Abstract
Cox1 has been suggested as a barcode marker for diatoms but it has not been tested intensively in any group of closely related species outside Sellaphora. We evaluated the use of cox1, LSU and rbcL for phylogeny and identification in the taxonomically problematic but ecologically important freshwater diatom Nitzschia palea, for which LSU rDNA sequences, mating and morphological data had already been published, and for which DNA and/or clonal isolates were still available; some new isolates were added. The previous and new information concur in suggesting that N. palea is a complex of several or many species. Where cox1 sequences could be obtained, they were more variable than LSU and rbcL and discriminated between lineages that differed in their morphologies, mating compatibilities, LSU sequences or rbcL sequences. Repeated failures for some strains to recover cox1 sequences from DNA that yielded LSU and rbcL suggest that cox1 will be impractical as a universal barcode marker in diatoms until better primers are designed. LSU and rbcL, though less variable than cox1, can be sequenced reliably in N. palea and, together, seem to show sufficient discrimination to be worth further consideration for species recognition.
Introduction
Nitzschia palea (Kützing) Grunow is a widely distributed freshwater species (see references provided in Finlay et al., Citation2002), which occurs in both lentic and lotic habitats, where it is a common and often abundant component of benthic diatom communities. Some morphologies of N. palea are said to be indicators of organic pollution (e.g. Lange-Bertalot, Citation1979; Khan, Citation1990) or tolerant of heavy metal pollution (e.g. Sabater, Citation2000; Lai et al., Citation2003), whereas others seem to prefer less polluted waters (e.g. Lange-Bertalot, Citation1980; Van Dam et al., Citation1994; Potapova & Charles, Citation2007). However, the discrimination between these varieties is not always clear (Archibald, Citation1983; Krammer & Lange-Bertalot, Citation1988), making N. palea a taxonomically problematic species complex. All individuals of the species have a similar linear-lanceolate shape, with rostrate to subcapitate apices, and all lack central raphe endings, but valve dimensions and pattern densities vary considerably, e.g. valve widths of 3–5 µm and stria densities of 30–50 in 10 µm. The type material of N. palea (from Tennstädt, Germany), as well as of its var. debilis (from Falaise, France), were examined by Trobajo & Cox (Citation2006). Using culture experiments, morphometrics, partial large-subunit ribosomal (LSU r-) DNA sequences and mating experiments, Trobajo et al. (Citation2009) showed that much of the morphological variation, especially in valve width and striation density, is probably genetically based, rather than being caused by phenotypic plasticity. Together, morphometric, LSU rDNA and mating data suggested that N. palea may be a complex of more than one biological species, which cannot be easily discriminated on the basis of morphology. Indeed, the array of different morphologies in N. palea seemed to form a continuum between some with narrow, very delicately structured valves and others that are wider and more coarsely structured. This, together with the delicacy of the cell wall structure and the scarcity of morphological characters in both LM and EM, makes DNA barcoding an especially attractive option for the identification (and also the discovery) of species in the ecologically important N. palea complex and in the group to which N. palea belongs, i.e. Nitzschia sect. Lanceolatae (Krammer & Lange-Bertalot, Citation1988).
Several recent studies have examined the effectiveness of short DNA sequences for characterizing known diatom species (as DNA ‘barcodes’) and for helping the discovery of hitherto undescribed species. The regions tested include a region near the 5′ end of the mitochondrial cox1 gene, choroplast-encoded rbcL, and nuclear small-subunit (SSU) and internal transcribed spacer (ITS) rDNA (Evans et al., Citation2007; Jahn et al., Citation2007; Moniz & Kaczmarska, Citation2009, Citation2010). To evaluate how well candidate markers perform in discriminating between closely related species, Evans et al. (Citation2007) used Sellaphora as a model group, because of the abundance of information about species limits and relationships within the genus (e.g. Evans et al., Citation2008). They found that cox1 performed better than the other genes tested (SSU and ITS rDNA, rbcL) because it (1) could be amplified and directly sequenced in most cases using a single pair of primers, (2) gave unambiguous reads (reflecting the rarity of intragenomic variants) and (3), for a span of 624 base-pairs (bp), gave divergences of c. 20 bp between isolates that were thought to represent sister species. Cox1 has also been found to be an effective identifier in Pseudochattonella (Riisberg & Edvardsen, Citation2008) and Fucus (Kucera & Saunders, Citation2008), which represent two other major groups of heterokonts (dictyochophytes and brown algae, respectively), and in the red algae (e.g. Le Gall & Saunders, Citation2010). In contrast, Moniz & Kaczmarska (Citation2009, Citation2010) have argued that partial ITS rDNA is the best candidate for universal barcoding of diatoms. However, as for some other diatom genera, direct sequencing of ITS rDNA is impractical in Nitzschia palea because of intragenomic variation (Trobajo et al., Citation2009). Moniz & Kaczmarska (Citation2009), Evans et al. (Citation2007) and Jahn et al. (Citation2007) did not evaluate partial (D1–D2) LSU rDNA as a barcode marker, despite the successful use of this marker in several species-level studies of diatoms (reviewed by Mann & Evans, Citation2007; Amato et al., Citation2007), including N. palea (Trobajo et al., Citation2009).
