
ABSTRACT
Whole genome sequencing datasets present the opportunity to not only study evolution in the target organism, but also the associated holobiont. The capacity to study epi-endobiotic kelp associations is improving substantially with the increased availability of high-throughput sequencing datasets. The goal of this study was to determine if shotgun sequencing libraries could be used to document epi- and endophyte/faunal species colonizing Alaria kelp sporophytes from Kamchatka (Russia), the Bay of Fundy (Atlantic Canada) and Nuuk (Greenland). Mitochondrial coxI and plastid rbcL reads were extracted and assembled from six Alaria whole genome sequencing datasets. In total, contigs representing 11 epi-endobiotic species were assembled, of which Chordariacean diversity dominated. Given the presence of a newly discovered phaeophycean coxI sequence lacking an rbcL counterpart, we secondarily tested our hypothesis that the coxI sequence belonged to a phaeophycean parasite. The entire read dataset was assembled for the Alaria specimen hosting the putative parasite, the mitochondrial genome was retrieved, and plastid scaffolds were annotated and screened for phylogenetic placement matching the coxI sequence. The mitochondrial genome of the candidate parasite displayed numerous atypical features, including duplicated genes and rearrangements, and clear signs of relaxed selection, in line with the notion this organism may have a deviant lifestyle. The plastid genome was recovered as several fragments and lacked genes for photosystem and cytochrome complexes and chlorophyll biosynthesis, confirming our hypothesis that the unknown phaeophycean represented a parasitic species. Furthermore, classification to order remained unclear for the phaeophycean parasite, suggesting this species could represent a newly discovered higher-level lineage. Our study showcases the utility of whole-genome sequencing datasets in revealing surprising aspects of the eukaryotic diversity inhabiting kelp holobionts.
Introduction
Improvements in sequencing techniques have profoundly shifted the landscape of genetic analysis, opening new applications in environmental and taxonomic biology (Bella et al., Citation2013; Oliveira et al., Citation2018). In particular, the use of taxonomically widespread genes as DNA barcodes combined with high-throughput sequencing (HTS) has enabled parallel species detection in environmental samples, an approach known as metabarcoding. Alternatively, shotgun metagenomic sequencing, wherein all genomic information is sequenced (i.e. whole genome sequencing), can be used to detect all species present in complex samples without the use of taxon specific primers. Our perception of biodiversity from pelagic and benthic marine systems has been greatly improved as a result of these advances in sequencing (e.g. Leray & Knowlton, Citation2014; Marcelino & Verbruggen, Citation2016; Tragin & Vaulot, Citation2019), and vast genomic resources are forthcoming (Delmont et al., Citation2020). High-throughput metabarcoding approaches are therefore leading towards a more complete, or ‘holistic’, overview of species communities, with implications for ecological and evolutionary understanding. While the realm of ‘dark taxa’ has largely been explored in fungi (Ryberg & Nilsson, Citation2018), it stands to reason the world of algae holds similar secrets.
One such dark realm is likely the epi- and endophyte/faunal communities that inhabit kelp. Such associations are expected to be common and widespread, as kelps form globally distributed marine forests and are among the most biodiverse ecosystems of arctic and temperate coastal regions (Bringloe et al., Citation2020). Many filamentous brown algae of the order Ectocarpales are known to live as kelp epi-endophytes, with some species named on the basis of these associations (e.g. Pedersen, Citation2011). Faunal associations are similarly well-documented (e.g. Ronowicz et al., Citation2008). Species-level taxonomy of microbiota inhabiting kelps, however, is often complicated by rare species, taxonomically challenging groups, and poorly understood and/or cryptic diversity (Wlodarska-Kowalczuk et al., Citation2009), meaning the precise nature of algal and faunal kelp inhabitants has remained a difficult area of study. Modern DNA-sequencing methods promise to reveal significant insight into kelp epi-endophyte/faunal associations. Whole genome sequencing datasets, while typically used to provide information about the host genome, could also provide insights into associated microbial diversity.
Alaria is a widely distributed northern hemisphere genus of kelp, with most species occurring in the North Pacific, while A. esculenta (Linnaeus) Greville extends through the Arctic and into the Atlantic (Kraan, Citation2020). Alaria is cultivated for direct consumption and biomedical applications in the Atlantic and is being developed for aquaculture practice in the Gulf of Alaska (Stekoll, Citation2019). Alaria esculenta is also among the Atlantic kelps forecast to retreat northwards with colder waters as climate change progresses (Assis et al., Citation2018). Climate change is also predicted to impact the microbiome, and consequently overall health, in some species of kelp (Qiu et al., Citation2019). Less clear are the potential impacts of climate change on the kelp holobiont, which includes the host and all its associated micro- (e.g. viruses, archaea, bacteria, microalgae, protozoa) and macrobiota (e.g. fungi, metazoa and seaweeds), partly because the focus of research has been on bacteria (van der Loos et al., Citation2019). Safeguarding Alaria, along with its ecological and economic value, requires a solid knowledge of functional diversity and capacity for adaptation, both for the host and associated holobiont. Shotgun metagenomic sequencing datasets are a critical asset towards this end.
Our first objective was to determine if epi- and endobiotic species colonizing Alaria kelp sporophytes could be documented by assembling DNA barcode markers obtained from shotgun sequencing libraries. In particular, we sought to assemble coxI and rbcL genes from our datasets given their widespread use in global DNA barcoding efforts (Hebert et al., Citation2003), the latter of which is additionally used for photosynthetic organisms (e.g. algae: Le Gall & Saunders, Citation2010). Given the presence of a newly discovered phaeophycean mitochondrial coxI contig lacking a plastid rbcL counterpart, we initiated a second objective of assembling both organellar genomes for this epi-endophytic species. We hypothesized the unknown phaeophycean represented a parasitic lineage; if true, we expected (1) to retrieve plastid genes not related to photosynthesis (e.g. ribosomal genes), whereas plastid genes related to photosynthesis should be absent (e.g. photosystem, electron transport, ATP synthases, chlorophyll biosynthesis); and (2) to detect signatures of relaxed selection in genes of the unknown phaeophycean, that is, higher rates of non-synonymous substitutions.
