Abstract
The scavenger receptor SR-BI plays an important role in the hepatic clearance of HDL cholesterol and other lipids, driving reverse cholesterol transport and contributing to protection against atherosclerosis in mouse models. We characterized the role of endocytosis in lipid uptake from HDL, mediated by the human SR-BI, using a variety of approaches to inhibit endocytosis, including hypertonic shock, potassium or energy depletion and disassembly of the actin cytoskeleton. Our studies revealed that unlike mouse SR-BI, human SR-BI-mediated HDL-lipid uptake was reduced by inhibition of endocytosis. This was not dependent on the cytoplasmic C-terminus of SR-BI. Monitoring the uptake of both the protein and lipid components of HDL revealed that although overall lipid uptake was decreased, the degree of selective lipid uptake was increased. These data suggest that that endocytosis is a dynamic regulator of SR-BI's selective lipid uptake activity.
Introduction
HDL plays an important role in protection against atherosclerosis in part by mediating reverse cholesterol transport from macrophage foam cells in atherosclerotic plaque, to the liver Citation[1]. Studies in gene-targeted and transgenic mice revealed that the multi-ligand receptor SR-BI (scavenger receptor, class B type I) plays an important role in HDL metabolism, control of plasma HDL-cholesterol levels and reverse cholesterol transport (Citation[1] and references therein). Elimination of SR-BI expression increases and hepatic overexpression reduces atherosclerosis in mouse models (Citation[1] and references therein). Mice deficient in both SR-BI and apoE develop severe occlusive coronary artery atherosclerosis and myocardial infarction and exhibit cardiac functional and conductance abnormalities prior to early death by 8 weeks of age Citation[2]. Elimination of SR-BI in bone marrow derived cells, including macrophages, also results in increased atherosclerosis but without altering plasma lipoprotein cholesterol levels Citation[3] (also Citation[1] and references therein). Thus, both hepatic and macrophage SR-BI protect against atherosclerosis, and reverse cholesterol transport may be only one of multiple pathways involved.
SR-BI is heavily glycosylated, fatty acylated and is localized to lipid rafts and/or caveolae in many but not all cell types Citation[4–9]. It undergoes internalization and recycling back to the cell surface. In polarized cells, cholesterol depletion induces SR-BI redistribution from the basal to apical membrane surface in a protein kinase A dependent manner Citation[9]. In differentiating 3T3-L1 adipocytes and HepG2 human hepatoma cells, recruitment of SR-BI to the cell surface from internal sites is induced by insulin and/or serum and is dependent on the phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B) signaling pathway Citation[10], Citation[11]. In a variety of cell types, HDL is internalized and undergoes resecretion or retro-endocytosis back to the cell surface, in an SR-BI-dependent manner Citation[12–14]. HDL-dependent cholesterol efflux mediated by murine SR-BI appears to involve HDL internalization and retroendocytosis Citation[15]. On the other hand, murine SR-BI-mediated lipid uptake from either HDL or LDL is not affected by inhibition of endocytosis, and can occur in a purified reconstituted system, containing only SR-BI in proteoliposomes devoid of other cellular facotors Citation[16–18]. This suggests that endocytosis is not required for murine SR-BI mediated lipid uptake from either HDL or LDL. The role of endocytosis in HDL lipid uptake mediated by the human (h) SR-BI, (also called CLA-1, or CD36 and LIMP-2 related antigen 1) is not known. Analysis of the role of endocytosis in human SR-BI-mediated lipid uptake from lipoproteins will provide insight into whether the lipid uptake pathway is conserved between mouse and human SR-BI and may help to identify functionally important regions of the molecule.
In this study, we examine the role of endocytosis in human SR-BI-mediated HDL-lipid uptake in transfected Chinese hamster ovary derived cells. We demonstrate that human SR-BI-mediated HDL-lipid uptake is sensitive to inhibition of endocytosis by hypertonic shock or depletion of potassium or energy. This suggests that endocytosis is normally required for HDL-lipid uptake mediated by human SR-BI. Despite the decrease in overall HDL lipid uptake when endocytosis was inhibited, there was an increase in the degree of selective lipid uptake, measured as the ratio of HDL lipid/HDL protein taken up. We also demonstrate that the endocytosis-dependent HDL lipid uptake was not affected by mutation or deletion of the cytoplasmic C-terminus of human SR-BI. These findings suggest that endocytosis is a dynamic regulator of SR-BI's selective lipid uptake activity.
