matrix conflict permutation tail probability test was performed using MATRIX.EXE. This test compares the incompatibility counts in the data set with those in random permutations of the characters among strains. The proportion of cases having as low or lower incompatibility counts than the original data set is interpreted as the probability that the observed amount of incompatibility is comparable to that resulting from random assignment of characters to terminal taxa, i.e. panmixia. JACTAX.EXE was then used to explore the possibility that one or a few strains are responsible for a large proportion of the incompatibilities in the data set (i.e. one or a few hybrid genotypes in a mostly clonal/allogamous group). Here, differences in incompatibility counts in the original data set are compared with those observed when each terminal taxon is excluded in turn from the counting. The taxon contributing the most to overall incompatibility in the data is then excluded and the analyses iterated, as long as the remaining data set contains incompatibilities. Thus, it can be determined whether the conflict in the data set is caused by one, or a few, individuals. In our case, this would mean that, although the population is basically clonal, rare sexual events lead to the presence of hybrid genotypes.
Results
Nuclear rDNA sequence variation
lists the rDNA variants found in each strain and general information about the sequences of this region is summarized in . The D1/D2 regions of the 28S rDNA were invariably 613 bp in strains from the Cyclotella species, excluding amplification primer binding sites. The PCR products from the Discostella strains (D. pseudostelligera [Hustedt] Houk & Klee and D. woltereckii [Hustedt] Houk & Klee) were 605 bp, that from Stephanodiscus hantzschii Grunow 603 bp.
Table 2. Summary of genetic variation observed within the rDNA groups of C. meneghiniana and other Cyclotella species investigated
With the exception of two C. meneghiniana strains (AT-Cm7 and CCMP 335; see , ), variants could be unambiguously resolved from sequences containing mixed base calls using a parsimony-based method (Clark, Citation1990). The sequence obtained from strain CCMP 335 contained eight mixed base calls. It could be decomposed in three different ways into two variants, one of which was identical to an unambiguously identified variant. The sequence from strain AT-cm7 contained 12 mixed bases. It could also be decomposed in two different ways. For subsequent analyses, the most commonly found already identified variant (D1) and the corresponding complementary variants (D4, D5, ) were used.
Within-morphospecies variation in the partial 28S rDNA sequences was small except for C. meneghiniana (). A single variant was observed in the three C. quillensis strains, as well as in the two C. choctawatcheeana strains. Polymorphisms were observed in all other species represented by more than one strain (). Two different variants were found (Cry, Cry2) in the three C. cryptica strains (CCMP 331, 332 and 333). Variant Cry, with a very similar variant (Cry3), was also found in another strain (CCMP 336), identified as C. meneghiniana by CCMP. Based on these results (see below), this strain probably belongs to C. cryptica. Up to four nucleotide differences separated pairs of the three variants found in these four strains. Four different variants were found in the five C. cf. scaldensis strains, with up to five nucleotide differences between them.
Sequence variation in this region within C. meneghiniana was much larger. Twenty-one different variants were observed, with 72 polymorphic positions (11.7%) among the C. meneghiniana strains. The number of nucleotide differences between pairs of these variants ranged from one to 32. However, within-strain sequence variation was much smaller. Fewer than three nucleotide differences were found between variants from single strains of C. meneghiniana, with three exceptions (). As described previously (Beszteri et al ., Citation2005a ), two divergent variants (A1, D1, differing at 22 positions) were found in strain G184. Two strains from group E (CCMP 335, AT-cm7) contained combinations of alleles differing at eight (variants D1, D5) and 12 positions (variants D1, D4), respectively.
Table 3. Variable positions in rDNA sequences in group D of C. meneghiniana. Variants are listed in the first column, and position numbers are given as heads of the other columns. Ambiguities that could not be resolved are present for the last two variants, CCMP 335 and AT-cm7
The partial 28S rDNA variants obtained from C. meneghiniana could be grouped into eight distinct groups based on their similarities and co-occurrences in single strains. (Strain G184 was excluded; the single co-occurrence of rDNA types A1 and D1 () has been tentatively interpreted as a hybrid.) With the exception of group D, each of these groups displayed no more than four polymorphic nucleotides (, ). The nucleotide differences among variants belonging to different groups ranged from 8 to 33. These groups were monophyletic, with strong bootstrap support (>97%), in phylogenetic analyses (). The only exception was again group D: the monophyly of this group had lower bootstrap support (63% NJ, 70% MP).