Materials and methods
Barcodes assembly
Young tissue of Alaria was collected from Kamchatka (Russia), near Nuuk (Southwest Greenland), and the Bay of Fundy (Atlantic Canada; see Supplemental table S1), and total genomic DNA was extracted using a modified CTAB protocol (Cremen et al., Citation2016). Extracted DNA was sent to GENEWIZ (Suzhou, China), where libraries were generated using the Illumina VAHTS Universal DNA Library prep kit and protocols, and sequenced on the NovaSeq System (paired-end, 150 bp reads, ~15–30 Gb of data/specimen). In total we sequenced one specimen of A. esculenta sensu lato (type crispa; Klochkova et al., Citation2018) (104 million paired-end reads), and five specimens of A. esculenta sensu stricto (41.5–130 million paired-end reads/specimen; Supplemental table S1). Epi-endophyte/faunal reads were extracted from raw read files in a multi-step process that first identified sequences corresponding to the DNA barcode markers coxI and rbcL, followed by filtering to exclude host reads (Alaria). First, a DNA barcode library was built based on Arctic marine organisms (Hardy et al., Citation2010), including Annelida, Arthropoda, Brachiopoda, Chaetognatha, Cnidaria, Echinodermata, Echiura, Mollusca, Nematoda and Sipuncula. Bryozoa, Porifera and the algal phyla Rhodophyta and Ochrophyta were added to the library, while Chordata and Cephalopoda were excluded as we did not reasonably expect these taxa to colonize kelp. Publicly available DNA barcode data (mitochondrial cox1 and plastid rbcL) were retrieved from the Barcode of Life Data System (BOLD; Ratnasingham & Hebert, Citation2013) and GenBank. In total, the library consisted of ~36,000 unique species records. Bowtie2 v.2.3.4 (Langmead & Salzberg, Citation2012) was used to map reads to within 20% of the indexed species data (using custom parameters: –local –score-min G,12,8 –al-gz). The mitochondrial and plastid genomes of the host Alaria samples were de novo assembled using NOVOPlasty v.3.7.2 using default settings (Dierckxsens et al., Citation2017; Supplementary table S1), which were used to filter/remove host reads from the epi-endophyte datasets (Bowtie2 custom parameters: –score-min L,-0.6,-0.12 –un-gz). The final epi-endophyte reads were assembled using default settings in Velvet v.1.2.10 (Zerbino & Birney, Citation2008). Identifications were assigned to contigs based on BOLD and GenBank searches, and non-target contigs (e.g. bacterial, human) were discarded. Final contigs were trimmed to regions with a minimum of 2× coverage and 100% consensus, and average read coverage was noted. The obtained cox1 and rbcL sequences were linked to one another based on presence/absence patterns within Alaria samples (i.e. markers can be linked if retrieved from the same individual of Alaria) and phylogenetic placement (i.e. consistent location in trees). Trees were built using available GenBank data and RAxML v.8.2.11 (Stamatakis, Citation2014) with a GTR GAMMA substitution model and partitioning according to the three codon positions.
Prediction testing
Given the presence of a newly discovered phaeophycean cox1 contig, we attempted to assemble its organellar genomes by assembling reads from the host sample (TTB000062 from Greenland; Supplemental table S1) using SPAdes v.3.13.0 with specified k-mer values of 21, 33, 55, 77, 99 and 109 (Nurk et al., Citation2013). The final assembly scaffolds are available via Figshare (https://doi.org/10.6084/m9.figshare.13140464), while the short reads can be assessed via the NCBI Short Read Archive (accession SAMN16729885). Scaffolds identified as the mitochondrial genome of the unknown phaeophycean were annotated using MFannot (Beck & Lang, Citation2010) and oriented by aligning with previously published brown algal mitochondrial genomes (Supplementary table S2). A phylogenetic tree at the amino acid level was built from mitochondrial genomes available on GenBank and the Sequence Read Archive using RAxML v.8.2.11 (Stamatakis, Citation2014) and a GTR GAMMA substitution model with partitioning across genes (16 genes, 4790 amino acid sites). A mitochondrial phylogenetic tree at the nucleotide level was also built incorporating data on all known brown algal orders and using RAxML with a GTR GAMMA substitution model and partitioning across genes (Supplementary table S2).
The assembly was further screened for plastid scaffolds corresponding to the unknown phaeophycean. A local database of available brown algal chloroplast genomes was built, and assembled scaffolds matching the database (BLAST e-value <1−40) were kept for further analysis (available at https://doi.org/10.6084/m9.figshare.13140464). These scaffolds were then annotated using MFannot. A dataset of brown algal chloroplast genomes representative of the various orders was constructed and annotated with MFannot for consistency; some chloroplast genomes for Chordales, Laminariales and Sphacelariales were derived from the SRA and assembled using default settings in NOVOPlasty v.3.7.2 (Supplementary table S2). Brown algal chloroplast coding genes (140 total) were extracted from the plastid scaffolds and from selected brown algal genomes and aligned in Geneious Prime v.2019.2.3 (Kearse et al., Citation2012). RAxML trees were constructed for each gene alignment with a GTR GAMMA substitution model. Plastid scaffolds belonging to the unknown phaeophycean were identified based on phylogenetic placement, that is, a long branch belonging to the Brown Algal Crown Radiation (BACR; Bringloe et al., Citation2020), but not matching with earlier diverging orders, the host (Alaria), or Ectocarpales (Chordariacean epi-endophytes); annotations are available via Figshare (https://doi.org/10.6084/m9.figshare.13140464). A phylogenetic tree at the amino acid level incorporating the unknown phaeophycean scaffolds was built using RAxML with a GTR GAMMA substitution model and partitioning across genes (47 genes and 9648 amino acid sites; Supplementary table S2).