Here we report molecular phylogenies of the N. palea complex based on cox1 and rbcL, using the same worldwide set of clones analysed for LSU rDNA by Trobajo et al. (Citation2009), together with four new isolates from Spain and Scotland. We examine the suitability of cox1, rbcL and LSU rDNA for barcoding species in the N. palea complex and put particular focus on three putative species (operational taxonomic units, OTUs) distinguished within the N. palea species complex by Trobajo et al. (Citation2009). One species (clade A) comprised a well-supported LSU rDNA clade of Japanese and Spanish strains. The other two putative species (mating groups 1 and 2) were recognized ‘biologically’, each one consisting of clones able to mate among themselves but reproductively isolated from clones belonging to the other group, and apparently also from clade A and all other clones included in the LSU rDNA analysis.
Materials and methods
New strains () were isolated and grown as described by Trobajo et al. (Citation2009) and Evans et al. (Citation2007). Among them were four new isolates of N. palea and two other species of Nitzschia sect. Lanceolatae, including N. capitellata, which is sister to N. palea (though with weak support) in trees based on LSU rDNA analyses (Trobajo et al., Citation2009). Two further Nitzschia species were added, including the type species of the genus, N. sigmoidea. Cultures were harvested and DNA extracted as described in Trobajo et al. (Citation2009). Membership of the three putative species (OTUs) recognized by Trobajo et al. (Citation2009) is specified in , as ‘N. palea LSU clade A’, ‘N. palea mating group 1’ and ‘N. palea mating group 2’.
Table 1. Isolation information for new Nitzschia strains sequenced for this study. Original strain designations correspond to the isolate names on the voucher slides held in Royal Botanic Garden of Edinburgh. See for explanation of ‘N. palea agg.’.
Table 2. GenBank accession numbers for the species and strains analysed. Accession numbers for sequences obtained for this study are shown in bold. Strain identifier refers to the names of the isolates as used in this paper. Within the Nitzschia palea species complex, clones are identified according to their membership of one of the three putative species (OTUs) recognized by Trobajo et al. (Citation2009), i.e. ‘LSU clade A’, ‘mating group 1’ and ‘mating group 2’, or, for clones not belonging to any of these groups, they are referred to as ‘N. palea agg.’.
Cox1
Cox1 amplifications were performed using the primers GazF2 (Saunders, Citation2005) and GazR2 (Lane et al., Citation2007). The PCR reaction volumes were 30 µl and contained 1.5 µl of DNA template, 1 × NH4 buffer, 2.5 mM MgCl2, 200 µM of dNTPs, 0.3 µM of each primer and 0.75 units of Taq (Bioline, London, UK). PCR conditions were one cycle of 3 min at 95°C; 35 cycles of 95°C for 30 s, 50°C for 1 min and 72°C for 1.5 min; and, finally, 72°C for 10 min. PCR products were purified and sequenced as described in Evans et al. (Citation2007). In isolates where sequencing failed, a variety of internal primers, including those detailed in Evans et al. (Citation2007) and some designed specifically for this study were used with unsuccessful results.
RbcL
The rbcL region was amplified using the primers DPrbcL1 and DPrbcL7 (Jones et al., Citation2005). The PCR reaction volumes were 50 µl and contained 2 µl of DNA template, 1 × NH4 buffer, 2.5 mM MgCl2, 200 µM of dNTPs, 0.3 µM of each primer and 1.25 units of Taq (Bioline, London, UK). PCR conditions were as described in Jones et al. (Citation2005). PCR products were purified using ExoSAP-IT (GE Healthcare Life Sciences, Little Chalfont, UK) and sequenced as described in Evans et al. (Citation2007), using the amplification primers and two internal primers (NDrbcL5 and NDrbcL1l: Evans et al., Citation2008).
LSU rDNA
LSU rDNA sequence data were from Trobajo et al. (Citation2009), who list the GenBank accessions, voucher specimens and source localities, and details for DNA extraction, amplification, etc.
Sequence alignment and phylogenetic analyses
Sequences were edited using CEQ software (Beckman Coulter) and aligned in Sequencher 4.5 (Gene Codes Coorporation, Ann Arbor, MI, USA).
The sequences analysed are listed in . Those for each gene were initially aligned using Clustal X (Thompson et al., Citation1997) and then by eye in BIOEDIT (Hall, Citation1999), both as raw nucleotides and translated into peptide sequences. Uncorrected p-distances were calculated using PAUP* (version 4.0b.10) (Swofford, Citation2003) by dividing the number of nucleotide differences by the total number of nucleotides compared.
Phylogenetic trees were rooted using other raphid diatoms as an outgroup (Sellaphora spp. for cox1 and Eunotia minor for rbcL). Maximum parsimony (MP), maximum likelihood (ML) and distance (NJ) analyses were performed using PAUP*. Gaps were treated as missing data and characters treated as multistate and unordered in all analyses. Parsimony and maximum likelihood analyses were performed using heuristic searches with 100 random addition replicates and a branch-swapping algorithm (tree–bisection–reconnection, TBR). The optimal models of sequence evolution (General Time Reversible with a proportion of invariable sites and a gamma distribution with four rate categories on the variable sites for rbcL ; and Tamura–Nei with a proportion of invariable sites and a gamma distribution for cox1) were selected according to the Akaike Information Criterion (Posada & Buckley, Citation2004) in Modeltest Version 3.06 (Posada & Crandall, Citation1998). Neighbour-joining analyses were performed (Saitou & Nei, Citation1987), using the same model as for ML (Hillis et al., Citation1996). Bootstrapping (Felsenstein, Citation1985) was performed using 1000 replicates for the MP and distance analyses and 100 for the ML.