Table 4. Matrix of pair-wise nucleotide differences of the nuclear 28S rDNA variants
Sequences from other morphospecies in the C. meneghiniana-complex (C. quillensis and C. cryptica) could not be clearly separated from sequences of C. meneghiniana. Pairwise differences between strains from the three different morphospecies (C. quillensis v. C. meneghiniana: 16–32 bp, C. cryptica v. C. meneghiniana: 13–29 bp) were comparable to those between the different C. meneghiniana subgroups (group A v. group B: 23–25 bp, group A v. group D: 13–24 bp, group A v. group G: 25–29 bp). Phylogenetic analyses () did not separate these three morphospecies into three clades either (see below).
Fig. 1. Phylogenies of the nuclear 28S rDNA (left) and plastid encoded psaA (right) variants. Trees were calculated by bootstrapping the alignments in 1,000 replicates and calculating neighbour-joining (NJ) trees with maximum-likelihood (ML) distances using the best-fit models chosen by the Akaike Information Criterion. Branches recovered with less than 60% bootstrap support were collapsed into polytomies, and branch lengths were estimated on the collapsed trees using ML. Lines connecting groups of 28S rDNA and psaA variants indicate that the corresponding variants were found in the same group of strains. A single co-occurrence is not shown, that of 28S rDNA variant D1 and psaA variant cmI, in the possibly non-clonal or hybrid strain G184 (see Beszteri et al . Citation2005a ). The trees were rooted using sequences from Stephanodiscus hantzschii (SHAN, shan) and the Discostella (DPS EU, dpseu) strains (see for rDNA and psaA variants found in strains) as outgroups. Numbers indicate bootstrap support from the NJ and maximum parsimony analyses.
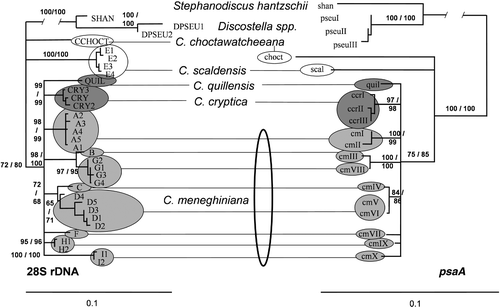
The phylogenetic relationships among the above groups of rDNA variants remained largely unresolved (), only a few nodes had bootstrap support above 60%. Variants of the Cyclotella (excluding Discotella) strains constituted a strongly supported clade, including a C. meneghiniana complex clade (BS: 100 in both MP and NJ). However, monophyly of C. meneghiniana excluding C. quillensis and C. cryptica was not supported. In the single best trees recovered from NJ and ML analyses, variants of C. quillensis and C. cryptica always grouped among C. meneghiniana variants, leaving the latter para- or polyphyletic. Also, in the strict consensus of the 92 most parsimonious trees obtained (length: 253 steps, consistency index: 0.727), C. quillensis (variant Quil) was sister to group A of C. meneghiniana. Thus, the optimal trees indicate the paraphyly of the C. meneghiniana LSU variants. Bootstrap analyses did not increase confidence in these results because most nodes within the C. meneghiniana complex had bootstrap values below 60% ().
Sequence variation in the psaA gene and congruence between the two data sets
In every case, the length of the sequenced regions was 715 nucleotides. No mixed base calls were observed in any of the sequences. Over the entire data set, including outgroups, there was variation in 133 positions, 96 within Cyclotella, 52 within the C. meneghiniana complex, and 45 within C. meneghiniana. Sixteen of the 238 amino acid positions were variable in the whole dataset, 14 within Cyclotella, seven within the C. meneghiniana complex and six within C. meneghiniana.