Regression analysis was used to determine the chance of not recovering a rbcL contig given the presence of a coxI contig in the unknown phaeophycean. Reads were mapped to the recovered coxI and rbcL contigs in all samples in order to consistently calculate read coverage using Bowtie2 v.2.3.4 (Langmead & Salzberg, Citation2012); reads were only mapped if they were an exact match. Given the presence of two coxI sequences of Phaeostroma in TTB000062 and only one rbcL contig (presumably because they were closely related species), coverage for these contigs was not included in the analysis. Regression analysis was performed on nine sets of values, and prediction intervals were calculated in Excel at 95, 99 and 99.9% intervals. The observed coverage for plastid scaffolds belonging to the unknown phaeophycean was also calculated via read mapping (as described above).
Signatures of relaxed selection in the unknown phaeophycean were tested using RELAX (default settings) in HyPhy (Wertheim et al., Citation2015). RELAX tests for the strength of natural selection in a specified branch(es) relative to reference branches in a phylogeny, summarized by the selection intensity parameter k, wherein values < 1 indicate relaxed selection and values > 1 indicate intensification. The null hypothesis of k = 1 was tested. Genes were first concatenated according to functional group, then alignments were masked for conserved blocks while maintaining codon positions using default parameters in Gblocks (Castresana, Citation2000). Single gene and concatenated (post masking) alignments can be accessed via Figshare (https://doi.org/10.6084/m9.figshare.13140464). RAxML trees were generated using a GTR GAMMA substitution model, with partitioning according to gene and codon position. Finally, gene alignments were analysed using the relax command in HyPhy 2.5.17, employing the universal genetic code (code 11, bacterial code, unavailable for analysis of plastid genes). Reference taxa are listed in Supplementary table S2. In order to focus the analyses on brown seaweeds and avoid long branch artefacts, the outgroup Ishege okamurae Yendo was excluded in the analyses for mitochondrial genes, and the outgroups Nanofrustulum shiloi (J.J.Lee, Reimer & McEnery) Round, Hallsteinsen & Paasche and Vaucheria litorea C.Agardh were excluded from the analyses of plastid genes.
Results
In total, contigs were resolved for 11 epi-endophyte/faunal species, of which seven were clear members of the brown algal family Chordariaceae (Ectocarpales; ; Supplementary table S3). A newly discovered brown algal coxI sequence was also detected, which lacked a corresponding rbcL contig (). We also detected one red algal endophyte (Acrochaetium alariae (Jónsson) Bornet), and one diatom (Thalassiosira sp., Supplementary table S3). Animal taxa were rare in the final contigs, with the exception of the epifaunal hydrozoan of the Leptomedusae Obelia longissima Pallas which occurred on all Greenland Alaria. The decapod Penaeus monodon Fabricius (tiger shrimp) was also detected in a sample from Greenland (TTB000062). Read-depth of the epi/endobiotic contigs was generally good (between 7× and 15× mean read coverage), and was noticeably scant in only a couple of cases (cox1 for A. alariae and rbcL for Laminariocolax aecidioides (Rosenvinge) A.F.Peters, with 2.5 and 3.4 mean read coverage, respectively; Supplementary fig. S1, table S3). In comparison, the host organelles always exceeded 200× coverage.
Fig. 1. Maximum-likelihood trees depicting cox1 and rbcL relationships among brown algal epi-endophytes detected on Alaria. Alignments are 658 and 1466 bp, respectively. Contigs linked to the same species are marked with the dashed lines. Bootstrap values are indicated at nodes; an * indicates full support, - indicates < 50% support
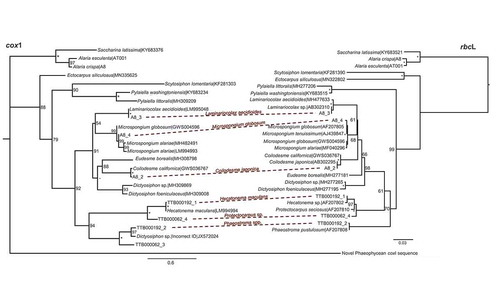
The presence of a newly discovered coxI phaeophycean sequence lacking an rbcL counterpart initiated our second objective of attempting to assemble its organellar genomes. The mitochondrial genome was resolved as four scaffolds that featured four copies of atp9 (two presumably functional, one truncated, and one a clear pseudogene), a pseudogene copy of rps13 (as evidenced by internal stop codons), potential loss of rps7, and tatC reading in the opposite direction upstream from its typical location in brown algae (reading in the reverse direction after nad1; ). Hypothetical proteins (ORFs) were also abundant, 8/9 of which were unique among available brown algal mitochondrial sequences. The mitochondrial genome was relatively large (~46 ka compared with ~38 ka typically reported in brown algal mitochondrial genomes). Phylogenetic analysis of 16 mitochondrial genes confidently placed the unknown phaeophycean near the base of the BACR (). A nucleotide analysis incorporating data from all brown algal orders further indicated the unknown phaeophycean appeared to belong to the BACR, though it did not clearly correspond to any of the known brown algal orders (Supplementary fig. S2).
