
ABSTRACT
Bacteria associated with microalgae strongly affect algal biomass and derived product yield and quality. Nevertheless, only a few studies have addressed the detailed phylogenetic characterization of bacterial communities associated with microalgae. In this study, the phycospheric bacterial communities associated with different Tetraselmis suecica F&M-M33 cultures, a green marine microalga with several industrial applications, were analysed using a metagenomic approach. The T. suecica F&M-M33 cultures used originated from the same ancestral microalgal non-axenic culture but were physically and geographically separated for years and maintained under different growing conditions. Despite the different history of the cultures, a ‘core’ bacterial community was identified, accounting for 70% of the total bacterial community and formed by at least 13 families. Among the ‘core’ operational taxonomic units (OTUs), 24 different genera were identified. Nevertheless, there was a high variability in the relative proportions of the taxa forming the ‘core’ community, indicating that the growing conditions and/or external contamination influence the relative abundance of these microorganisms. Our study allowed the identification of persistent taxa that may be used to deepen the knowledge of the complex relationship between T. suecica and its associated bacteria.
Introduction
The ‘phycosphere’, a name analogous to the more commonly-known ‘rhizosphere’, is a zone rich in nutrients that surrounds microalgal cells (Bell & Mitchell, Citation1972). During growth, microalgae produce and release several molecules (exudates) such as amino acids, peptides and sugars (Grossart & Simon, Citation2007; Thornton, Citation2014), which are utilized by bacteria. It has been observed that nutrient availability plays a key role in determining the relationship between algae and bacteria. Bacteria may either stimulate or inhibit algal growth, and their interactions may range from parasitism to mutualism (Ramanan et al., Citation2016). In this way, complex interactions are established between bacteria and microalgae (Segev et al., Citation2016).
Recently, several studies have attempted to describe the bacterial communities associated with different algal species (Hold et al., Citation2001; Green et al., Citation2004; Nicolas et al., Citation2004; Makridis et al., Citation2006; Sapp et al., Citation2007; Lakaniemi et al., Citation2012; Le Chevanton et al., Citation2013). However, the factors driving the association mechanisms between bacteria and algae remain unclear, and there are more questions than answers (Ramanan et al., Citation2016).
In addition to its importance in basic research, the study of microalgal bacterial communities may also furnish new knowledge for microalgae biotechnological applications. Metagenomic studies have begun to describe the microbial communities associated with microalgae (Wirth et al., Citation2015; Krohn-Molt et al., Citation2017; Sambles et al., Citation2017). Nevertheless, scant information is available on their stability and resilience. Recently, it was shown that the succession of bacteria associated with Nannochloropsis salina, grown for one month in semi-continuous cultures in an outdoor open system, was influenced not only by environmental conditions but also by the seeded bacterial community (Geng et al., Citation2016). Different microalgae select specifically their bacterial community composition; however, the level of specificity may vary (Krohn-Molt et al. Citation2017). For example, it was shown that bacterial communities associated with Chlorella are more variable than those associated with Scenedesmus and Micrasterias (Krohn-Molt et al. Citation2017). To the best of our knowledge, little information is available about the Tetraselmis suecica/bacteria relationship (Nicolas et al., Citation2004; Biondi et al., Citation2017). Tetraselmis is a green marine microalga suitable for various applications; it is used mainly as food for bivalve molluscs or larval stages of crustaceans and for the pseudo-green water technique (Tredici et al., Citation2009). Because of its high protein and polyunsaturated fatty acid content, dried T. suecica inclusion in fish diet (particularly for European sea bass) represents an attractive potential application (Cardinaletti et al., Citation2018). Tetraselmis suecica biomass cultivation has also gained attention regarding the production of biofuel, proteins and bioactive compounds such as polyunsaturated fatty acids, α-tocopherol, chlorophyll, β-carotene and polyphenols (Schwenzfeier et al., Citation2011; Bondioli et al., Citation2012; Yao et al., Citation2012; Perez-Lopez et al., Citation2014). Considering that the microbiota associated with microalgae may influence both the yield and quality of biomass, evaluating the structure and diversity of bacterial communities associated with T. suecica cultures is fundamental.
This study aimed to evaluate the structure and diversity of bacterial communities associated with T. suecica using a metagenomic approach, a useful tool to study complex microbial communities. Metagenomic analysis was applied to T. suecica F&M-M33 cultures, originating from an ancestral inoculum and sub-cultured for years under different laboratory and growing conditions, to determine the potential presence of a ‘core’ microbiota shared by all cultures.
Materials and methods
Tetraselmis suecica F&M-M33 cultures
Tetraselmis suecica F&M-M33 was obtained from the Culture Collection of Microalgae and Cyanobacteria of Fotosintetica & Microbiolgica S.r.l., a spin-off company of the University of Florence (Italy). The strain, obtained from an aquaculture facility and maintained in the collection for more than 20 years, was used as an inoculum for all cultures.
Samples LAB1 and LAB2 were collected from two laboratory flask cultures that were kept static and without CO2 bubbling at 22–25°C for a minimum of 3 months, and were periodically subcultured: LAB1 under constant illumination (80 µmol photons m–2 s–1) provided by metal halide lamps and LAB2 under diffuse natural light (light passing through the laboratory window). LAB1 and LAB2 samples were collected in the stationary phase in November 2016 and February 2017, respectively.
Sample SES was collected in September 2016 from an outdoor culture grown in a 40 l vertical GWP®-III photobioreactor (Chini Zittelli et al., Citation2013a) at the experimental facilities of Fotosintetica & Microbiologica S.r.l. in Sesto Fiorentino (Florence, Italy). The culture was mixed by air bubbling, thermo-regulated by circulating cold water in an internal heat exchanger when the temperature exceeded 28°C and CO2 was provided based on the pH values (set at 7.8–8.0). The samples were collected in the early stationary phase.
One culture of T. suecica F&M-M33 was sent to the microalgae production plant of Archimede Ricerche S.r.l. in Camporosso (Imperia, Italy) for mass production about a decade ago. Sample CAM was obtained in July 2016 from a culture grown in the laboratory at the Archimede Ricerche S.r.l. plant in a 0.5 l bubble tube and used as an inoculum for an outdoor culture in a GWP®-I photobioreactor (Rodolfi et al., Citation2009) kept in a greenhouse. The culture was grown under continuous illumination (150 µmol photons m–2 s–1) at 25–27°C. CO2 was provided as needed, in a mixture with air, based on the set pH. The sample was collected at the end of the active growth period.