Patterns of variation within the morphospecies investigated were similar to the nuclear LSU sequences, with most morphospecies showing little or no variation, with the exception of C. meneghiniana. Within the latter, 45 nucleotide (six amino acid) positions were variable. However, sequence variation among single strains within the C. meneghiniana groups delimited on the 28S rDNA sequences was also minor (). With the exception of groups A (two psaA alleles displaying three nucleotide differences) and D (two psaA alleles differing at a single nucleotide position), a single psaA variant was found for each group.
Two psaA variants (ccrI, ccrII, differing at four nucleotide positions) were found in the three strains labelled C. cryptica (CCMP 331, 332 and 333). A third variant (ccrIII, differing from ccrI at one, and from ccrII at three nucleotide positions) was found in strain CCMP 336, labelled as C. meneghiniana in the culture collection, but with strong affinities to the C. cryptica strains in its nuclear ribosomal sequences (see above). Whereas pair-wise differences among psaA variants ccrI, ccrII and ccrIII ranged from one to four, the smallest number of substitutions separating any of them from variants found in C. meneghiniana strains was 13. Thus, the psaA data reinforced the conclusion that strain CCMP 336 belongs to C. cryptica rather than to C. meneghiniana.
Results of phylogenetic analyses were similar to those obtained from the LSU rDNA data: the monophyly of Cyclotella was strongly supported (BS 100% in both MP and NJ), as was monophyly of the C. meneghiniana complex (BS: 74% NJ, 84% MP), but the branching order of clades within the latter remained unresolved. In the strict consensus of the 80 most parsimonious trees obtained, the only clades recovered within the C. meneghiniana complex were the same as those shown in . As with the nuclear rDNA data set, neither mono-, nor paraphyly of C. meneghiniana was supported by high bootstrap values.
Most importantly, strict association between the nuclear rDNA and the psaA variants was observed. Within most groups identified on partial 28S rDNA sequence variation, identical psaA sequences were obtained from each strain (). Exceptions were the C. cryptica strains (three psaA types with four variable positions), and groups A (two psaA types, cmI and cmII, differing at three positions) and D (cmV and cmVI, with a single nucleotide difference) of C. meneghiniana (, ). Even in these cases, the differences between the psaA variants obtained from a single rDNA group were much smaller than the differences separating them from any other variant (see ).
AFLP
Two hundred and thirty-two AFLP markers were scored for the 12 strains included in the AFLP analysis using three primer pairs; 174 of the loci were polymorphic. All 12 fingerprints were unique. Only 61 (Daverden population, seven strains) and 79 (Nienburg population, five strains) of the 174 polymorphic markers showed polymorphism within the populations. Fifty-nine of the variable loci represented fixed differences between the two populations (i.e. the same, presence or absence, allele was found in each strain from one population, and the other allele in each strain of the other population). The loci showing fixed differences are (by definition) also phylogenetically compatible with each other. A matrix incompatibility permutation tail test showed highly significantly (p < 0.01) low phylogenetic incompatibility (i.e. deviation from panmictic expectations) for the combined data set. However, the number of incompatible character pairs in the data set was substantial (2,943), and neither of the two populations deviated from panmictic expectations when analysed separately. The number of incompatible pairs was 97 (in the Daverden population, permutation tail probability: 0.89) and 217 (in the Nienburg population, permutation tail probability: 0.25), respectively. Thus, the two populations each showed patterns of genetic variation indistinguishable from that expected assuming random mating within the populations.
We also tested whether all strains contributed a comparable amount to the phylogenetic incompatibility in the two populations, or whether this was due to the inclusion of one or a few hybrid strains. The contribution of individual strains to incompatibility within a population was therefore assessed by sequentially omitting them from the data set, and comparing incompatibility counts of data sets with and without each strain. The strain that appeared to contribute the most to incompatibility was removed, and the test repeated until no further incompatibilities remained in the data set. The results showed that in both populations, the removal of the first strain substantially decreased the amount of phylogenetic incompatibility in the data (not shown). However, a complete lack of incompatibilities could not be reached in either population. Some incompatibility persisted even when only four strains remained. (Four terminal taxa constitute the limit of phylogenetic incompatibility testing in a data set with non-polarized characters – i.e. by definition, no incompatibilities can be present in a data set of three taxa).