Fig. 2. Annotated mitochondrial genomes of the putative phaeophycean parasite and host Alaria esculenta. Notable differences between genomes are highlighted in red. The contigs resolved for the putative Phaeophycean parasite are indicated with black lines
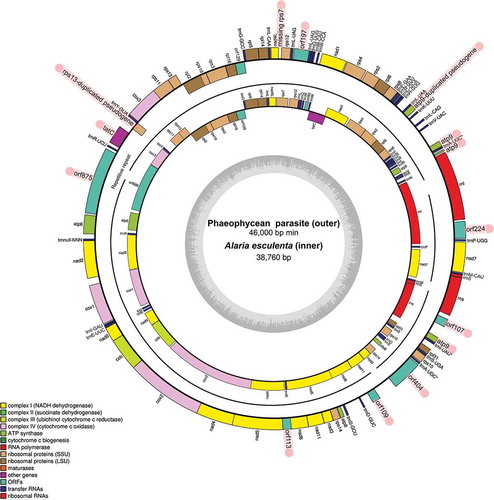
Fig. 3. Maximum-likelihood trees depicting placement of the unknown phaeophycean, along with plastid genes recorded for the unknown phaeophycean. The mitochondrial tree is based on 16 translated genes (4790 amino acid sites); the plastid tree is based on 47 translated genes (9648 amino acid sites). Trimmed from the trees are the root clades Ishige okamurae (Ishigeales) for the mitochondrial tree, and Nanofrustulum shiloi (Bacillariophyceae) and Vaucheria litorea (Xanthophyceae) from the plastid tree. Nodes lacking a bootstrap value are fully supported, while values are depicted for nodes with <95% support. Red crosses indicate genes that appear to be absent from the plastid genome of the unknown Phaeophycean, while green checkmarks indicate confirmed genes. GenBank and SRA accessions are available in Supplementary table S2

Scanning for a plastid corresponding to the unknown Phaeophycean revealed 12 scaffolds. Only a subset of the expected plastid genes appeared to be present. Importantly, all the photosystem, cytochrome b/f complex, chlorophyll biosynthesis and carbon fixation genes were absent, as were most of the ycf genes (). In contrast, nearly the full complement of ribosomal genes was recovered, as were genes for ATP synthase with the exception of atpE. InterProScan predicted the expected domains in nearly all genes, but did not predict the expected domains in atpF, while only membrane subunit b was predicted in atpG. Genes belonging to the host (Alaria) and Chordariacean epi-endophytes (three species) were consistently detected in all the plastid gene trees for TTB000062. To rule out low coverage driving the absence of plastid genes in the assembly, we used regression analysis to predict the coverage of rbcL given the known coxI coverage in the newly discovered phaeophycean. Regression analysis predicted the plastid coverage for the unknown phaeophycean to be 30×, with a 99% prediction interval of 12.8× and 46.9× coverage, and a 99.9% prediction interval of 3.5× and 56× coverage (Supplementary fig. S1). The observed coverage of the unknown phaeophycean plastid genes was 11.8× with two standard deviations of 7.5× and 16× coverage (Supplementary fig. S1). Phylogenetic analysis of the unknown phaeophycean plastid genes further supported placement within the BACR ().
HyPhy analyses detected significantly low k values (parameter for selection intensity) in nearly all the newly discovered phaeophycean genes selected for analysis (), the only exception being cytochrome oxidase c subunits.
Table 1. HyPhy results testing for signatures of relaxed selection in mitochondrial and plastid functional gene groups of the putative parasitic brown alga
Discussion
Whole genome data has presented novel opportunities to not only explore target (host) genomes, but also the diversity and genomes of the accompanying holobiont. We sought to infer the epi-endobiont of A. esculenta, a key habitat forming kelp in temperate and Arctic waters. Besides highlighting the utility of whole genome sequencing for inferring eukaryotic species present in/on kelp, our results also revealed a brown alga we argue represents a newly discovered parasitic lineage.
Detecting epi-endobiotic diversity
Among the epi-endophytes detected in the specimens of Alaria, the Chordariaceae were particularly prevalent (, Supplementary table S3). The species detected differed between our Russian and Atlantic/Arctic host specimens, and Phaeostroma pustulosum was the only widespread species in the Atlantic/Arctic region. Many of the taxa reported are known epiphytes, including Laminariocolax aecidioides (Heesch & Peters, Citation1999), Microspongium (Gauna et al., Citation2015; Múrua et al., Citation2018), Phaeostroma spp. (Pedersen, Citation1981, Citation2011), Protectocarpus (Bjaerke & Fredriksen, Citation2003), and Coilodesme japonica (Deshmuke & Tatewaki, Citation2001). Some of these species are clearly adapted for an endophytic life strategy, as in L. aecidioides which inserts itself into the sporophyte of brown algae (typically kelp) using a germ tube (Heesch & Peters, Citation1999). Microspongium alariae is also known to undergo morphological changes in an endophytic association with A. esculenta, suggesting evolution of a host-specific relationship (Múrua et al., Citation2018). Interestingly, Protectocarpus speciosus and Microspongium globosum are known to form dense coatings together (Kuckuck, Citation1955), as do various epiphytic species from Greenland (Pedersen, Citation2011), implying that interactions among Chordariaceaen species facilitate concurrent occupation of host kelp tissue. The detection of known epi-endophytes attests to the accuracy of our methods and the utility of purposing whole genome data towards establishing knowledge about epi-endobiont diversity.
Epiphyte/faunal associations with Alaria outside the brown algae were also detected. The centric diatom Thalassiosira was identified, and despite its pelagic nature, weak connections with algal hosts have been recorded (Tanaka, Citation1986). It is possible the detection of Thalassiosira here was due to its large overall abundance throughout marine ecosystems (Round et al., Citation1990), or a seasonal bloom. Obelia longissima, on the other hand, is likely a common Arctic epiphyte, as evidenced by its presence on all the Greenland Alaria sequenced, and the reported presence of hydrozoan species on up to 27% of Arctic Laminaria digitata by Ronowicz et al. (Citation2008). This can be attributed to the life history of O. longissima, which includes a sessile colonial stage (Tyler-Walters, Citation2003). The red alga Acrochaetium alariae was also detected on A. esculenta s.l. from Russia, and as the name implies, this species is described on the basis of its tendency to grow in/on species of Alaria. Curiously, we also detected the decapod species Penaeus monodon in libraries generated from a Greenland specimen of Alaria, despite field and lab procedures wherein young tissue (close to stipe) free of obvious epiphytes was taken. This is a widely distributed tropical species, introduced to several areas of the globe, including the Gulf of Mexico (Fuller et al., Citation2014) and as far north as the UK and France, though it apparently has not become established in Europe (Rodríguez & Suárez, Citation2001). The cold northern waters where specimens of Alaria were collected seemingly precludes the presence of P. monodon. We therefore interpreted these sequences as lab contamination, though we could not trace this to an obvious source, and the contamination was not ubiquitous across samples. Whether the presence of P. monodon on specimens of Alaria is a lab artefact or biologically accurate remains unknown.