All cultures were grown in F medium (Guillard & Ryther, Citation1962), CAM and SES without vitamin addition. Artificial seawater (Adriatic Sea International, Rimini, Italy), used for medium preparation, was sterilized by autoclaving at 121°C for 20 min for LAB1 and LAB2 cultures. However, for SES and CAM cultures, the artificial seawater was filtered through 1 µm filters (Domnick Hunter, Durham, UK). Stock nutrient solutions (nitrate, phosphate, iron, micronutrients (zinc, cobalt, manganese, molybdenum, copper)) for LAB1, LAB2 and SES cultures were prepared in deionized water and autoclaved before addition to seawater, while stock nutrient solution for CAM was prepared by dissolving salts in sterile water and was not autoclaved. Vitamin solution (thiamine, biotin, B12) for LAB cultures was sterilized by filtration (0.2 μm pore size) before addition to the culture medium. After collection, the algal culture samples were stored at −80°C.
Isolation and identification of bacterial strains
Bacterial strains were isolated from T. suecica F&M-M33 cultures following the procedure described in Biondi et al. (Citation2017). Briefly, serial dilutions of the microalgal culture samples were prepared using a sterile saline solution (NaCl, 9 g l−1). Aliquots of 100 μl of each culture dilution were spread on Marine Agar (Difco Marine Broth 2216, Sparks, Maryland, USA) plates, which were incubated at 27°C. Colonies that differed morphologically (colour, shape and edge) were picked and streaked on Marine Agar plates (Laboratorios CONDA, Madrid, Spain). The strains were then identified by 16S rDNA sequencing. One millilitre of a liquid culture, grown overnight in Marine Broth (Laboratorios CONDA) at 27°C under 200 rpm agitation, was centrifuged at 16168 × g (Centrifuge 5415 D; Eppendorf, Milan, Italy) for 1 min. Bacterial pellets were frozen at −80°C for 20 min and then resuspended in 375 µl of a solution containing 30 mM NaCl and 2 mM EDTA (pH 8). The cell suspension was transferred into tubes containing glass beads and 125 µl of extraction buffer (500 mM NaCl, 50 mM Tris-HCl, 50 mM EDTA, 4% SDS, pH 8). Samples were homogenized using a MM300 disrupter (Retsch, Haan, Germany) for 2 min at 30 cycles s–1 and were then incubated for 10 min at 70°C. After 1 h of incubation at 4°C, the samples were centrifuged for 1 min at 16168 × g and the supernatants were recovered.
The extracted DNA was used as a target for PCR amplification of the 16S rDNA gene using primers 63f and 1397r (Marchesi et al., Citation1998). Amplicons were sequenced at the Interdepartmental Centre for Agricultural, Chemical and Industrial Biotechnology (CIBIACI, Florence, Italy). The 16S rDNA sequence chromatograms were checked and edited to verify the absence of ambiguous peaks. The 16S rRNA gene amplicon sequence data are available at GenBank database: accession numbers MH475102–MH475135.
Total DNA extraction from T. suecica F&M-M33 cultures
Total DNA was extracted following the CTAB buffer protocol (Pini et al., Citation2012) combined with mechanical disruption with glass beads. One millilitre of algal culture was centrifuged at 16 168 × g (Centrifuge 5415 D; Eppendorf) for 1 min, the supernatant was discharged, and the collected biomass was frozen at −80°C for 20 min. The pellet was resuspended in 1 ml of CTAB buffer (2% (w/v) CTAB, 1.4 M NaCl, 0.2% (v/v) β-mercaptoethanol, 20 mM EDTA, 100 mM Tris-HCl, pH 8.0) and was transferred to a 2 ml tube containing glass beads (0.75–1 mm in diameter; Retsch), shaken for 2 min at 30 cycles s–1 using a MM300 disrupter (Retsch) and then incubated for 1 h at 65°C. After chloroform-isoamyl alcohol extraction, the aqueous phase was collected, and the nucleic acid was precipitated with isopropanol. DNA was washed twice with 70% ethanol and resuspended in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). For each sample, DNA was extracted from three different aliquots and then was combined and stored at −20°C.
PCR–Denaturing Gradient Gel Electrophoresis (DGGE)
Three primer sets were used to amplify three different variable regions of the 16S rDNA gene: V1–V3 region (63F-R518GC), V3–V5 region (357F-R907GC) and V6–V8 region (F968GC-1401R) (Yu & Morrison, Citation2004). PCR was carried out using a T100 Thermal Cycler (Bio-Rad Laboratories, Hertfordshire, UK) and in a 25 µl final volume containing 1× Flexi PCR buffer (Promega Italia, Milan, Italy), 1.5 mM MgCl2, 250 µM deoxynucleotide triphosphates (dNTPs), 400 nM each primer, and 1U GoTaq®Flexi DNA polymerase (Promega). Amplifications were performed under the following conditions: an initial denaturation for 4 min at 95°C, followed by 35 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 45 s and a final extension step at 72°C for 10 min. DGGE analysis was carried out by loading amplicons on a 6% polyacrylamide gel (acrylamide/bis 37.5:1), with a 47–60% denaturing gradient (100% denaturant consisting of 40% v/v deionized formamide, 7 M urea). Electrophoresis was performed using the Dcode DGGE System (Bio-Rad) at 60°C for 18 h and at a constant voltage of 85 V. After electrophoresis, the gels were stained with SYBR®Gold (Molecular Probes, Eugene, Oregon, USA). The gel images were digitally captured under UV light using the ChemiDoc XRS apparatus (Bio-Rad) and were analysed using GelCompareII software v 4.6 (Applied Maths, Saint-Martens-Latem, Belgium). Each band was considered representative of a single bacterial group, and the band intensity corresponded to its relative abundance. Richness was measured counting the number of bands in each DGGE profile, and the Shannon-Weiner diversity index was calculated as – ∑pi log2 (pi), where pi represents the relative abundance of a given fragment in the DGGE profile (Pastorelli et al., Citation2011).