Discussion
Cryptic diversity in Cyclotella meneghiniana
The data obtained in this study strongly support the hypothesis that C. meneghiniana does not constitute a genetically homogeneous species. The following facts indicate the presence of genetic structure within C. meneghiniana: (1) within-species sequence variation substantially exceeded average within-strain variation in the nuclear rDNA segment sequenced. Groups of rDNA variants were clearly distinct (, ). Even group D, showing the most variation, was monophyletic in phylogenetic analyses (). Whereas within-strain variation was encountered in the nuclear rDNA sequences, variants belonging to different groups never occurred together except for one single possible hybrid strain (G184), discussed previously (Beszteri et al ., Citation2005a ). (2) groups of strains defined on their nuclear rDNA genotypes had identical or close to identical psaA sequences, whereas psaA sequence differences between rDNA groups were always larger ().
Sequence variation at both loci within the C. meneghiniana groups was comparable to variation observed within the other morphospecies studied here (). In the case of the D1/D2 regions of the 28S rDNA, this was also comparable to the degree of intraspecific variation observed in other algal groups (John et al ., Citation2003; Sarno et al ., Citation2005).
The observed genetic structure within C. meneghiniana cannot be explained by geographic isolation. Strains belonging to different genotypic groups occurred in sympatry in several cases (, Beszteri et al ., Citation2005a ). Strains from two localities (Geeste Estuary, Bremerhaven, 20 strains prefixed G, and River Weser by Hasenbüren, six strains prefixed AT-10, , Beszteri et al ., Citation2005b ) showed large genetic diversity; strains from other localities (River Weser by Nienburg and Daverden) all belonged to genotypic group A. On the other hand, strains belonging to several genotypic groups could be found at different geographic localities (e.g. group C – Northern Germany and Luxembourg; group D – Rivers Ems and Weser, the Köttinger See, Germany, and Loch Leven, Scotland).
Wider sampling revealed four new genotypic groups (groups F, G, H, I) in addition to the four groups (A–D) discovered in our previous, single-population study (Beszteri et al ., Citation2005b ). We have found only one representative of each of the new genotypic groups H and I, suggesting that additional sampling from new geographic localities might reveal the existence of more cryptic groups within C. meneghiniana.
Clonality or cryptic allogamous species?
The above results raise the question whether the observed variation pattern can be explained by the complete lack of sexual reproduction in C. meneghiniana, or by the presence of multiple sexual species within it. Although patterns of ribosomal DNA variation might be of some use for answering this question (Beszteri et al ., Citation2005a ), they are not ideal for this purpose because of the low amount of variation revealed within the groups. AFLP fingerprints have two important advantages in this context: they show more variation, and they provide information about genetic variation at multiple loci. Using multilocus genotypes, phylogenetic incompatibility among loci can be explored. In the case of strict clonality or selfing (autogamy), genome-wide linkage and phylogenetic compatibility is expected among loci, whereas, if sexual reproduction occurs (even occasionally), this linkage is expected to break down. In the case of the dominant AFLPs, allele frequency-based tests of linkage disequilibrium are inappropriate, and our low sample size also precludes the use of such tests. However, tests based on phylogenetic compatibility among loci are less sensitive to these factors and they allowed us to test the hypothesis that allogamy is absent.
The AFLP data showed high degrees of incompatibility among loci within both populations. The data strongly suggest that, at least occasionally, allogamous sexual reproduction occurred in the recent past of these populations, reshuffling genetic variation among lineages. This supports the conclusion that C. meneghiniana is a complex of cryptic allogamous sexual species rather than a strictly autogamous complex. Our small sample size does not allow us to draw strong conclusions about the frequency of such sexual events. We also do not know whether some or all of the other genotypic groups revealed by our study show allogamous reproduction. Extended population sampling will be required to clarify further the prevalence and frequency of sexual reproduction in these groups.
Population differentiation on a small geographic scale or even more cryptic species?