Detecting a novel phaeophycean parasite
The most exciting finding was a newly discovered mitochondrial genome belonging to an unknown organism inhabiting a specimen of A. esculenta from Greenland. Alignment of available brown algal mitochondrial genomes reveals a largely conserved genome architecture and set of genes, present even in earlier diverging orders (i.e. Ishegeales, Dictyotales, Sphacelariales; Liu et al., Citation2018; Starko et al., in review; Supplementary table S2). The unprecedented presence of gene duplications and rearrangements (listed in Results) therefore stood in stark contrast to the conserved nature of brown algal mitochondrial genomes reported to date (). The high saturation of substitutions, depicted as long branches in the phylogenetic trees (), further suggested unique evolutionary pressures were acting on this genome. HyPhy results indicated selection was significantly relaxed in the unknown phaeophycean (), which accounted for the elevated substitution rate and supported our hypothesis that this phaeophycean was parasitic. In the red algal parasite Pterocladiophila hemisphaerica, elevated substitution rates in the organellar genomes lead to similarly long branches (Preuss et al., Citation2020). The search for an accompanying plastid genome provided further insight, as it revealed this lineage lacks nearly all genes needed for photosynthesis (). Overall, our hypothesis that the unknown epi-endophyte represented a parasitic phaeophyte appeared to be validated.
The inference of parasitism on the basis of missing photosynthetic genes could justifiably be met with criticism, as the absence of evidence is not highly regarded as evidence of absence. This criticism can be dispelled, however, on several fronts: (1) the presence of plastid genes matching phylogenetic placement of the newly discovered mitochondrial genome is strong support that a corresponding plastid genome is present; (2) our regression analysis indicated the chances of not detecting genes for photosynthesis in this plastid genome, if they were present, were less than 1/1000 (Supplementary fig. S1); and (3) the genes of the parasitic organism were clearly evolving under relaxed selection (), as expected in parasitic lineages (Preuss et al., Citation2020). Within this context, the absence of genes related to photosynthesis rather serves as good evidence for a parasitic lifestyle. Indeed, coverage of the phaeophycean parasite’s plastid scaffolds averaged 11.8×, greater than in most of the other brown algal rbcL contigs resolved (Supplementary fig. S1), and as such, genes for photosynthesis could not have escaped detection. We note further that two of the contigs presented here (Nodes 40500, 81423) should likely be merged, as evidenced by a matching 24 bp segment at the beginning and end of the sequences (respectively). Assembly errors at the junction between these two scaffolds (causing the broken assembly) were likely caused by the presence of a tRNA gene, wherein the conserved motif likely led to errors during co-assembly of multiple brown algal plastid genomes. Indeed, mapping reads back to the merged scaffolds confirmed they probably represent the same continuous strand of DNA (i.e. consistent coverage of overlapping reads in the junction). This finding is notable given the presence of rps14 and rpl20 on either side of the junction, which typically flank a block of photosynthetic genes containing rbcL, rbcS, psaA, and psaB. Merging the two scaffolds therefore suggests these genes for photosynthesis are missing from the plastid genome, though we note that genome architecture can vary dramatically across brown algal orders (Starko et al., in review).
Phylogenetic placement of the phaeophycean parasite remained mysterious, though we suggest it is a member of a radiation event relatively late in the evolution of brown algae. Several orders diverged early in the evolution of the brown algae, including Dictyotales and Sphacelariales (Bringloe et al., Citation2020; ). Most brown algal orders stem from a radiation that began in the early Cretaceous, dubbed the Brown Algal Crown Radiation (BACR). Our phylogenetic analysis consistently placed the parasitic lineage among later diverging orders (). Further supporting this placement, the few mitochondrial gene rearrangements characteristic of earlier diverging orders were not present in the unknown phaeophycean mitochondrial genome, that is, the positions of atp8, atp9, rps10 and rpl31 were consistent with members of the BACR (Starko et al., in review; ). We do caution against interpreting bootstrap values and accompanying phylogenetic associations with the parasitic lineage (e.g. grouping of plastid genes with Fucales in ), as its position among the BACR was inconsistent across analyses, probably due to long-branch attraction artefacts (Supplementary figs S1 and S3; Preuss et al., Citation2020). It is worth asking whether this species may represent a new order of brown algae. An in-depth investigation to visually confirm and report on any unique morphological features will be needed for formal taxonomic classification and should be carefully scrutinized against previous work on Greenland brown algal epi-endophytes (Pedersen, Citation2011). Further analysis of Alaria, among other kelp sporophytes, will help determine the range of the phaeophycean parasite, would potentially reveal other related species, and provide further evolutionary context for understanding the transition to parasitism.