For Bayesian analyses, the posterior probability distribution was estimated using Metropolis-Coupled Markov Chain Monte Carlo as implemented in MrBayes (ver. 3.1.2: Ronquist & Huelsenbeck, Citation2003). The GTR (6 substitution types) model for DNA sequence evolution was used. Rate heterogeneity among sites was modelled with a gamma distribution, estimated with four discrete rate categories, and a parameter for the proportion of invariant sites. The default priors from MrBayes were used (with flat dirichlet prior settings for base frequencies and substitution rate matrices and uniform prior settings for rate parameters). We performed two independent runs for 1 000 000 generations per run, sampling every 100 generations. Each run used four chains, one cold and three heated, with heating temperature adjusted to 0.1 to facilitate mixing of the chains. The first 100 000 generations were sampled before the chain reached stationary phase (‘burn-in’) and were discarded for the calculation of the 50% majority-rule consensus tree and posterior probabilities (PP), which were based on pooled samples from stationary phase of the two runs.
We made a combined alignment containing 17 Nitzschia palea clones, for which we had both LSU and cox1 sequences, together with two chimera sequences obtained from the combination of the LSU and cox1 GenBank sequences of Nitzschia frustulum (LSU: AF417671; cox1: AB020225), and Cylindrotheca closterium (LSU: AF417666; cox1: AB020222). To test for data set congruence, we used the partition homogeneity test executable in PAUP (this test is equivalent to the incongruence length difference test of Farris et al. (Citation1995); for convenience, it will be referred to as the ILD in this paper). We conducted 2000 replicates of the ILD test using simple sequence addition and TBR. Parsimony-uninformative characters were removed before running the test. No tests of incongruence were made between the LSU rDNA or cox1 and rbcL data sets because of the much smaller size of the rbcL set.
Results
Cox1 was sequenced for 22 N. palea isolates (). Among these were 18 of the 25 clones isolated by Trobajo et al. (Citation2009) for which they obtained LSU rDNA sequences, and four new clones from Catalonia (New Spain1 and New Spain2) and Scotland (New Scot1 and New Scot2). For six further clones out of those examined by Trobajo et al., namely Belgium1–4, Spain A4 or Spain A1, repeated attempts, using different primers, failed to recover cox1. The Belgium1–4 and Spain A4 failures were particularly unfortunate because of the information about relationships within N. palea revealed by these clones from mating and molecular (LSU) data (Trobajo et al., Citation2009). A further attempt to obtain sequences for these clones, by using the degenerate primers of Ehara et al. (Citation2000) to amplify the whole cox1 gene, followed by cloning, also failed (Dr M. Hollingsworth, personal communication).
The lengths of the cox1 sequences recovered were 430–665 bp, the shortest being from Spain A2 and Japan A1 (430 and 486 bp, at the 5′ end of the amplified region, respectively), which were not included in the phylogenetic analyses. Most N. palea sequences were >600 bp and were readily aligned, with no indels. Nitzschia capitellata Spain yielded an even shorter sequence than any of the N. palea isolates (305 bp, at the 3′ end of the amplified region) and was therefore omitted from the phylogenetic analysis. However, five other Bacillariaceae were included, comprising three new sequences and two from GenBank (). Two Sellaphora species were added, as a more distant outgroup of raphid diatoms. In order to keep the maximum number of clones in the analysis without reducing too much the lengths of the sequences over which comparisons were made, we truncated the alignment at the 5′ and 3′ ends to 495 bp and this was the basis for all our analyses.
Over the truncated cox1 alignment, three pairs of isolates were identical: Spain A5–Japan B1, India–Sri Lanka2, and New Spain1–New Spain2. Divergences among other N. palea isolates ranged up to 65 bp (13%), for New Scot2 vs UK ().
Table 3. p distances (right-top) and bp differences (left-bottom) between N. palea cox1 sequences (alignment of 495 bp). Clades with good support or identical sequences are outlined by a solid line. Shaded boxes: p distance and bp difference for the maximally different N. palea clones (UK vs New Scot2).