Illumina MiSeq sequencing and data processing
For each sample, the V3–V4 region of the 16S rDNA gene was amplified using primers Pro341f and Pro805R (Takahashi et al., Citation2014) and barcodes were added to the forward primer. Amplicons for each library were purified and mixed in equal proportion. Illumina MiSeq v3 chemistry 300 base paired-end (PE) amplification and sequencing were performed at BMR Genomics (Padova, Italy). Reads were merged with FLASH v1.2.11 (Magoc & Salzberg, Citation2011) using the following parameters: −m 20, −M 280, Phred score default of 33. Sequences were then trimmed to discard primers with Prinseq-lite (Schmieder & Edwards, Citation2011) and sequences shorter than 200 bp were filtered out. Chimeras were removed using USEARCH 6.1 (Edgar et al., Citation2011). De novo OTU picking was performed using Swarm (Mahe et al., Citation2014) within QIIME 1.9.1 (Caporaso et al., Citation2010) and Silva 132 (Yilmaz et al., Citation2014) as the reference database. Representative sequences (most abundant) for each OTU were aligned and an OTU table was constructed using correctly aligned sequences. Sequences identified as chloroplasts were removed from further analysis. OTUs representing less than 0.005% of the total read abundance were discarded (Bokulich et al., Citation2013). UPGMA dendrograms were constructed using a rarefied (14890 OTUs) OTU table with the Dice and Bray–Curtis indices (1000 replicates) within PAST (Hammer et al., Citation2001). The 16S rRNA gene amplicon sequence data are available at the National Centre for Biotechnology Information Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) bioproject number PRJNA474805, SRA accession SRP149916.
Results
Isolation and identification of bacteria from T. suecica F&M-M33 cultures
Based on the different morphological characteristics of the colony (colour, shape and edge), 34 bacterial strains () were isolated from T. suecica F&M-M33 cultures: 10, 11 and 13 bacterial strains were isolated from LAB1, SES and CAM cultures, respectively. LAB2 was not sampled because it was considered to be, in the design phase of the experiments, similar to LAB1.
Table 1. Identification of bacterial isolates through 16S rDNA sequencing. Strains were isolated from LAB1, SES and CAM Tetraselmis suecica F&M-M33 cultures
16S rDNA sequencing of isolates led to the identification of 23 different taxa () belonging to 20 different genera subdivided in eight orders. The highest number of genera was retrieved within the Rhodobacterales order: Labrenzia, Mameliella, Marin-ovum, Pseudoceanicola, Roseivivax, Roseovarius and Stappia.
The genera Muricauda, Marinobacter and Roseivivax were isolated from all cultures analysed. Mameliella was found in LAB1 and SES, while Nitratireductor and Labrenzia were found in SES and CAM.
Selection of 16S rDNA primers to be used in microbiota analysis
The most promising 16S rDNA region for metagenomic analysis was selected based on the PCR–DGGE results. The diversity of bacterial communities, determined using the Richness and Shannon–Weiner indices calculated in each DGGE profile, is reported in Supplementary table S1 for each primer set used. The Richness index ranged from 8 to 19, while the Shannon–Weiner index ranged from 2.048 to 2.891 (Supplementary table S1). The V3–V5 and V6–V8 regions allowed the description of a higher diversity of the analysed communities than the V1–V3 region. Except for CAM samples, the V3–V5 region showed the highest values of the Richness and Shannon–Weiner diversity indices (Supplementary table S1). Therefore, considering the results obtained herein and those reported in the literature (Sanchez et al., Citation2007), the V3–V5 region of the 16S rRNA gene was selected for the metagenomic analysis of bacterial microbiota associated with T. suecica F&M-M33.
Metagenomic analysis of bacterial communities associated with laboratory and outdoor cultures of T. suecica F&M-M33
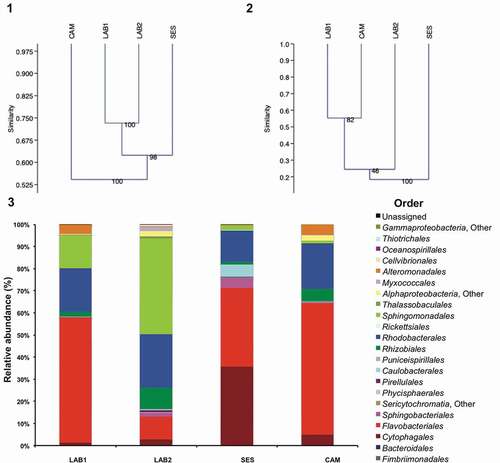
The composition of the microbial communities associated with four non-axenic T. suecica F&M-M33 cultures was analysed at different taxonomic levels. Illumina MiSeq v3 sequencing, which was performed on the variable region V3–V4 of 16S rDNA, produced 643 870 reads. A total of 133 418 sequences was obtained (ranging from around 14 895 to 47 714 sequences per library), allowing the identification of 479 OTUs with a range from 239 to 327 per sample. Rarefaction curves showed a high sequencing coverage for all the samples (Supplementary fig. S1). The UPGMA dendrogram obtained with Dice coefficient similarity, which is based on the presence–absence of OTUs, indicated samples LAB1 and LAB2 as the most similar, clustering with 73% similarity, while CAM was the most different, with 55% similarity to all the others (). Applying the Bray–Curtis index (based on the OTU relative abundance), the UPGMA dendrogram showed a different tree architecture than that obtained using the Dice coefficient. LAB1 and CAM were more similar to the other samples, clustering together and sharing ~55% similarity (). LAB2 shared ~25% similarity with LAB1 and CAM; SES was the most different showing less than 20% similarity to the other samples ().
Figs 1–3. Bacterial communities associated with four different cultures of Tetraselmis suecica F&M-M33 derived from the same starter culture. The same microalgal strain was grown in different conditions for a decade: LAB1 and LAB2 were derived from the same maintenance culture sampled at two different times; SES was a culture grown in a GWP®-III photobioreactor; and CAM was a culture obtained from a 0.5-l bubble tube. Fig. 1. Hierarchical cluster analysis (UPGMA, Dice coefficient of similarity) based on the occurrences of OTUs in the four T. suecica F&M-M33 cultures. Fig. 2. Hierarchical cluster analysis (UPGMA, Bray–Curtis coefficient of similarity) based on the relative abundances of OTUs in the four T. suecica F&M-M33 cultures. Fig. 3. Relative abundance of the orders of the bacterial communities associated with four different cultures of T. suecica F&M-M33.
Phylogenetic analysis of identified OTUs from T. suecica cultures
Eighty-six per cent of the sequences were identified to the genus level. The microbial communities associated with T. suecica were formed by at least 53 genera, 18 orders and five different phyla (, Supplementary tables S2, S4 and S6). Most of the genera retrieved (42) belong to Proteobacteria, particularly to three Alphaproteobacteria orders (): Rhodobacterales (13), Rhizobiales (10) and Sphingomonadales (7). Sandaracinus was the only genus observed belonging to Deltaproteobacteria. Four genera belonged to Gammaproteobacteria: Marinobacter, Haliea, Pseudohaliea and Alcanivorax (Supplementary table S6). Nine genera belonging to Bacteroidetes were identified; among them, Muricauda, Taeseokella and Arenibacter were more abundant (Supplementary table S6).