The most unexpected result of this study was the substantial differentiation between the two sets of strains used in the AFLP analyses. The samples from which these cultures were isolated were taken on the same day from the same river, at sites about 40 km apart, within about 2 hours. Of the 174 polymorphic loci, 59 showed a fixed difference between the two groups of strains, and a further 64 loci were compatible with this grouping. Only 29 parsimony informative loci showed variation patterns that were incompatible with the split between the two populations. Although sample sizes were very small, these genetic differences between strains from the two localities were unexpectedly large.
Further, more intensive population sampling is required to be able to interpret this substantial genetic differentiation. Until then, we can only speculate whether it can be explained by strong divergent selective pressure between the two localities, or by the possibility that these groups of strains (group A) contain more than one biological species. Patterns of variation at both loci are congruent with the latter idea (cryptic species): psaA variants in strains from the two populations also showed three fixed mutational differences (). With the exception of variant A2, which occurred in both groups, their 28S rDNA variants were also different. Shared presence of variant A2 could be interpreted as a polymorphism retained from the common ancestor of the two species.
The above alternative – large intraspecific genetic differentiation or the presence of cryptic species – seems to be a recurrent outcome of studies involving high resolution genotyping of diatom populations. The first detailed population genetic study of a diatom revealed seasonal alternation between highly differentiated populations in Skeletonema costatum Greville using multilocus enzyme electrophoresis (Gallagher, Citation1980). We now know that this morphospecies is in fact a complex of several genetically distinct entities (Sarno et al ., Citation2005). However, it is still unclear whether the summer and winter populations detected by Gallagher were representatives of one or more of these semi-cryptic species. Another study, performed with the taxonomically unproblematic diatom Ditylum brightwellii (West) Grunow using microsatellite markers, detected genetically distinct populations in different parts of a single estuarine system (Rynearson & Armbrust, Citation2004). In this case too, it remains unclear whether the substantial differentiation was caused by differential growth of distinct species (reproductively isolated groups), or whether different subsets of the genetic variation present in a single species were favoured in the different environments. Based on the recurrence of this question in the few population genetic investigations on diatoms to date, and with the widening application of high resolution molecular markers, one can predict that diatomists will increasingly be confronted with this problem in the future.
Taxonomic conclusions: Cyclotella cryptica, C. quillensis and C. meneghiniana
As noted, one of the morphospecies included in our study, C. cryptica is known to be able to produce valve morphology typical of C. meneghiniana when grown at low salinities and after auxospore production (Schultz, Citation1971). Other workers (Desikachary & Rao, Citation1973; Hoops & Floyd, Citation1979) also reported that C. meneghiniana cultures can produce the C. cryptica morphology, and smaller morphological changes in C. meneghiniana cultures caused by salinity changes have also been shown (Hoops & Floyd, Citation1979; Tuchman et al ., Citation1984). We did not investigate the effects of salinity on valve morphologies, but future attempts to identify morphological differences between the apparently cryptic taxa within the C. meneghiniana complex will need to consider this factor.
Our results indicated that strains identified as C. cryptica were genetically homogeneous and different from C. meneghiniana. Although their rDNA and psaA variants were not more different from other C. meneghiniana strains than the latter from each other, they formed a distinct group within the C. meneghiniana complex in both data sets (). A comparable pattern was observed by Bourne (Citation1992) in her plastid DNA RFLP study: a clade of three C. cryptica strains grouped well within the deeper clade including strains identified as C. meneghiniana. CCMP 336 has probably been misidentified. Based on its nuclear rDNA and psaA sequence, we list it as C. cryptica, although according to the CCMP catalogue it is a strain of C. meneghiniana. Our results justify the taxonomic separation of C. cryptica from C. meneghiniana: even though morphologies produced by the two species may be indistinguishable (probably depending on growth conditions), they seem to be genetically distinct (and reproductively isolated).
The situation with C. quillensis is similar. The morphological differences between this species and C. meneghiniana are also slight (Battarbee & Keister, Citation1982; Håkansson & Kling, Citation1994) but C. quillensis showed no variation in either sequenced region, and was genetically distinct from all other groups. Its separation from C. meneghiniana and C. cryptica is justified, but it is part of the well-supported, larger Cyclotella clade including C. meneghiniana and C. cryptica.