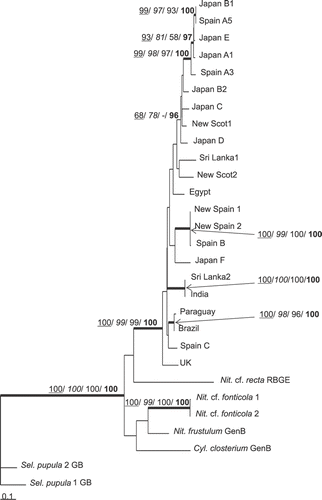
In the cox1 trees (), N. palea strains formed a well supported clade in which many terminals lay on separate nonzero-length branches, reflecting the possession by each of autapomorphic sites. Few nodes of the cox1 trees were well supported with all types of analysis (MP, ML, NJ, Bayesian), the few being a clade containing Japan B1, Japan E, Japan A1, Spain A3 and Spain A5; a clade containing Spain B, New Spain1 and New Spain2; and a clade containing Paraguay and Brazil. A clade containing Spain C, as well as Paraguay and Brazil, was recovered by all kinds of analysis using the unpartitioned nucleotide matrix, but received low support. However, this clade did receive rather good support (0.98 and 0.96 posterior probabilities) in the three Bayesian analyses using codon-partitioned models (not shown). The two new N. palea strains from Scotland (New Scot1 and New Scot2) were distant from any other strains and from each other (, ).
Fig. 1. ML tree based on cox1 gene, illustrating the phylogeny of N. palea sequences. The tree was rooted using two Sellaphora pupula sequences. Maximum parsimony, neighbour joining and maximum likelihood bootstrap values above 50% and BI posterior probabilities above 95% (MP/ NJ/ ML/ BI) are shown on the branches. Internal nodes supported by bootstrap values above 80% and posterior probabilities above 95% in at least two types of analyses are highlighted as thick lines.
To check the quality of the material of the clones that failed to yield cox1 sequences and to further explore relationships among N. palea, rbcL was amplified and sequenced from 13 isolates of N. palea and three of N. capitellata (). These sequences, together with six further representatives of the Bacillariaceae and four other raphid diatoms, were used for the rbcL phylogenetic analyses.
The lengths of the new rbcL sequences recovered varied from 1355 to 1483 bp, sometimes including the region beyond the stop codon. The sequence for Japan B1 contained a nine-nucleotide (three-codon) insertion, which was removed from the alignment before analysis, as were four further positions where the amino acid sequence indicated likely errors in some sequences. After removing these positions, as well as two fragments at 5′ and 3′ ends to avoid gaps resulting from use of some short GenBank sequences, the truncated alignment used in the analyses was 1334 bp long. Among N. palea strains, the four Belgium clones were identical for the alignment, as were the pairs Paraguay–Brazil and Spain A5–Japan B1 (). Several well-supported clades were also found: N. capitellata–N. palea; all N. capitellata isolates; all N. palea isolates; and Spain C–Spain A4–Paraguay–Brazil (). Among the three clones Spain C, Paraguay and Brazil, the maximum divergence was 8 bp (0.6%) (), whereas it was 46 bp (6%) with cox1. The group of Japan B1, Japan E, Japan A1, Spain A3 and Spain A5, which comprised a well-supported clade in cox1 analyses, formed a paraphyletic group (ML, BI topologies) or weakly supported clade (MP, NJ topologies) in rbcL analyses ().
Fig. 2. ML tree based on rbcL gene, illustrating the phylogeny of N. palea sequences. The tree was rooted using Eunotia minor. Maximum parsimony, neighbour joining and maximum likelihood bootstrap values above 50% and BI posterior probabilities above 95% (MP/ NJ/ ML/ BI) are shown on the branches. Internal nodes supported by bootstrap values above 80% and posterior probabilities above 95% in at least two types of analyses are highlighted as thick lines.
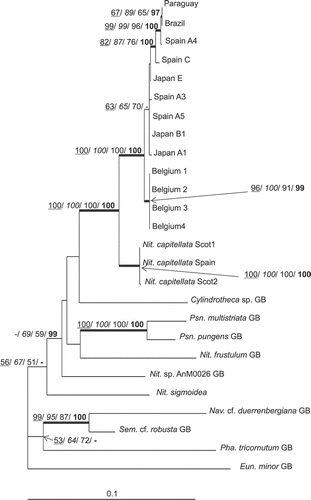
Table 4. p distances (right-top) and bp differences (left-bottom) between N. palea rbcL sequences (alignment of 1334 bp). Two clades with moderate to good support are outlined by solid lines, the remaining isolates, which formed a paraphyletic group, by a dashed line.
The ILD test suggested no critical significant topological incongruence between LSU and cox1 data sets (P = 0.248).
Discussion
Nitzschia palea genetic diversity as revealed by three molecular markers
In groups of organisms where species relationships and boundaries are already firmly established from many independent data, evaluation of identification aids can be straightforward, based on how well and how easily each aid discriminates between existing taxa. Few diatoms currently allow such testing, apart from Pseudo-nitzschia (Amato et al., Citation2007) and Sellaphora (Evans et al., Citation2007), because of a general lack of corroboration of species boundaries established from a very limited range of morphological characters. Therefore, before evaluating different genes as barcode markers in N. palea, it is important to check whether data from these genes conflict with the results of our previous study of N. palea, which led (on the basis of some correspondence between morphometrics, mating experiments and LSU rDNA sequence variation) to the delineation of three putative species and a number of molecular lineages represented by only one or two clones. Within N. palea, the relationships indicated by cox1 analyses are broadly consistent with those revealed by LSU (Trobajo et al., Citation2009). When rooted by other Bacillariaceae and more distantly related pennate diatoms, N. palea isolates form asymmetrical raked trees with both genes. The tree topologies do not differ significantly and there is good support from both cox1 and LSU for a clade, called ‘clade A’ by Trobajo et al. (Citation2009), comprising five clones from Japan and Spain (JapanA1, B1 and E; SpainA3 and A5). The Paraguay and Brazil clones, and also SriLanka2 and India, are identical or almost identical in both LSU and cox1 (5 bp difference between Paraguay and Brazil in cox1), and Japan B1 and Spain A5 are identical not only in cox1 and LSU but also in rbcL. These data illustrate wide dispersal of organisms that have diverged little with respect to fast-evolving genes (i.e. genes that evolve sufficiently rapidly to be commonly used in studies of species-level relationships or as barcode markers).