Fig. 6. Relative abundance of genera forming the ‘core’ bacterial community of Tetraselmis suecica F&M-M33 cultures. The same microalgal strain was grown in different conditions for a decade: LAB1 was a laboratory maintenance culture sampled in autumn; LAB2 was a laboratory maintenance culture sampled in winter; SES was a culture grown in a GWP®-III photobioreactor; CAM was a culture obtained from a 0.5-l bubble tube. Genera whose abundance was less than 1% in all samples were collapsed at higher taxonomic ranks.
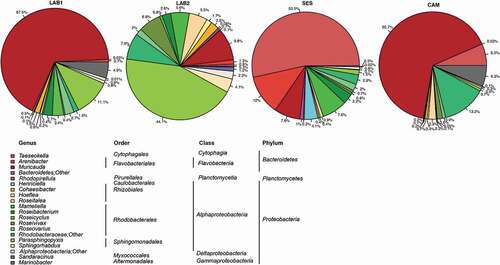
Analysing the relative frequency of the identified genera in all samples, more than 98% of the total microbiota belonged to Proteobacteria or Bacteroidetes (). LAB2 was the only sample where the dominating genus, Parasphingopyxis (36%), was affiliated with Proteobacteria. In LAB1 and CAM, Muricauda was the dominating genus (52% and 47%, respectively); in SES, the dominating genus was Taeseokella (35%).
Tetraselmis suecica ‘core’ microbiota
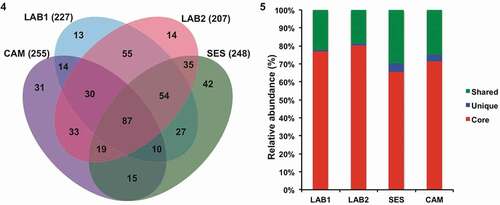
Analysis of OTUs across all four samples revealed a set of OTUs that were always present, named ‘core’. The ‘core’ bacterial communities were formed by 87 OTUs (), accounting for 77, 80, 66 and 72% of the total sequences retrieved from LAB1, LAB2, SES and CAM, respectively (). Additionally, 19–30% of sequences were shared among two or three samples, while unique OTUs (detected in one sample only) accounted for 0.7–5% of the sequences in LAB1 (13 OTUs), LAB2 (13 OTUs), CAM (31 OTUs) and SES (42 OTUs) (Supplementary tables S7–S11).
Figs 4–5. Tetraselmis suecica F&M-M33 ‘core’ bacterial community. The same microalgal strain was grown in different conditions for a decade: LAB1 and LAB2 were derived from the same maintenance culture sampled at two different times; SES was a culture grown in a GWP®-III photobioreactor; and CAM was a culture obtained from a 0.5-l bubble tube. Fig. 4. Bacterial OTU distributions in the four samples. Fig. 5. Relative abundance (percentage) of unique, shared and ‘core’ sequences retrieved.
The ‘core’ bacterial communities were represented by OTUs affiliated with 24 genera belonging to Proteobacteria, Bacteroidetes and Planctomycetes (). Nevertheless, the relative composition of the four bacterial ‘core’ communities was different, reflecting what was observed for the total bacterial communities associated with T. suecica cultures. The predominant genus in the core communities was Muricauda for LAB1 and CAM (67.5% and 65.7%, respectively) and Taeseokella for SES (53.5%), both genera belonging to Bacteroidetes. The most abundant genus in LAB2 was Parasphingopyxis (Proteobacteria) (44.1%). The genus Muricauda, which was dominant in LAB1 and CAM cultures, was present at 9.8 and 7.8% in LAB2 and SES, respectively (). Taeseokella was the third most abundant genus in CAM; however, in LAB1 and LAB2, its relative abundance was below 1%. Parasphingopyxis represented more than 10% of the sequences only in LAB2, where it was the dominant genus, and in LAB1.
The other genera whose relative abundances were higher than 5% in at least one T. suecica culture, were Arenibacter, Roseitalea, Mameliella, Roseivivax and Marinobacter. Marinobacter was the only genus of the core community belonging to Gammaproteobacteria; its relative abundance was 6.3% in CAM, 4.9% in LAB1 and 0.3% in SES and LAB2. A high relative abundance of the genus Arenibacter was observed in CAM (12%). The relative abundance of Roseitalea (Rhizobiales) and Roseivivax (Rhodobacterales) was above 5% in the LAB2 culture only, and that of Mameliella (Rhodobacterales) was above 5% in LAB2 and SES ().
The only genus belonging to Planctomycetes was Rhodopirellula, whose relative abundance was below 1% in all samples.
Discussion
In large-scale microalgal cultivation, axenic conditions are unrealistic to achieve and maintain; thus microalgae are constantly co-cultivated with bacteria. Bacteria may be either derived from the bacterial community associated with the microalga or from contamination in the culturing system (Tredici, Citation2004; Chini Zittelli et al., Citation2013a, Citationb).
The present study aimed to evaluate which bacterial taxa are persistently associated with T. suecica regardless of different environmental/growth conditions. Thus, we used samples collected from cultures originating from the same original T. suecica F&M-M33 starter culture, but kept for approximately a decade in different laboratories (except for LAB2, which was kept for only a few months in a different laboratory). Moreover, the sampled cultures were grown in different culture conditions (e.g. different culture system, light regime).
Differences in T. suecica F&M-M33 bacterial community structures, as observed in the UPGMA dendrograms, were mainly due to the relative abundances of the bacterial OTUs and, to a lesser extent, the presence or absence of certain OTUs. Indeed, the laboratory-maintained cultures, which showed almost 73% similarity with respect to the OTU presence/absence, had very different compositions in terms of the relative abundances of the different taxa, which may be related to the different light conditions of the two specimens (continuous artificial light for LAB1, diffuse natural light for LAB2).
Despite the different histories of the cultures, a ‘core’ bacterial community was identified, constituted by 87 OTUs. All taxa identified were related to bacteria commonly found in the marine environment, and all the OTUs belonged to the phyla Proteobacteria or Bacteroidetes, with the exception of Rhodopirellula which belongs to Planctomycetes.