Materials and methods
Materials
Plasmids peGFP-C1 and pDsRed-N1 were from BD Clontech (Palo Alto, CA, USA). The Quickchange site-directed mutagenesis kit was from Stratagene (Palo Alto, CA, USA). Plasmid, pmRFP, containing monomeric red fluorescent protein (mRFP) Citation[19] was from Dr R. Tsien (University of California, San Diego, USA). The eGFP-clathrin light chain expression plasmid Citation[20] was from Dr J. H. Keen (Thomas Jefferson University, Philadelphia, USA). Anti-SR-BI495 antibody, and the ldlA7 and ldlA[mSR-BI] cell lines were from Dr M. Krieger (Massachusetts Institute of Technology, Cambridge, USA). Rabbit anti-SR-BI-C-terminus polyclonal antibody NB 101–400 was from Novus Biologicals (Littleton, CO, USA). The mouse anti-GFP monoclonal antibody was from Stressgen Bioreagents Inc. (Cedarlane Laboratories Inc, Hornby ON, Canada). Polyclonal rat saposin A antibody was from CR Morales (McGill University, Montreal Canada) and cathepsin A antibody was from Y Suzuki (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). Alexa 596-transferrin, 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI), TA-cloning vector and Lipofectin transfection reagent were from Invitrogen Canada Inc (Burlington, ON, Canada). All other reagents were from sources previously described Citation[23].
Methods
Generation of expression plasmids
For peGFP-hSR-BI, (human SR-BI tagged on its N-terminus with enhanced green fluorescent protein (eGFP)), PCR was used to introduce Sal1 and Kpn1 restriction sites on either end of the human SR-BI cDNA, which was inserted into the TA cloning vector, released with Sal1 and Kpn1 and ligated to peGFP-C1 similarly digested. Sequence alterations arising from PCR Citation[21] were corrected by site-directed mutagenesis. Plasmid pmRFP-hSR-BI (hSR-BI tagged on its N-terminus with mRFP) was generated from peGFP-hSR-BI by replacing the coding sequence of eGFP with that for mRFP. Plasmid phSR-BI (encoding untagged hSR-BI) was generated by inserting hSR-BI cDNA with its stop codon into pDsRed-N1. The mutant plasmids peGFP-hSR-BI Y471D, peGFP-hSR-BI Y471F and peGFP-hSR-BIΔC (containing a stop codon after the codon encoding I464) were generated from peGFP-hSR-BI by site-directed mutagenesis. All plasmids were sequenced (MOBIX core facility, McMaster University).
Cell lines and culture conditions
LdlA7 Chinese hamster ovary (CHO) mutant cells, lacking functional LDL receptor, and ldlA7 cells stably overexpressing mouse Citation[22] or human SR-BI (tagged or untagged) were cultured as described previously Citation[4], Citation[23]. LdlA7 cells were Lipofectin-transfected with appropriate plasmids and exposed to G418 (0.5 mg/ml) selection. EGFP-hSR-BI- or mRFP-hSR-BI–transfected cells were subjected to fluorescence activated cell sorting (FACS) for expressing cells. Cells transfected with untagged human SR-BI, as well as ldlA[mSR-BI] cells were incubated with DiI-HDL (5 µg/ml) for 2 h at 37°C and subjected to FACS to enrich for cells expressing functional SR-BI. In each case, cells with the highest levels of fluorescence were collected, pooled and expanded.
HDL preparation and labeling
HDL was prepared from human plasma by KBr density gradient ultracentrifugation and was labeled with DiI according to standard protocols Citation[22], Citation[24]. DiI/alexa 488-double labeled HDL was prepared as described previously Citation[23]. Newborn calf lipoprotein deficient serum (NCLPDS) was prepared by KBr density gradient centrifugation of newborn calf serum Citation[25]. Maleyl-BSA was prepared as described Citation[26].