Reports of parasitic brown algae are rare. As far as we know, the only reported parasitic brown alga is Herpodiscus durvilleae, which is of the order Sphacelariales and is an obligate parasite of Durvillaea antarctica (Heesch et al., Citation2008). Parasitism is inferred in H. durvilleae on the basis of grey plastids, indicating loss of photosynthetic capacity, though a functional copy of rbcL remains present (Heesch et al., Citation2008). Plastids in other parasitic algae ubiquitously show a reduction in genome size and loss of photosystem genes, while housekeeping genes are generally retained (i.e. genes used in translation and transcription such as ribosomal proteins; ; Salomaki et al., Citation2015; Preuss et al., Citation2020). Genes related to amino acid biosynthesis, fatty acid biosynthesis, and transport may also be retained, such as in the red algal parasite Choreocolax polysiphoniae (Salomaki et al., Citation2015). Curiously, ATP synthases appeared to be present in the brown algal parasite detected here, though atpE was missing and not all expected domains were predicted for atpF and atpG (). These genes represent the last step in photosynthesis, forming the membrane-bound ATP synthase complex that uses a proton gradient to convert ADP to ATP. Lacking the photosystem and cytochrome complexes to generate the proton gradient, it is difficult to envision a function for ATP synthase orthologues in the phaeophycean parasite. The absence of atpE, which forms the epsilon subunit of the F1 complex, would presumably decouple the F1 and F0 rotors, and thus decouple ATP synthesis and proton transport. Conservation of these genes in the absence of photosynthesis suggests the ATP synthase complex might have evolved an alternative function in the phaeophycean parasite. Here, relaxed selection intensity was inferred for these genes rather than an intensification of positive selection as would be expected of this scenario (). Interestingly, the genes for ATP synthase are also conserved in the reduced plastid of the heterotrophic green alga Prototheca wickerhamii (Knauf & Hachtel, Citation2002).
Novel approaches are needed to fully extract biological information from accumulating marine genomic resources. Our objectives not only showcase the utility of whole genome sequencing to reveal eukaryotic diversity of the kelp holobiont, but also the underlying biology of these diminutive species. Some details related to methodology warrant consideration for future work. In at least one case, rbcL reads from two species of Phaeostroma appeared to be present in a specimen of Alaria (TTB000062; ). As the reads could not be manually teased apart, nor resolved through various assembly attempts, we were unable to resolve the rbcL contig for the second species of Phaeostroma. We also faced the inherent challenge of linking up results from multiple markers within host samples, an issue that is probably exacerbated when sampling older kelp tissue (i.e. more time for colonization by multiple species). New analytical pipelines are needed to automate and overcome these challenges, and ultimately upscale and enhance species detection and genome assembly from environmental samples. Another consideration for rigorous ecological study of kelp epi-endophytes/fauna using HTS data is the threshold for detection, which will vary according to sampling methods (habits of the collector, the amount of tissue taken for DNA and the position of that tissue on the specimen), integrity of the sample prior to DNA extraction, the extraction procedures themselves, sequencing methods, amount of DNA reads produced, and downstream analysis. Here, we expect our methods only detected the most abundant and obvious taxa (in fact, low coverage diatom contigs were also evident in our assembly of TTB000062, indicating epi-endophytes remained uncharacterized). Our findings also highlight the importance of characterizing epi-endophyte/faunal communities for population genomic work of kelp, the read data from which need to be removed or avoided prior to analysis to avoid the risk of conflating biological signals from multiple species (particularly abundant organellar DNA).
In conclusion, our study showcases how whole genome sequencing data can be used to reveal epi-endophyte/faunal associations with kelp, and indicates unique eukaryotic lineages remain to be characterized, even in the well-studied brown algae. The identified putative phaeophycean parasite mitochondrial genome and plastid scaffolds, along with annotations, can be accessed via GenBank (Supplementary table S1 and/or https://doi.org/10.6084/m9.figshare.13140464), and are expected to facilitate further analysis of brown algal organellar genome evolution. The host mitochondrial and plastid genomes are additionally provided for A. esculenta s.l. (MT767059/MT767060) and A. esculenta s.s. (MT767061/MT767062). Our analysis here was an admittedly passive assessment of epi-endophyte/faunal diversity. A dedicated study incorporating multiple host species, temporal sampling and broad geographic scope is certain to reveal further novel diversity with implications for our understanding of brown algal evolution. Moreover, as HTS data become increasingly available, so too will our knowledge of kelp epi-endophyte/faunal associations.
Supplementary table S1. New GenBank accessions for epi-endophyte sequences, including whole organellar genome data in Alaria and the phaeophycean parasite. s.l. = sensu lato, s.s. = sensu stricto.
Supplementary table S2. GenBank accession information for sequences used in phylogenetic analyses.
Supplementary table S3. Epi-endophytes detected in specimens of Alaria.
Supplementary figure S1. Regression analysis of coxI and rbcL contig coverage for epi-endophytes detected in shotgun sequencing libraries for Alaria.
Supplementary figure S2. Maximum-likelihood tree depicting placement of the phaeophycean parasite.
Author contributions
T. T. Bringloe: conceived the study, conducted the analysis, wrote the manuscript; R. Sauermann: conceived the study, conducted the analysis, wrote the manuscript; D. Krause-Jensen: provided specimens and edited the manuscript; B. Olesen: provided specimens and edited the manuscript; A. Klimova: provided specimens and edited the manuscript; T. A. Klochkova: provided specimens and edited the manuscript; H. Verbruggen: conceived the study, conducted the analysis and edited the manuscript.