Muricauda was the dominant taxon in algal cultures exposed to continuous artificial light (LAB1 and CAM). It is well known that microalgal cultures under continuous illumination modify their metabolism with respect to the light/dark cycle. Light also stimulates dimethylsulphoniopropionate (DMSP) synthesis (Galí et al., Citation2011), an organosulphur compound produced by phytoplankton and seaweeds that has an important implication in global climate and hydrological cycles (Sunda et al., Citation2002). An increase in the DMSP concentration may result in the stimulation of specific groups of bacteria. Flavobacteria, such as Muricauda, may oxidize dimethyl sulphide (DMS) to dimethyl sulphoxide during chemoheterotrophic growth in the presence of labile organic matter (Green et al., Citation2011). As O2 produced during photosynthesis may increase heterotrophic respiration of DMS (Franchini & Steinke, Citation2017) a longer light period may have favoured DMSP synthesis and its consumption by bacteria. Therefore, we may hypothesize that the high relative abundance of Muricauda in LAB1 and CAM samples might be due to DMSP/DSM production by Tetraselmis (Groene, Citation1995; Van Bergeijk & Stal, Citation2001). All Muricauda strains isolated (grown on marine agar) were characterized by an intense yellow colour. Genes encoding proteins related to carotenoid biosynthesis, such as zeaxanthin and cryptoxanthin, have been identified in Muricauda ruestringensis and M. lutaonensis genomes (Oh et al., Citation2015). Zeaxanthin is well-reported as an antioxidant compound (Prabhu et al., Citation2013) and may improve Muricauda resistance to reactive oxygen species (ROS) (Lubart et al., Citation2011).
Taeseokella was the most abundant genus in the ‘core’ community of the SES culture, followed by Arenibacter which was present at a relative abundance higher than 10%. The only species described thus far is T. kangwonensis, which was isolated from a freshwater sample (Joung et al., Citation2015); herein, for the first time, members of this genus have been found to be associated with a microalga. Taeseokella kangwonensis is related to Lacihabitans soyangensis HME6675T (92.6% 16S rRNA gene sequence similarity), Leadbetterella byssophila 4M15T (89.0%), Fluviimonas pallidilutea TQQ6T (89.7%) and Emticicia oligotrophica GPTSA100-15T (89.8%). However, among them, T. kangwonensis is the only strain able to grow in the presence of high levels of NaCl (up to 5%) (Joung et al., Citation2015). Arenibacter has been identified in the bacterial communities associated with Chlorella, Gymnodinium and Scrippsiella (Green et al., Citation2004, Citation2011; Makridis et al., Citation2012), but its role remains unclear.
In the LAB2 ‘core’ community, the genus showing the highest relative abundance was Parasphingopyxis, which was also present at a high level in LAB1. The genus Parasphingopyxis includes two species: P. lamellibrachiae, isolated from a marine annelid worm (Uchida et al., Citation2012), and P. algicola, isolated from a marine red alga (Jeong et al., Citation2017). The Parasphingopyxis genus description was recently amended, including its ability to grow in the presence of 6 % (w/v) NaCl (Jeong et al., Citation2017).
Within Proteobacteria, only few OTUs belonging to Delta- and Gammaproteobacteria were identified. Marinobacter was the only genus identified belonging to Gammaproteobacteria within the ‘core’ community. It is important to note that Marinobacter flavimaris strains were isolated from all the T. suecica F&M-M33 cultures. Several Marinobacter strains are siderophore producers (i.e. vibrioferrin); in the dinoflagellate Scrippsiella trochoidea, Marinobacter strains promote iron assimilation by facilitating its photochemical redox cycling (Amin et al., Citation2009). Marinobacter isolates may also enhance the growth of the diatom Pseudo-nitzschia (Sison-Mangus et al., Citation2014) and Marinobacter was found to be the dominant group in different cultures of the green alga Ostreococcus tauri grown under different conditions (Lupette et al., Citation2016). Therefore, Marinobacter strains could play an important role in the iron cycle associated with microalgae growth.
In all the cultures analysed, a constant presence of bacteria belonging to the order Rhodobacterales was observed (ranging from 11% to 26% in the ‘core’ community; 14–24% in the whole community). In particular, Roseivivax, Roseovarius, Roseicyclus and Mameliella were among the five most abundant genera observed in at least one sample; these fall within the Roseobacter group. The Roseobacter group (Simon et al., Citation2017) is widespread in the marine environment, often associated with microalgae and involved in important marine biogeochemical cycles (Buchan et al., Citation2005). Members of the Roseobacter group have gained specific features related to the phycospheric environment such as desulphonation (Simon et al., Citation2017), and vitamin and indole-3-acetic acid production (Amin et al., Citation2015). Most of the strains isolated herein were identified as members of Rhodobacterales, belonging to the genera Labrenzia, Mameliella, Marinovum, Pseudooceanicola, Roseivivax, Roseovarius and Stappia, and two strains which were identified at family rank (Rhodobacteraceae). Therefore, both metagenomics analysis and cultivation approaches showed that the Rhodobacterales clade was not only a significant presence but was also characterized by a wide diversity.
A low presence of Planctomycetes, represented by the genus Rhodopirellula, was also observed in the ‘core’ bacterial community. Rhodopirellula strains have been found in association with macroalgae (Bengtsson & Ovreas, Citation2010; Lage & Bondoso, Citation2011). In R. baltica, more than 100 different genes encoding sulphatase proteins have been identified (Glockner et al., Citation2003), indicating the possibility for it to metabolize complex sulphated polysaccharides usually present in the cell walls of marine algae such as Tetraselmis (Raposo et al., Citation2013). Its presence could therefore be useful to scavenge cell wall residues of dead algal cells.
In summary, the results obtained provided deep insight into the T. suecica F&M-M33 associated bacterial community and show the existence of a bacterial ‘core’ community mainly formed by marine bacteria. Even if this ‘core’ community was strongly influenced by environmental/culturing conditions in terms of the relative abundance of the different groups, the presence of the same OTUs/taxa in cultures separated for more than a decade supports the hypothesis of an archetypal bacterial community persistently associated with T. suecica. All the cultivable bacteria isolated herein and identified using a classic approach were also observed in the sequencing data except for Bacillus, Mesorhizobium and Marinovum. Indeed, within the isolated bacteria, some of the dominant taxa, as well as bacteria accounting for less than 0.01% (i.e. Pseudoceanicola, Labrenzia and Alcanivorax), were present; on the other hand, sequences belonging to Bacillus and Mesorhizobium were observed in non-filtered sequencing data (data not shown).
The obtained isolates belonging to the ‘core’ bacterial community of T. suecica may be a starting point to study the interaction between microalgae and bacteria and to increase the production and/or quality of T. suecica biomass. Particularly intriguing is the presence, among the most abundant taxa, of Taeseokella and Parasphingopyxis, two recently described genera. Little information is available on these bacteria, and Taeseokella strains have never before been observed to be associated with microalgae. Thus, elucidating their functions in this ecosystem might provide valuable data to better understand what drives the T. suecica microbiota assembly.