In LSU trees (Trobajo et al., Citation2009), Spain B did not group with any other clone and lay on a separate branch. In contrast, with cox1 Spain B is very similar to New Spain1 and New Spain2 (2 bp difference) and forms a well-supported clade with them. Interestingly, all three of these came from the same catchment (the Ter River, Catalonia, NE Spain) but were isolated at different times from different locations.
Although the relationships among N. palea clones indicated by LSU and cox1 data are not dissimilar, the sequence divergences differ markedly. For example, for Paraguay vs Japan B1 or vs Spain A5, the divergences are 1.2% for LSU, but 10.7% for cox1. Furthermore, for some pairs of strains, cox1 revealed differences where LSU did not, such as between Paraguay and Brazil (1.0% different in cox1) or between Spain A3 and Spain A5 or Japan B1 (4.6% different in cox1). Percentage sequence divergences in rbcL were even lower than in LSU, e.g. 0.8% for Paraguay vs Japan B1 or vs Spain A5, but absolute numbers of bp differences were similar because of the greater length of the rbcL alignment.
Comparisons with other diatom species complexes
For sets of Sellaphora clones determined (from morphology and mating experiments) to belong to the same species, intra-species cox1 divergences ranged from 0–0.8% (Evans et al., Citation2007, : S. capitata, S. bacillum and S. blackfordensis). Between pairs of Sellaphora isolates that might represent sister species (from mating and molecular data), divergences were 2.9–3.7% (S. capitata–S. pupula “afro”, S. blackfordensis–S. pupula “pseudocapitate”, and S. bisexualis–S. pupula “upland elliptical”, Evans et al., Citation2007, with taxon names updated in Mann et al., Citation2008, Citation2009). Hence, the 5 bp (1%) cox1 difference between Paraguay and Brazil (which are sexually compatible: Trobajo et al., Citation2009) is close to the range of intra-species differences expected in N. palea if Sellaphora is a good model for other raphid diatoms. On the other hand, most of the divergences between clones of N. palea exceed the minimal inter-species divergences in Sellaphora, which is consistent with our previous suggestion (Trobajo et al., Citation2009) that N. palea may be a complex of several species. Moreover, among the clade A clones, Spain A3 differs more from the others in cox1 (JapanA1, B1 and F, and Spain A5) than sister species in Sellaphora (4.6% vs 2.9–3.7%), which could indicate even further taxon differentiation within N. palea than was shown by the LSU data. For comparison, intraspecies cox1 divergences are generally only 0–0.3% in animals, red algae and brown algae (Kucera & Saunders, Citation2008, p. 1070).
However, rbcL data indicate that extrapolation from Sellaphora and interpretation of divergences is not straightforward. In Sellaphora, the 5′ region of cox1 selected as a candidate barcode region evolves faster than rbcL, as in N. palea, but the relative rates differ: divergences of cox1 are around ten times greater than those of rbcL in N. palea (see above, ‘Nitzschia palea genetic diversity as revealed by three molecular markers’, and and ), but only around five times greater in Sellaphora (Evans et al., Citation2007, fig. 3). Further, rbcL differences for N. palea groups known to be reproductively isolated are at most 1% (e.g. between Belgium1–4 and Paraguay–Brazil–Spain A4: ), which is less than the minimal inter-species difference found in Sellaphora (1.6% between S. blackfordensis and S. pupula ‘pseudocapitate’). Thus the absolute rates of evolution of one or both genes (rbcL and cox1) must differ between Nitzschia palea and Sellaphora.
Amato et al. (Citation2007) examined diversity and relationships among seven Pseudo-nitzschia species, using morphological data, mating experiments, and four molecular markers (rbcL, partial LSU, and ITS1 and ITS2). P-distances were not given, but measurements for closest relatives in ML trees (Amato et al., Citation2007, , ) indicate that rbcL does not evolve more slowly than LSU in Pseudo-nitzschia, in contrast to N. palea. Amato et al. also showed that rbcL discriminated between two species (P. cuspidata and P. pseudodelicatissima) that have identical LSU sequences.