Supplemental Material
Download MS Word (387.3 KB)Acknowledgements
We thank the collectors of Bay of Fundy Alaria samples: Marie Dankworth, Cody Brooks, Josh Evans and Dr Gary Saunders. We additionally thank Dr Gary Saunders for providing cox1 and rbcL data for C. californica and M. globosum, and Kavitha Uthanumallian for providing guidance in the HyPhy analysis. We further thank the Greenland ecosystem monitoring programme (g-e-m.dk) ‘Nuuk Basis’, which provided logistic support for sampling in Greenland. We also recognize the Traditional Inhabitants of both ceded and unceded territory on which this research was conducted, including the Passamaquoddy Tribe of the Wabanaki confederation (Atlantic Canada), the Kalaallit Inuit (Greenland), the Ainu, Chuvans, Evens, Itelmen and Koryaks tribes (Kamchatka Peninsula) and the Wurundjeri, Boonwurrung, Taungurong, Dja Dja Wurrung and Wathaurung people of the Kulin Nation (Melbourne, Australia). We also acknowledge that gains in contemporary knowledge invariably build on a history of race, gender, and sexual orientation discrimination.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary information
The following supplementary material is accessible via the Supplementary Content tab on the article’s online page at https://doi.org/10.1080/09670262.2021.1882704
Additional information
Funding
References
- Assis, J., Araújo, M.B. & Serrão, E.Á. (2018). Projected climate changes threaten ancient refugia of kelp forests in the North Atlantic. Global Change Biology, 24: e55–e66.
- Beck, N. & Lang, B. (2010). MFannot, organelle genome annotation webserver. http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl.
- Bella, J., Bao, Y., Gloor, G., Burton, J. & Reid, G. (2013). High throughput sequencing methods and analysis for microbiome research. Journal of Microbiological Methods, 95: 401–414. https://doi.org/10.1016/j.mimet.2013.08.011
- Bjærke, M.R. & Fredriksen, S. (2003). Epiphytic macroalgae on the introduced brown seaweed Sargassum muticum (Yendo) Fensholt (Phaeophyceae) in Norway. Sarsia North Atlantic Marine Science, 88: 353–364.
- Bringloe, T.T., Starko, S., Wade, R.M., Vieira, C., Kawai, H., Clerck, O., Cock, J.M., Coelho, S.M., Destombe, C., Valero, M., Neiva, J., Pearson, G.A., Faugeron, S., Serrão, E.A. & Verbruggen, H. (2020). Phylogeny and evolution of the brown algae. Critical Reviews in Plant Sciences, 39: 281–321.
- Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17: 540–552.
- Cremen, M.C.M., Huisman, J.M., Marcelino, V.R. & Verbruggen, H. (2016). Taxonomic revision of Halimeda (Bryopsidales, Chlorophyta) in south-western Australia. Australian Systematic Botany, 29: 41–54.
- Delmont, T.O., Gaia, M., Hinsinger, D.D., Fremont, P., Guerra, A.F., Eren, A.M., Vanni, C., Kourlaiev, A., d’Agata, L., Clayssen, Q., Villar, E., Labadie, K., Cruaud, C., Poulain, J., Da Dilva, C., Wessner, M., Noel, B., Aury, J-M., Tara Oceans Coordinators, de Vargas, C., Bowler, C., Karsenti, E., Pelletier, E., Wincker, P. & Jaillon, O. (2020). Functional repertoire convergence of distantly related eukaryotic plankton lineages revealed by genome-resolved metagenomics. bioRxiv: https://doi.org/10.1101/2020.10.15.341214.
- Deshmukhe, G.V. & Tatewaki, M. (2001). Phenology of brown alga Coilodesme japonica (Phaeophyta, Dictyosiphonales) with respect to the host-specificity along Muroran coast, North Pacific Ocean, Japan. Indian Journal of Marine Science, 30: 161–165.
- Dierckxsens, N., Mardulyn, P. & Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data, Nucleic Acids Research, 45: e18.
- Fuller, P.L., Knott, D.M., Kingsley-Smith, P.R., Morris, J.A., Buckel, C.A., Hunter, M.E. & Hartman, L.D. (2014). Invasion of Asian tiger shrimp, Penaues monodon Fabricius, 1798, in the western north Atlantic and Gulf of Mexico. Aquatic Invasions, 9: 59–70.
- Gauna, M.C., Cáceres, E.J. and Parodi, E.R. (2015). Spatial and temporal variability in algal epiphytes on Patagonian Dictyota dichotoma (Dictyotales, Phaeophyceae). Aquatic Botany, 120: 338–345.
- Hardy, S.M., Carr, C.M., Hardmann, M., Steinke, D., Corstorphine, E. & Mah, C. (2010). Biodiversity and phylogeography of Arctic marine fauna: insights from molecular tools. Marine Biodiversity, 41: 195–210.
- Hebert, P.D.N., Cywinska, A., Ball, S.L. & deWaard, J.R. (2003). Biological identifications through DNA barcodes. Proceedings of the Royal Society of London B, 270: 313–321.
- Heesch, S., & Peters, A.F. (1999). Scanning electron microscopy observation of host entry by two brown algae endolithic in Laminaria saccharina (Laminariales, Phaeophyceae). Phycological Research, 47: 1–5.
- Heesch, S., Peters, A.F., Broom, J.E. & Hurd, C.L. (2008). Affiliation of the parasite Herpodiscus durvillaeae (Phaeophyceae) with the Sphacelariales based on DNA sequence comparisons and morphological observations. European Journal of Phycology, 43: 283–295.
- Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P. & Drummond, A. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28: 1647–1649.
- Klochkova, T.A., Klimova, A.V. & Klochkova, N.G. (2018). Infraspecies forms of Alaria esculenta (Laminariales, Ochrophyta) in the marine flora of Eastern Kamchatka: first revision. Bulletin оf Kamchatka State Technical University, 43: 74–86.
- Knauf, U. & Hachtel, W. (2002). The genes encoding subunits of ATP synthases are conserved in the reduced plastid genomes of the heterotrophic alga Prototheca wickerhamii. Molecular Genetics and Genomics, 267: 492–497.
- Kraan, S. (2020). Concise review of the genus Alaria Greville, 1830. Journal of Applied Phycology. https://doi.org/10.1007/s10811-020-02222-0.
- Kuckuck, P. (1955). Ectocarpaceen-Studien III Protectocarpus nov. gen. Helgoländer wissenschaftliche Meeresuntersuchungen, 5: 119–140.