Supplementary information
The following supplementary material is accessible via the Supplementary Content tab on the article’s online page at https://doi.org/10.1080/09670262.2019.1606940
Supplementary methods S1. 16Sr DNA amplification.
Supplementary table S1. Richness (number of bands) and diversity (Shannon–Weiner index) of the DGGE banding profiles of 16S rDNA from bacterial communities associated with Tetraselmis suecica F&M-M33. V1–V3, V3–V5 and V6–V8 indicate the 16S rDNA regions amplified by primer pairs used in the analysis. The same microalgal strain was grown in different conditions, for a decade: LAB1 and LAB2 were derived from the same maintenance culture sampled at two different times (autumn and winter respectively); SES was a culture grown in a GWP®-III photobioreactor; and CAM was a culture obtained from a 0.5-l bubble tube.
Supplementary table S2. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the total community for each sample at the phylum level.
Supplementary table S3. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the total community for each sample at the class level.
Supplementary table S4. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the total community for each sample at the order level.
Supplementary table S5. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the total community for each sample at the family level.
Supplementary table S6. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the total community for each sample at the genus level.
Supplementary table S7. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the core community for each sample at the phylum level.
Supplementary table S8. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the core community for each sample at the class level.
Supplementary table S9. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the core community for each sample at the order level.
Supplementary table S10. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the core community for each sample at the family level.
Supplementary table S11. QIIME taxa table of the Tetraselmis suecica F&M-M33 microbiota composition of the core community for each sample at the genus level.
Supplementary fig. S1. Sample-based rarefaction curves representing the number of observed OTUs at different sequencing depths. The same microalgal strain was grown in different conditions for a decade: LAB1 was derived from a laboratory maintenance culture sampled in autumn = red; LAB2 was derived from the same maintenance culture sampled in winter = green; SES was a culture grown in a GWP®-III photobioreactor = blue; and CAM was a culture obtained from a 0.5-I bubble tube.
Author contributions
C. Viti and L. Giovannetti conceived the project. E. Piampiano, F. Pini, L. Giovannetti and C. Viti drafted the manuscript. N. Biondi collected the samples. E. Piampiano, F. Pini and R. Pastorelli analysed the samples and compiled the data. C. Viti and L. Giovannetti provided material support and funding for this project. All authors commented on and contributed to the final manuscript.
TEJP-2018-0067-File008.docx
Download MS Word (216.7 KB)TEJP-2018-0067-File007.xlsx
Download MS Excel (41.1 KB)TEJP-2018-0067-File006.docx
Download MS Word (14.9 KB)TEJP-2018-0067-File005.docx
Download MS Word (14.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Amin, S.A., Green, D.H., Hart, M.C., Kupper, F.C., Sunda, W.G. & Carrano, C.J. (2009). Photolysis of iron-siderophore chelates promotes bacterial-algal mutualism. Proceedings of the National Academy of Sciences USA, 106: 17071–17076.
- Amin, S.A., Hmelo, L.R., van Tol, H.M., Durham, B.P., Carlson, L.T., Heal, K.R., Morales, R.L., Berthiaume, C.T., Parker, M.S., Djunaedi, B., Ingalls, A.E., Parsek, M.R., Moran, M.A. & Armbrust, E.V. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature, 522: 98–101.
- Bell, W. & Mitchell, R. (1972). Chemotactic and growth response of marine bacteria to algal extracellular products. Biological Bulletin, 143: 265–277.
- Bengtsson, M.M. & Ovreas, L. (2010). Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiology, 10: 261. doi:https://doi.org/10.1186/1471-2180-10-261.
- Biondi, N., Cheloni, G., Tatti, E., Decorosi, F., Rodolfi, L., Giovannetti, L., Viti, C. & Tredici, M.R. (2017). The bacterial community associated with Tetraselmis suecica outdoor mass cultures. Journal of Applied Phycology, 29: 67–78.
- Bokulich, N.A., Subramanian, S., Faith, J.J., Gevers, D., Gordon, J.I., Knight, R., Mills, D.A. & Caporaso, J.G. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature Methods, 10: 57–59.
- Bondioli, P., Della Bella, L., Rivolta, G., Zittelli, G.C., Bassi, N., Rodolfi, L., Casini, D., Prussi, M., Chiaramonti, D. & Tredici, M.R. (2012). Oil production by the marine microalgae Nannochloropsis sp. F&M-M24 and Tetraselmis suecica F&M-M33. Bioresource Technology, 114: 567–572.
- Buchan, A., Gonzalez, J.M. & Moran, M.A. (2005). Overview of the marine Roseobacter lineage. Applied and Environmental Microbiology, 71: 5665–5677.
- Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., Fierer, N., Pena, A.G., Goodrich, J.K., Gordon, J.I., Huttley, G.A., Kelley, S.T., Knights, D., Koenig, J.E., Ley, R.E., Lozupone, C.A., McDonald, D., Muegge, B.D., Pirrung, M., Reeder, J., Sevinsky, J.R., Turnbaugh, P.J., Walters, W.A., Widmann, J., Yatsunenko, T., Zaneveld, J. & Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7: 335–336.
- Cardinaletti, G., Messina, M., Bruno, M., Tulli, F., Poli, B.M., Giorgi, G., Chini Zittelli, G., Tredici, M. & Tibaldi, E. (2018). Effects of graded levels of a blend of Tisochrysis lutea and Tetraselmis suecica dried biomass on growth and muscle tissue composition of European sea bass (Dicentrarchus labrax) fed diets low in fish meal and oil. Aquaculture, 485: 173–182.
- Chini Zittelli, G., Biondi, N., Rodolfi, L. & Tredici, M.R. (2013a). Photobioreactors for mass production of microalgae. In Handbook of Microalgal Culture (Richmond, A. & Hu, Q., editors), 225–266. Wiley Hoboken, NJ.
- Chini Zittelli, G., Rodolfi, L., Bassi, N., Biondi, N. & Tredici, M.R. (2013b). Photobioreactors for microalgal biofuel production. In Algae for Biofuels and Energy (Borowitzka, M.A. & Moheimani, N.R., editors), 115–131. Springer, Dordrecht.
- Edgar, R.C., Haas, B.J., Clemente, J.C., Quince, C. & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27: 2194–2200.
- Franchini, F. & Steinke, M. (2017). Quantification of dimethyl sulfide (DMS) production in the sea anemone Aiptasia sp. to simulate the sea-to-air flux from coral reefs. Biogeosciences, 14: 5765–5774.