Diatom barcoding
Cox1 not only discriminated between all of the putative Sellaphora species present in the work of Evans et al. (Citation2007), but was also successfully amplified and sequenced, and gave unambiguous reads in almost all of the 33 clones studied. Therefore, cox1 was suggested as a strong candidate for diatom barcoding. In N. palea, cox1 seems to discriminate well among lineages, but in a sizeable minority of clones, including some key strains (e.g. Belgium1–4 and Spain A4) for which there is information about reproductive biology, cox1 completely failed to amplify or gave short reads. This mirrors difficulties in obtaining cox1 sequences in other pennate and centric diatoms (Evans et al., Citation2007; Moniz & Kaczmarska, Citation2009) and reduces the number of comparisons that can be made with other potential barcode markers in N. palea. In , we summarize comparisons of intra- and interspecific divergence for the three putative species delineated by Trobajo et al. (Citation2009) in the N. palea complex, both in relation to each other and in relation to the remaining clones studied by Trobajo et al. In addition, we offer two circumscriptions of ‘mating group 2’, one including and one excluding Spain C, because of the close relationship between Spain C and the Group 2 clones (Paraguay, Brazil and Spain A4) indicated by rbcL data. It should be noted, however, that inclusion of Spain C with Group 2, though phylogenetically consistent with the rbcL data (), receives no significant support from the cox1 () or LSU rDNA data and is inconsistent with the reproductive isolation apparent between Spain C and the other three clones in our earlier study (Trobajo et al., Citation2009). For all three genes (cox1, LSU rDNA and rbcL), intraspecific divergences are no greater than interspecific divergences and could allow any of these regions to be used as barcode markers, providing that Spain C is not treated as conspecific with the Group 2 clones (Paraguay, Brazil and Spain A4). However, none of the genes studied can yet be claimed to be ideal, because there is no clear evidence of a barcode gap (a discontinuity between the levels of intraspecific and interspecific divergence such as was found in the red algae studied by Le Gall & Saunders, Citation2010), only a threshold.
Table 5. Percentage divergence of LSU (alignment of 498 bp), cox1 (alignment of 495 bp) and rbcL (alignment of 1334 bp) among Nitzschia palea isolates. Cells filled in grey indicate intra-OTU divergences.
ITS-2 rDNA is another region that has proved effective for the recognition and identification of diatom species (e.g. Amato et al., Citation2007; Vanormelingen et al., Citation2007; Moniz & Kaczmarska, Citation2009, Citation2010). However, because divergent copies of ITS can be present and abundant within single individuals, sequencing sometimes requires prior cloning, for example in N. palea (Trobajo et al., Citation2009) and at least in some species of Achnanthes (E. Clavero, unpublished data), Eunotia (Vanormelingen et al., Citation2007), Navicula (A. Poulíčková, personal communication), Pseudo-nitzschia (D’Alelio et al., Citation2009) and Sellaphora (Behnke et al., Citation2004). For such taxa the effect is that direct sequencing currently yields poor quality or unreadable sequences, which will undermine the use of ITS regions for barcoding until the predominant copy can be reliably and easily sequenced without cloning. Riisberg & Edvardsen (Citation2008) also found ITS to be less suitable than other markers in the dictyochophyte Pseudochattonella, both because of intragenomic variation (making cloning essential) and because ITS sequences from P. farcimen Eikrem, Edvardsen & Throndsen and P. verruculosa (Y. Hara & Chihara) Eikrem did not assort according to species, nor even according to individual isolate. In contrast, although LSU (Beszteri et al., Citation2005) and even SSU rDNA (Alverson & Kolnick, Citation2005) can also display intragenomic variation in diatoms, this seems rarely to cause any practical difficulties for direct sequencing (e.g. Ellegaard et al., Citation2008).
LSU and rbcL are less variable than cox1 within the N. palea complex and seem to be imperfect for species discovery in this group, because of the apparent persistence of ancestral sequences and the regular absence of reciprocal monophyly (though these may be an artefact of limited sampling). However, these genes show some discrimination of putative species, here and elsewhere (e.g. Amato et al., Citation2007; Cerino et al., Citation2005; Evans et al., Citation2007, Citation2008), especially if taken together, and they deserve further consideration as molecular identifiers (barcodes), although rbcL, if used in its entirety, is over-long (c. 1400 bp) for a conventional barcode. Furthermore, LSU and rbcL were easily amplified and sequenced in all N. palea tested, using currently available primers, and previous studies indicate that this is true for other diatoms (references in Mann & Evans, Citation2007). Cox1 may currently seem impractical as a universal barcode marker in diatoms, because of the amplification and sequencing failures, which may be caused by introns of the kind detected by Ehara et al. (Citation2000). However, its virtues (high signal for a short c. 600 bp sequence, low or no intra-genomic variation, ability to discriminate between closely related species) argue that cox1 deserves further efforts to develop better primers, to provide the universality that any barcode must have.
Acknowledgements
This study was supported by an EU Framework 6 Synthesys award to Rosa Trobajo. We are very grateful to Dr Michelle Hollingsworth (RBGE) for her attempts, unfortunately unsuccessful, to clone and sequence the whole cox1 gene, which we hoped might reveal why our PCR primers failed for some N. palea isolates. We are grateful to two anonymous reviewers, whose constructive comments helped us to improve this manuscript.
Related Research Data
References
- Alverson , AJ and Kolnick , L . 2005 . Intragenomic nucleotide polymorphism among small subunit (18S) rDNA paralogs in the diatom genus Skeletonema (Bacillariophyta) . J. Phycol. , 41 : 1248 – 1257 .
- Amato , A , Kooistra , WHCF , Levialdi Ghiron , JH , Mann , DG , Pröschold , T and Montresor , M . 2007 . Reproductive isolation among sympatric cryptic species in marine diatoms . Protist , 158 : 193 – 207 .