In general, our results (Beszteri et al . Citation2005a , this paper) give some support to taxonomic concepts based on frustule ultrastructure but, by providing new insight into patterns of intraspecific variation, they also contribute to the debate over the separation of C. cryptica from C. meneghiniana, and the apparent cryptic diversity within C. meneghiniana. However, several questions remain. Are all cryptic groups in C. meneghiniana allogamous? Are the cryptic species globally distributed? Which ecological factors lead to the emergence of reproductive isolation, and which ensure their co-existence? More practically, do the cryptic groups deserve taxonomic separation, and at what level? Might some of them be good indicator species, unlike C. meneghiniana sensu lato, which occurs over a wide range of environmental conditions?
Currently, little is known about the relative rates of morphological, molecular and physiological divergence following speciation, or about the global distribution of cryptic species. In addition to the need for similar studies to clarify species limits in diatoms, it is also necessary to integrate such work with ecological, population genetic and biogeographical studies to understand the significance of cryptic species for diatom ecology and evolution.
Acknowledgements
Thanks to E. Hegewald, É. Ács, E. Theriot and A. Alverson (US National Science Foundation grant DEB 0111883) for providing cultures, A. Garcia-Sáez for discussions, W. Kooistra for a helpful review of a previous version of this manuscript, and E. Cox and two anonymous reviewers for their constructive suggestions. This work was in part supported by the project “Algaterra” of the German Ministry of Education and Research (BMBF 01LC0026, http://www.algaterra.org).
Related Research Data
References
- Ajzenberg , D , Banuls , AL , Tibayrenc , M and Darde , ML . 2002 . Microsatellite analysis of Toxoplasma gondii shows considerable polymorphism structured into two main clonal groups . Int. J. Parasitol. , 32 : 27 – 38 .
- Battarbee , RW and Keister , CM . 1982 . “ The frustular morphology and taxonomic relationships of Cyclotella quillensis Bailey ” . In Proceedings of the 7th Diatom Symposium , Edited by: Mann , DG . 173 – 184 . Koenigstein, , Germany : Otto Koeltz .
- Beszteri , B , Ács , É. and Medlin , LK . 2005a . Ribosomal DNA sequence variation among sympatric strains of the Cyclotella meneghiniana complex (Bacillariophyceae) reveals cryptic diversity . Protist , 156 : 317 – 333 .
- Beszteri , B , Ács , É. and Medlin , LK . 2005b . Conventional and geometric morphometric studies of valve ultrastructural variation in two closely related Cyclotella species (Bacillariophyceae) . Eur. J. Phycol. , 40 : 73 – 88 .
- Bourne , CEM . 1992 . “ Chloroplast DNA structure, variation and phylogeny in closely related species of Cyclotella ” . In PhD Dissertation , University of Michigan .
- Bourne , CEM , Palmer , JD and Stoermer , EF . 1992 . Organization of the chloroplast genome of the freshwater centric diatom Cyclotella meneghiniana . J. Phycol. , 28 : 347 – 355 .
- Büchel , C . 2003 . Fucoxanthin-chlorophyll proteins in diatoms: 18 and 19 kDa subunits assemble into different oligomeric states . Biochemistry (Mosc) , 42 : 13027 – 13034 .
- Clark , A . 1990 . Inference of haplotypes from PCR-amplified samples of diploid populations . Mol. Biol. Evol. , 7 : 111 – 122 .
- Desikachary , TV and Rao , VNR . 1973 . Studies on Cyclotella meneghiniana Kütz. III. The frustule . Proc. Ind. Acad. Sci. , 77B : 78 – 91 .
- Doyle , JJ and Doyle , JL . 1990 . Isolation of plant DNA from fresh tissue . Focus , 12 : 13 – 15 .
- Drebes , G . 1977 . “ Sexuality ” . In The Biology of Diatoms , Edited by: Werner , D . 250 – 283 . Oxford, UK : Blackwell Scientific Publications .