- Galí, M., Saló, V., Almeda, R., Calbet, A. & Simó, R. (2011). Stimulation of gross dimethylsulfide (DMS) production by solar radiation. Geophysical Research Letters 38. doi: https://doi.org/10.1029/2011GL048051.
- Geng, H.F., Sale, K.L., Tran-Gyamfi, M.B., Lane, T.W. & Yu, E.T. (2016). Longitudinal analysis of microbiota in microalga Nannochloropsis salina cultures. Microbial Ecology, 72: 14–24.
- Glockner, F.O., Kube, M., Bauer, M., Teeling, H., Lombardot, T., Ludwig, W., Gade, D., Beck, A., Borzym, K., Heitmann, K., Rabus, R., Schlesner, H., Amann, R. & Reinhardt, R. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proceedings of the National Academy of Sciences USA, 100: 8298–8303.
- Green, D.H., Llewellyn, L.E., Negri, A.P., Blackburn, S.I. & Bolch, C.J.S. (2004). Phylogenetic and functional diversity of the cultivable bacterial community associated with the paralytic shellfish poisoning dinoflagellate Gymnodinium catenatum. FEMS Microbiology Ecology, 47: 345–357.
- Green, D.H., Shenoy, D.M., Hart, M.C. & Hatton, A.D. (2011). Coupling of dimethylsulfide oxidation to biomass production by a marine Flavobacterium. Applied and Environmental Microbiology, 77: 3137–3140.
- Groene, T. (1995). Biogenic production and consumption of dimethylsulfide (DMS) and dimethylsulfoniopropionate (DMSP) in the marine epipelagic zone: a review. Journal of Marine Systems, 6: 191–209.
- Grossart, H.P. & Simon, M. (2007). Interactions of planktonic algae and bacteria. Effects on algal growth and organic matter dynamics. Aquatic Microbial Ecology, 47: 163–176.
- Guillard, R. & Ryther, J. (1962). Studies of marine planktonic diatoms: I. Cyclotella nana Hustedt and Detonula confervacea (Cleve) Gran. Canadian Journal of Microbiology, 8: 229–239.
- Hammer, Ø., Harper, D.A.T. & Ryan, P.D. (2001). PAST: Paleontological Statistics software packages for education and data analysis. Palaeontologia Electronica, 4: XIX–XX.
- Hold, G.L., Smith, E.A., Birkbeck, T.H. & Gallacher, S. (2001). Comparison of paralytic shellfish toxin (PST) production by the dinoflagellates Alexandrium lusitanicum NEPCC 253 and Alexandrium tamarense NEPCC 407 in the presence and absence of bacteria. FEMS Microbiology Ecology, 36: 223–234.
- Jeong, S.E., Kim, K.H., Baek, K. & Jeon, C.O. (2017). Parasphingopyxis algicola sp. nov., isolated from a marine red alga Asparagopsis taxiformis and emended description of the genus Parasphingopyxis Uchida et al. 2012. International Journal of Systematic and Evolutionary Microbiology, 67: 3877–3881.
- Joung, Y., Hong, S., Kim, H., Kang, H., Farrance, C.E. & Joh, K. (2015). Taeseokella kangwonensis gen. nov., sp. nov., isolated from a freshwater reservoir. International Journal of Systematic and Evolutionary Microbiology, 65: 4309–4314.
- Krohn-Molt, I., Alawi, M., Forstner, K.U., Wiegandt, A., Burkhardt, L., Indenbirken, D., Thiess, M., Grundhoff, A., Kehr, J., Tholey, A. & Streit, W.R. (2017). Insights into microalga and bacteria interactions of selected phycosphere biofilms using metagenomic, transcriptomic, and proteomic approaches. Frontiers in Microbiology, 8: 1941. doi: https://doi.org/10.3389/Fmicb.2017.01941.
- Lage, O.M. & Bondoso, J. (2011). Planctomycetes diversity associated with macroalgae. FEMS Microbiology Ecology, 78: 366–375.
- Lakaniemi, A.M., Intihar, V.M., Tuovinen, O.H. & Puhakka, J.A. (2012). Growth of Chlorella vulgaris and associated bacteria in photobioreactors. Microbial Biotechnology, 5: 69–78.
- Le Chevanton, M., Garnier, M., Bougaran, G., Schreiber, N., Lukomska, E., Berard, J.B., Fouilland, E., Bernard, O. & Cadoret, J.P. (2013). Screening and selection of growth-promoting bacteria for Dunaliella cultures. Algal Research, 2: 212–222.
- Lubart, R., Lipovski, A., Nitzan, Y., & Friedmann, H. (2011). A possible mechanism for the bactericidal effect of visible light. Laser Therapy, 20: 17–22.
- Lupette, J., Lami, R., Krasovec, M., Grimsley, N., Moreau, H., Piganeau, G. & Sanchez-Ferandin, S. (2016). Marinobacter dominates the bacterial community of the Ostreococcus tauri phycosphere in culture. Frontiers in Microbiology, 7: 1414. doi: https://doi.org/10.3389/fmicb.2016.01414.
- Magoc, T. & Salzberg, S.L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27: 2957–2963.
- Mahe, F., Rognes, T., Quince, C., de Vargas, C. & Dunthorn, M. (2014). Swarm: robust and fast clustering method for amplicon-based studies. PeerJ, 2: e593. doi: https://doi.org/10.7717/peerj.593.
- Makridis, P., Costa, R.A. & Dinis, M.T. (2006). Microbial conditions and antimicrobial activity in cultures of two microalgae species, Tetraselmis chuii and Chlorella minutissima, and effect on bacterial load of enriched Artemia metanauplii. Aquaculture, 255: 76–81.
- Makridis, P., Ferreira, T., Kokou, F., Tsigenopoulos, C.S. & Divanach, P. (2012). Quantitative and qualitative aspects of bacterial communities associated with cultures of Chlorella minutissima. Journal of the World Aquaculture Society, 43: 571–578.
- Marchesi, J.R., Sato, T., Weightman, A.J., Martin, T.A., Fry, J.C., Hiom, S.J., Dymock, D. & Wade, W.G. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Applied and Environmental Microbiology, 64: 795–799.
- Nicolas, J.L., Corre, S. & Cochard, J.C. (2004). Bacterial population association with phytoplankton cultured in a bivalve hatchery. Microbial Ecology, 48: 400–413.
- Oh, J., Choe, H., Kim, B.K. & Kim, K.M. (2015). Complete genome of a coastal marine bacterium Muricauda lutaonensis KCTC 22339(T). Marine Genomics, 23: 51–53.