- Archibald , REM . 1983 . The diatoms of the Sundays and Great Fish Rivers in the Easter Cape Province of South Africa . Biblioth. Diatomol. , 1 : 1 – 362 .
- Behnke , A , Friedl , T , Chepurnov , VA and Mann , DG . 2004 . Reproductive compatibility and rDNA sequence analyses in the Sellaphora pupula species complex (Bacillariophyta) . J. Phycol. , 40 : 193 – 208 .
- Beszteri , B , Acs , E and Medlin , LK . 2005 . Ribosomal DNA sequence variation among sympatric strains of the Cyclotella meneghiniana complex (Bacillariophyceae) reveals cryptic diversity . Protist , 156 : 317 – 333 .
- Cerino , F , Orsini , L , Sarno , D , Dell’Aversano , C , Tartaglione , L and Zingone , A . 2005 . The alternation of different morphotypes in the seasonal cycle of the toxic diatom Pseudo-nitzschia galaxiae . Harmful Algae , 4 : 33 – 48 .
- D’Alelio , D , Amato , A , Kooistra , WHCF , Procaccini , G , Casotti , R and Montresor , M . 2009 . Internal transcribed spacer polymorphism in Pseudo-nitzschia multistriata (Bacillariophyceae) in the Gulf of Naples: recent divergence or intraspecific hybridization? . Protist , 160 : 9 – 20 .
- Ehara , M , Inagaki , Y , Watanabe , KI and Ohama , T . 2000 . Phylogenetic analysis of diatom cox1 genes and implications of a fluctuating GC content on mitochondrial genetic code evolution . Curr. Genet. , 37 : 29 – 33 .
- Ellegaard , M , Godhe , A , Härnström , K and McQuoid , M . 2008 . The species concept in a marine diatom: LSU rDNA-based phylogenetic differentiation in Skeletonema marinoi/dohrnii (Bacillariophyceae) is not reflected in morphology . Phycologia , 47 : 156 – 167 .
- Evans , KM , Wortley , AH and Mann , DG . 2007 . An assessment of potential diatom “barcode” genes (cox1, rbcL, 18S and ITS rDNA) and their effectiveness in determining relationships in Sellaphora (Bacillariophyta) . Protist , 158 : 349 – 364 .
- Evans , KM , Wortley , AH , Simpson , GE , Chepurnov , VA and Mann , DG . 2008 . A molecular systematic approach to explore diversity within the Sellaphora pupula species complex (Bacillariophyta) . J. Phycol. , 44 : 215 – 231 .
- Farris , JS , Källersjö , M , Kluge , AG and Bult , C . 1995 . Constructing a significance test for incongruence . Syst. Biol. , 44 : 570 – 572 .
- Felsenstein , J . 1985 . Confidence limits on phylogenies: an approach using the bootstrap . Evolution , 39 : 783 – 791 .
- Finlay , BJ , Monaghan , EB and Maberly , SC . 2002 . Hypothesis: the rate and scale of dispersal of freshwater diatom species is a function of their global abundance . Protist , 153 : 261 – 273 .
- Hall , TA . 1999 . BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT . Nucl. Acids Symp. Ser. , 41 : 95 – 98 .
- Hillis , DM , Mable , BK and Moritz , C . 1996 . “ Applications of molecular systematics ” . In Molecular Systematics , Edited by: Hillis , DM , Moritz , C and Mable , BK . 515 – 544 . Sunderland, MA : Sinauer Associates .
- JAHN, R., ZETZSCHE, H., REINHARDT, R. & GEMEINHOLZER, B. (2007). Diatoms and DNA barcoding: a pilot study on an environmental sample. In Proceedings of the 1st Central European Diatom Meeting (Kusber, H.-W. & Jahn, R., editors), 63–68. Botanic Garden and Botanical Museum Berlin-Dahlem (FU-Berlin), Germany [doi: 10.3372/cediatom.113]
- Jones , HM , Simpson , GE , Stickle , AJ and Mann , DG . 2005 . Life history and systematics of Petroneis (Bacillariophyta), with special reference to British waters . Eur. J. Phycol. , 40 : 43 – 71 .
- Khan , I . 1990 . Assessment of water-pollution using diatom community structure and species distribution – a case-study in a tropical river basin . Int. Rev. Gesamten Hydrobiol. , 75 : 317 – 338 .
- KRAMMER, K. & LANGE-BERTALOT, H. (1988). Bacillariophyceae. 2. Teil: Bacillariaceae, Epithemiaceae, Surirellaceae. In Süsswasserflora von Mitteleuropa (Ettl, H., Gerloff, J., Heynig, H. & Mollenhauer, D., editors), Vol. 2/2. G. Fischer, Stuttgart, Germany
- Kucera , H and Saunders , GW . 2008 . Assigning morphological variants of Fucus (Fucales, Phaeophyceae) in Canadian waters to recognized species using DNA barcoding . Botany (Botanique) , 86 : 1065 – 1079 .
- Lai , SD , Chen , PC and Hsu , HK . 2003 . Benthic algae as monitors of heavy metals in various polluted rivers by energy dispersive X-ray spectrometer . J. Environ. Sci. Health, Part A: Toxic/Hazard. , 38 : 855 – 866 .