- Field , CB , Behrenfeld , MJ , Randerson , JT and Falkowski , P . 1998 . Primary production of the biosphere: integrating terrestrial and oceanic components . Science , 281 : 237 – 240 .
- Finlay , BJ , Monaghan , EB and Maberly , SC . 2002 . Hypothesis: the rate and scale of dispersal of freshwater diatom species is a function of their global abundance . Protist , 153 : 261 – 273 .
- Gallagher , JC . 1980 . Population genetics of Skeletonema costatum (Bacillariophyceae) in Narragansett Bay . J. Phycol. , 16 : 464 – 474 .
- Giri , BS and Devi , NA . 1992 . Inhibition of cytokinesis by lead (Pb2+) in a centric diatom . Ind. J. Exp. Biol. , 30 : 201 – 214 .
- Håkansson , H . 2002 . A compilation and evaluation of species in the genera Stephanodiscus, Cyclostephanos and Cyclotella with a new genus in the family Stephanodiscaceae . Diatom Res. , 17 : 1 – 139 .
- Håkansson , H and Chepurnov , V . 1999 . A study of variation in valve morphology of the diatom Cyclotella meneghiniana in monoclonal cultures: effect of auxospore formation and different salinity conditions . Diatom Res. , 14 : 251 – 272 .
- Håkansson , H and Kling , H . 1994 . Cyclotella agassizensis nov. sp. and its relationship to C. quillensis Bailey and other prairie Cyclotella species . Diatom Res. , 9 : 289 – 301 .
- Hoops , HJ and Floyd , GL . 1979 . Ultrastructure of the centric diatom Cyclotella meneghiniana: vegetative cell and auxospore development . Phycologia , 18 : 424 – 435 .
- Houk , V and Klee , R . 2004 . The stelligeroid taxa of the genus Cyclotella (Kützing) Brébisson (Bacillariophyceae) and their transfer into the new genus Discostella gen. nov . Diatom Res. , 19 : 203 – 228 .
- Iyengar , MOP and Subrahmanyan , R . 1944 . On reduction division and auxospore formation in Cyclotella meneghiniana Kütz . J. Ind. Bot. Soc. , 23 : 125 – 152 .
- John , U , Fensome , RA and Medlin , LK . 2003 . The application of a molecular clock based on molecular sequences and the fossil record to explain biogeographic distributions within the Alexandrium tamarense “species complex” (Dinophyceae) . Mol. Biol. Evol. , 20 : 1015 – 1027 .
- John , U , Groben , R , Beszteri , B and Medlin , L . 2004 . Utility of Amplified Fragment Length Polymorphisms (AFLP) to analyse genetic structures within the Alexandrium tamarense species complex . Protist , 155 : 169 – 179 .
- Knowlton , N . 1993 . Sibling species in the sea . Ann. Review Ecol. Syst. , 24 : 189 – 216 .
- Knowlton , N . 2000 . Molecular genetic analyses of species boundaries in the sea . Hydrobiologia , 420 : 73 – 90 .
- Latala , A and Surosz , W . 1999 . Growth of four planktonic algae from brackish water in the presence of heavy metals . Polish Archives of Hydrobiology , 46 : 131 – 154 .
- Lewandowska , J and Kosakowska , A . 2004 . Effect of iron limitation on cells of the diatom Cyclotella meneghiniana Kützing . Oceanologia , 46 : 269 – 287 .
- Lohr , M and Wilhelm , C . 2001 . Xanthophyll synthesis in diatoms: quantification of putative intermediates and comparison of pigment conversion kinetics with rate constants derived from a model . Planta , 212 : 382 – 391 .
- Mann , DG . 1989 . The species concept in diatoms: evidence for morphologically distinct, sympatric gamodemes in four epipelic species . Pl. Syst. Evol. , 164 : 215 – 237 .
- Mann , DG . 1999 . The species concept in diatoms . Phycologia , 38 : 437 – 495 .
- Medlin , LK , Elwood , HJ , Stickel , S and Sogin , ML . 1991 . Morphological and genetic variation within the diatom Skeletonema costatum (Bacillariophyceae): Evidence for a new species, Skeletonema pseudocostatum . J. Phycol. , 27 : 514 – 524 .