- Pastorelli, R., Landi, S., Trabelsi, D., Piccolo, R., Mengoni, A., Bazzicalupo, M. & Pagliai, M. (2011). Effects of soil management on structure and activity of denitrifying bacterial communities. Applied Soil Ecology, 49: 46–58.
- Perez-Lopez, P., Gonzalez-Garcia, S., Ulloa, R.G., Sineiro, J., Feijoo, G. & Moreira, M.T. (2014). Life cycle assessment of the production of bioactive compounds from Tetraselmis suecica at pilot scale. Journal of Cleaner Production, 64: 323–331.
- Pini, F., Frascella, A., Santopolo, L., Bazzicalupo, M., Biondi, E.G., Scotti, C. & Mengoni, A. (2012). Exploring the plant-associated bacterial communities in Medicago sativa L. BMC Microbiology, 12: 78. doi: https://doi.org/10.1186/1471-2180-12-78.
- Prabhu, S., Rekha, P.D., Young, C.C., Hameed, A., Lin, S.Y. & Arun, A.B. (2013). Zeaxanthin production by novel marine isolates from coastal sand of India and its antioxidant properties. Applied Biochemistry and Biotechnology, 171: 817–831.
- Ramanan, R., Kim, B.H., Cho, D.H., Oh, H.M. & Kim, H.S. (2016). Algae-bacteria interactions: evolution, ecology and emerging applications. Biotechnology Advances, 34: 14–29.
- Raposo, M.F., de Morais, R.M. & Bernardo de Morais, A.M. (2013). Bioactivity and applications of sulphated polysaccharides from marine microalgae. Marine Drugs, 11: 233–252.
- Rodolfi, L., Chini Zittelli, G.., Bassi, N., Padovani, G., Biondi, N., Bonini, G. & Tredici, M.R. (2009). Microalgae for oil: strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnology and Bioengineering, 102: 100–112.
- Sambles, C., Moore, K., Lux, T.M., Jones, K., Littlejohn, G.R., Gouveia, J.D., Aves, S.J., Studholme, D.J., Lee, R. & Love, J. (2017). Metagenomic analysis of the complex microbial consortium associated with cultures of the oil-rich alga Botryococcus braunii. Microbiology Open, 6: 1–9.
- Sanchez, O., Gasol, J.M., Massana, R., Mas, J. & Pedros-Alio, C. (2007). Comparison of different denaturing gradient gel electrophoresis primer sets for the study of marine bacterioplankton communities. Applied and Environmental Microbiology, 73: 5962–5967.
- Sapp, M., Schwaderer, A.S., Wiltshire, K.H., Hoppe, H.G., Gerdts, G. & Wichels, A. (2007). Species-specific bacterial communities in the phycosphere of microalgae? Microbial Ecology, 53: 683–699.
- Schmieder, R. & Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27: 863–864.
- Schwenzfeier, A., Wierenga, P.A. & Gruppen, H. (2011). Isolation and characterization of soluble protein from the green microalgae Tetraselmis sp. Bioresource Technology, 102: 9121–9127.
- Segev, E., Wyche, T.P., Kim, K.H., Petersen, J., Ellebrandt, C., Vlamakis, H., Barteneva, N., Paulson, J.N., Chai, L., Clardy, J. & Kolter, R. (2016). Dynamic metabolic exchange governs a marine algal-bacterial interaction. Elife, 5: e17473. doi: https://doi.org/10.7554/eLife.17473.
- Simon, M., Scheuner, C., Meier-Kolthoff, J.P., Brinkhoff, T., Wagner-Dobler, I., Ulbrich, M., Klenk, H.P., Schomburg, D., Petersen, J. & Goker, M. (2017). Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME Journal, 11: 1483–1499.
- Sison-Mangus, M.P., Jiang, S., Tran, K.N. & Kudela, R.M. (2014). Host-specific adaptation governs the interaction of the marine diatom, Pseudo-nitzschia and their microbiota. ISME Journal, 8: 63–76.
- Sunda, W., Kieber, D.J., Kiene, R.P., & Huntsman, S. (2002). An antioxidant function for DMSP and DMS in marine algae. Nature, 418: 317–320.
- Takahashi, S., Tomita, J., Nishioka, K., Hisada, T. & Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE, 9: e105592. doi: https://doi.org/10.1371/journal.pone.0105592.
- Thornton, D.C.O. (2014). Dissolved organic matter (DOM) release by phytoplankton in the contemporary and future ocean. European Journal of Phycology, 49: 20–46.
- Tredici, M. R., Biondi, N., Ponis, E., Rodolfi, L. & Chini Zittelli, G. (2009). Advances in microalgal culture for aquaculture feed and other uses. In New Technologies in Aquaculture: Improving Production Efficiency, Quality and Environmental Management (Burnell G. & Allan G., editors), 610–676. Woodhead Publishing, Cambridge.
- Tredici, M.R. (2004). Mass production of microalgae: photobioreactors. In Handbook of Microalgal Culture Applied Phycology and Biotechnology (Richmond A., editor), 178–214. Blackwell, Hoboken, NJ.
- Uchida, H., Hamana, K., Miyazaki, M., Yoshida, T. & Nogi, Y. (2012). Parasphingopyxis lamellibrachiae gen. nov., sp. nov., isolated from a marine annelid worm. International Journal of Systematic and Evolutionary Microbiology, 62: 2224–2228.
- Van Bergeijk, S.A. & Stal, L.J. (2001). Dimethy-lsulfoniopropionate and dimethylsulfide in the marine flatworm Convoluta roscoffensis and its algal symbiont. Marine Biology, 138: 209–216.
- Wirth, R., Lakatos, G., Maroti, G., Bagi, Z., Minarovics, J., Nagy, K., Kondorosi, E., Rakhely, G. & Kovacs, K.L. (2015). Exploitation of algal-bacterial associations in a two-stage biohydrogen and biogas generation process. Biotechnology Biofuels, 8: 59. doi: https://doi.org/10.1186/S13068-015-0243-X.
- Yao, C.H., Ai, J.N., Cao, X.P., Xue, S. & Zhang, W. (2012). Enhancing starch production of a marine green microalga Tetraselmis subcordiformis through nutrient limitation. Bioresource Technology, 118: 438–444.
- Yilmaz, P., Parfrey, L.W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., Schweer, T., Peplies, J., Ludwig, W. & Glockner, F.O. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Research, 42: D643–648.
- Yu, Z.T. & Morrison, M. (2004). Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Applied and Environmental Microbiology, 70: 4800–4806.