- Lane , CE , Lindstrom , SC and Saunders , GW . 2007 . A molecular assessment of north east Pacific Alaria species (Laminariales, Phaeophyceae) with reference to the utility of DNA barcoding . Mol. Phylogenet. Evol. , 44 : 634 – 648 .
- Lange-Bertalot , H . 1979 . Toleranzgrenzen und Populationsdynamik benthischer Diatomeen bei unterschiedlich starker Abwasserbelastung . Arch. Hydrobiol., Suppl. , 56 : 184 – 219 .
- Lange-Bertalot , H . 1980 . New species, combinations and synonyms in the genus Nitzschia . Bacillaria , 3 : 41 – 77 .
- Le Gall , L and Saunders , GW . 2010 . DNA barcoding is a powerful tool to uncover algal diversity: a case study of the Phyllophoraceae (Gigartinales, Rhodophyta) in the Canadian flora . J. Phycol. , 46 : 374 – 389 .
- Mann , DG and Evans , KM . 2007 . “ Molecular genetics and the neglected art of diatomics ” . In Unravelling the Algae – The Past, Present and Future of Algal Systematics , Edited by: Brodie , J and Lewis , J . 231 – 265 . Boca Raton, Florida : CRC Press .
- Mann , DG , Evans , KM , Chepurnov , VA and Nagai , S . 2009 . Morphology and formal description of Sellaphora bisexualis, sp. nov. (Bacillariophyta) . Fottea , 9 : 199 – 209 .
- Mann , DG , Thomas , SJ and Evans , KM . 2008 . Revision of the diatom genus Sellaphora: a first account of the larger species in the British Isles . Fottea , 8 : 15 – 78 .
- Moniz , MBJ and Kaczmarska , I . 2009 . Barcoding diatoms: is there a good marker? . Mol. Ecol. Res. , 9 ( Suppl. 1 ) : 65 – 74 .
- Moniz , MBJ and Kaczmarska , I . 2010 . Barcoding of diatoms: nuclear encoded ITS revisited . Protist , 161 : 7 – 34 .
- Posada , D and Buckley , TR . 2004 . Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests . Syst. Biol. , 53 : 793 – 808 .
- Posada , D and Crandall , KA . 1998 . Modeltest: testing the model of DNA substitution . Bioinformatics , 14 : 817 – 818 .
- Potapova , M and Charles , DF . 2007 . Diatom metrics for monitoring eutrophication in rivers of the United States . Ecol. Indic. , 7 : 48 – 70 .
- Riisberg , I and Edvardsen , B . 2008 . Genetic variation in bloom-forming ichthyotoxic Pseudochattonella species (Dictyochophyceae, Heterokonta) using nuclear, mitochondrial and plastid DNA sequence data . Eur. J. Phycol. , 43 : 413 – 422 .
- Ronquist , F and Huelsenbeck , JP . 2003 . MRBAYES 3: Bayesian phylogenetic inference under mixed models . Bioinformatics , 19 : 1572 – 1574 .
- Sabater , S . 2000 . Diatom communities as indicators of environmental stress in the Guadiamar River, S-W. Spain, following a major mine tailings spill . J. Appl. Phycol. , 12 : 113 – 124 .
- Saitou , N and Nei , M . 1987 . The neighbor-joining method: a new method for reconstructing phylogenetic trees . Mol. Biol. Evol. , 4 : 406 – 425 .
- Saunders , GW . 2005 . Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future applications . Phil. Trans. R. Soc. , B 360 : 1879 – 1888 .
- Swofford , DL . 2003 . PAUP* Phylogenetic Analysis Using Parsimony (*and Other Methods) , Sunderland, MA : Sinauer Associates .
- Thompson , JD , Gibson , TJ , Plewniak , F , Jeanmougin , F and Higgins , DG . 1997 . The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools . Nucl. Acids Res. , 24 : 4876 – 4882 .
- Trobajo , R , Clavero , E , Chepurnov , VA , Sabbe , K , Mann , DG , Ishihara , S and Cox , EJ . 2009 . Morphological, genetic and mating diversity within the widespread bioindicator Nitzschia palea (Bacillariophyceae) . Phycologia , 48 : 443 – 459 .
- Trobajo , R and Cox , EJ . 2006 . “ Examination of type material of Nitzschia frustulum, N. palea and N. palea var. debilis ” . In Proceedings of the 18th International Diatom Symposium , Edited by: Witkowski , A . 431 – 445 . Bristol, UK : Biopress .
- Van Dam , H , Mertens , A and Sinkeldam , J . 1994 . A coded checklist and ecological indicator values of freshwater diatoms from the Netherlands . Neth. J. Aquatic. Ecol. , 28 : 117 – 133 .
- Vanormelingen , P , Chepurnov , VA , Mann , DG , Cousin , S , Sabbe , K and Vyverman , W . 2007 . Congruence of morphological, reproductive and ITS rDNA sequence data in a member of the Eunotia bilunaris (Bacillariophyta) species complex from New Zealand and Tasmania . Eur. J. Phycol. , 42 : 61 – 79 .