- Langmead, B. & Salzberg, S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods, 9: 357–359.
- Le Gall, L. & Saunders, G.W. (2010). DNA barcoding is a powerful tool to uncover algal diversity: a case study of the Phyllophoraceae (Gigartinales, Rhodophyta) in the Canadian flora. Journal of Phycology, 46: 374–389.
- Leray, M. & Knowlton, N. (2014). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proceedings of the National Academy of Sciences USA, 112: 2076–2081.
- Liu, F., Zhang, Y., Bi, Y., Chen, W. & Moejes, F.W. (2018). Understanding the evolution of mitochondrial genomes in Phaeophyceae inferred from mitogenome of Ishige okamurae (Ishigeales) and Dictyopteris divaricata (Dictyotales). Journal of Molecular Evolution, 87: 16–26.
- Marcelino, V.R., & Verbruggen, H. (2016). Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Scientific Reports, 6: 31508.
- Murúa, P., Küpper, F.C., Muñoz, L.A., Bernard, M. & Peters, A.F. (2018). Microspongium alariae in Alaria esculenta: a widely-distributed non-parasitic brown algal endophyte that shows cell modifications within its host. Botanica Marina, 61: 343–354.
- Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., Prjibelsky, A., Pyshkin, A., Sirotkin, A., Sirotkin, Y., Stepenauskas, R., McLean, J., Lasken, R., Clingenpeel, S.R., Woyke, T., Tesler, G., Alekseyev, M.A. & Pevzner, P.A. (2013). Assembling genomes and mini-metagenomes from highly chimeric reads. In Research in Computational Molecular Biology (Deng, M., Jiang, R., Sun, F. & Zhang, X., editors). Springer, Berlin.
- Oliveira, M.C., Repetti, S.I., Iha, C., Jackson, C.J., Díaz-Tapia, P., Lubiana, K.M.F., Cassano, V., Costa, J.F., Cremen, M.C.M., Marcelino, V.R. & Verbruggen, H. (2018). High-throughput sequencing for algal systematics. European Journal of Phycology, 53: 256–272.
- Pedersen, P.M. (1981). Culture studies on the rare brown alga Phaeostroma longisetum comb. nov. and its common relative P. pustulosum from Greenland. Nordic Journal of Botany, 1: 271–276.
- Pedersen, P.M. (2011). Grønlands havalger. Epsilon.dk. (In Danish.)
- Preuss, M., Verbruggen, H. & Zuccarello, G.C. (2020). The organelle genomes in the photosynthetic red algal parasite Pterocladiophila hemisphaerica (Florideophyceae, Rhodophyta) have elevated substitution rate and extreme gene loss in the plastid genome. Journal of Phycology, 56: 1006–1018.
- Qiu, Z., Coleman, M.A., Provost, E., Campbell, A.H., Kelaher, B.P., Dalton, S.J., Thomas, T., Steinberg, P.D. & Marzinelli, E.M. (2019). Future climate change is predicted to affect the microbiome and condition of habitat-forming kelp. Proceedings of the Royal Society B: Biological Sciences, 286: 20181887.
- Ratnasingham, S. & Hebert, P.D.N. (2013). A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS ONE, 8: e66312.
- Rodríguez, G. & Suárez, H. (2001). Anthropogenic dispersal of decapod crustaceans in aquatic environments. Interciencia, 26: 282–288.
- Ronowicz, M., Wlodarska-Kowalczuk, M. & Kuklinski, P. (2008). Factors influencing hydroids (Cnidaria: Hydrozoa) biodiversity and distribution in arctic kelp forest. Journal of Marine Biological Association of the United Kingdom, 88: 1567–1575.
- Round, F.E., Crawford, R.M. & Mann, D.G. (1990). The Diatoms: Biology and Morphology of the Genera. Cambridge University Press, New York.
- Ryberg, M. & Nilsson, R.H. (2018). New light on names and naming of dark taxa. MycoKeys, 30: 31–39.
- Salomaki, E.D., Nickles, K.R. & Lane, C.E. (2015). The ghost plastid of Choreocolax polysiphoniae. Journal of Phycology, 51: 217–221.
- Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30: 1312–1313.
- Starko, S., Bringloe, T.T., Gomez, M.S., Darby, H., Graham, S.W., Martone, P.T. (in review). Genomic rearrangements and sequence evolution across brown algal organelles.
- Stekoll, M.S. (2019). The seaweed resources of Alaska. Botanica Marina, 62: 227–235.
- Tanaka, N. (1986). Adhesive strength of epiphytic diatoms on various seaweeds. Bulletin of the Japanese Society of Scientific Fisheries, 52: 817–821.
- Tragin, M. & Vaulot, D. (2019). Novel diversity within marine Mamiellophyceae (Chlorophyta) unveiled by metabarcoding. Scientific Reports, 9: 5190.
- Tyler-Walters, H. (2003). A hydroid (Obelia longissima). In Marine Life Information Network: Biology and Sensitivity Key Information Reviews (Tyler-Walters, H. & Hiscock, K., editors). Plymouth: Marine Biological Association of the United Kingdom.
- Van der Loos, L.M., Eriksson, B.K. & Salles, J.F. (2019). The macroalgal holobiont in a changing sea. Trends in Microbiology, 27: 635–650.
- Wertheim, J.O., Murrell, B., Smith, M.D., Pond, S.L.K. & Scheffler, K. (2015). RELAX: detecting relaxed selection in a phylogenetic framework. Molecular Biology and Evolution, 32: 820–832.
- Wlodarska-Kowalczuk, M., Kuklinski, P., Ronowicz, M., Legezynska, J. & Gromisz, S. (2009). Assessing species richness of macrofauna associated with macroalgae in Arctic kelp forests (Hornsund, Svalbard). Polar Biology, 32: 897–905.
- Zerbino, D.R. & Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, 18: 821–829.