- Mes , THM . 1998 . Character compatibility of molecular markers to distinguish asexual and sexual reproduction . Mol. Ecol. , 7 : 1719 – 1727 .
- Posada , D . 1998 . Modeltest 3.06. , Provo, , USA : Department of Zoology, Brigham Young University .
- Rao , VNR . 1970 . Studies on Cyclotella meneghiniana Kütz. I. Sexual reproduction and auxospore formation . Proc. Ind. Acad. Sci. , 72 : 285 – 287 .
- Rao , VNR . 1971 . Studies on Cyclotella meneghiniana Kütz. II. Induction of auxospore formation . Phykos , 10 : 84 – 98 .
- Rao , VNR . 1996 . Size dependent reproductive behaviour in Cyclotella meneghiniana (Bacillariophyta) . Nova Hedw. Beih. , 112 : 235 – 238 .
- Round , FE , Crawford , RM and Mann , DG . 1990 . The Diatoms: Biology and Morphology of the Genera , Cambridge, UK : Cambridge University Press .
- Rynearson , TA and Armbrust , EV . 2004 . Genetic differentiation among populations of the planktonic marine diatom Ditylum brightwellii (Bacillariophyceae) . J. Phycol. , 40 : 34 – 43 .
- Sarno , D , Kooistra , WHCF , Medlin , LK , Percopo , I and Zingone , A . 2005 . Diversity in the genus Skeletonema (Bacillariophyceae). II. An assessment of the taxonomy of S. costatum-like species with the description of four new species . J. Phycol. , 41 : 151 – 176 .
- Schoeman , FR and Archibald , REM . 1980 . The diatom flora of Southern Africa. National Institute for Water Research, Council for Scientific and Industrial Research, Special Report 34
- Scholin , C , Herzog , M , Sogin , M and Anderson , D . 1994 . Identification of group- and strain-specific genetic markers for globally distributed Alexandrium (Dinophyceae). 2. Sequence analysis of a fragment of the LSU rRNA gene . J. Phycol. , 30 : 999 – 1011 .
- Schultz , ME . 1971 . Salinity-related polymorphism in the brackish-water diatom Cyclotella cryptica . Can. J. Bot. , 49 : 1285 – 1289 .
- Schultz , ME and Trainor , FR . 1968 . Production of male gametes and auxospores in the centric diatoms Cyclotella meneghiniana and C. cryptica . J. Phycol. , 4 : 85 – 88 .
- Stoermer , EF and Smol , JF . 1999 . The Diatoms. Applications for the Environmental and Earth Sciences , Königstein, , Germany : Koeltz Scientific Books .
- Swofford , DL . 1998 . PAUP*. Phylogenetic Analysis Using Parsimony (* and other methods). Version 4.0b10 , Massachusetts, , USA : Sinauer Associates Sunderland .
- Tedrow , O , Julius , ML and Schoenfuss , HL . 2002 . The effects of biogenically active compounds on Cyclotella meneghiniana (Bacillariophyta) . J. Phycol. , 38 ( S1 ) : 34 – 35 .
- Tibayrenc , M and Ayala , FJ . 2002 . The clonal theory of parasitic protozoa: 12 years on . Trends Parasitol. , 18 : 405 – 410 .
- Tuchman , ML , Theriot , E and Stoermer , EF . 1984 . Effects of low level salinity concentrations on the growth of Cyclotella meneghiniana Kütz. (Bacillariophyta) . Arch. Protistenkd. , 128 : 319 – 326 .
- Whitton , BA and Rott , E . 1996 . Use of Algae for Monitoring Rivers , Innsbruck, , Austria : Studia Student GmbH. .
- Wilkinson , M . 1995 . PICA 95: Software and Documentation, School of Biological Sciences , Bristol, UK : University of Bristol .
- Yoon , HS , Hackett , JD and Bhattacharya , D . 2002 . A single origin of the peridinin- and fucoxanthin-containing plastids in dinoflagellates through tertiary endosymbiosis . Proc. Natl. Acad. Sci. USA , 99 : 11724 – 11729 .