Cell surface biotinylation
Cells, cultured in media containing 3% NCLPDS, were released from dishes by mild trypsinization, washed and allowed to recover for 20 min at 37°C in Ham's F12 medium without additions. Cells were subjected to energy depletion (see below) and cell surface proteins were biotinylated by treatment of cells in suspension with sulfo-NHS-biotin as described previously Citation[23]. Preparation of cell lysates, capture of biotinylated proteins and analysis by SDS-PAGE and immunoblotting were as described previously Citation[4], Citation[23].
Treatment of cells and analysis of lipoprotein lipid and protein uptake
Potassium depletion Citation[27], exposure to hypertonic (0.45 M) sucrose Citation[28], and treatment with 10 µM of either cytochalasin D or colchicine were done as described previously Citation[23]. Cells were treated with Ro31-8220 or wortmannin for 10 min at 37°C with 1 ml of Ham's F12 containing 0.5% BSA without additions. For energy depletion, cells were washed with Hank's balanced salt solution (HBSS) either without (control) or with (energy depletion) 5 mM NaN3 and 50 mM 2-deoxyglucose for 30 min at 37°C Citation[17]. Labeled lipoproteins (to 5 µg/ml), with or without competitors (unlabeled HDL, AcLDL or maleyl-BSA, each to 200 µg/ml), were added and incubations were continued for 2 h at 37°C.
Cell association of fluorescent lipoproteins and flow cytometry
Cell association of DiI or alexa488 after incubation at 37°C, or cell binding after incubation at 4°C with fluorescently labeled lipoproteins were measured using either a Cytofluor 4000 fluorescence plate reader (Perkin Elmer Life Sciences) or by flow cytometry. For fluorescence detection using the fluorescence plate reader, cells were cultured in Falcon Optilux-Plus black walled, clear bottom 96 well dishes (BD Biosciences, Franklin Lakes, NJ USA). Cells were treated and incubated with labeled lipoproteins as described above. DiI fluorescence was measured using 530±25 nm excitation and 590±35 nm emission filters. Cell lysates were prepared with PBS containing 0.1% Triton X-100 and the protein content in each well was assayed. Flow cytometric analysis of DiI or alexa 488 fluorescence was as described previously Citation[23]. The geometric mean fluorescence value of the population of cells was taken as a measure of the level of uptake (DiI or alexa 488) or eGFP-hSR-BI expression for each sample. Specific uptake was determined as the difference between total uptake (in the absence of competitors) and non-specific uptake (determined by including a 40-fold excess of either unlabeled HDL, AcLDL, or maleyl-BSA as competitors). In all cases, untransfected ldlA7 cells were included as controls. Cell sorting was performed as described above using a BD Biosciences FACSVantage instrument.
Fluorescence microscopy
Live cell confocal microscopy was performed using either a Perkin Elmer Life and Analytical Sciences Ultraview, or a Quorum Technologies Inc Wave FX spinning disc confocal microscopy system. Briefly, ldlA[eGFP-hSR-BI] cells (50,000/dish) were seeded in medium containing 3% NCLPDS and cultured for two days on glass-bottom, 35 mm dishes (Bioptics, Beaver Falls, PA). Cells were treated for 1 h with 70 µM cycloheximide and imaged at 37°C.
Immunofluorescence staining of fixed, permeabilized cells was performed as described previously Citation[4]. For transferrin uptake, cells were cultured on poly-D-lysine coated glass slides, treated as described above and incubated at 37°C for 30 min with 50 µg/ml alexa 594-transferrin. Cells were fixed with 2.5% paraformaldehyde and stained with DAPI prior to mounting. Cells were imaged by epifluorescence microscopy using a Zeiss Axiovert 200 M wide-field inverted microscope or by confocal microscopy using a Zeiss LSM 510 laser scanning confocal microscope (Carl Zeiss Canada). Images were processed either using Axiovision or Simple PCI imaging software.
Results
Differential sensitivities of human and mouse SR-BI to protein kinase inhibitors and inhibition of endocytosis
We compared the sensitivity of human and murine SR-BI-mediated HDL lipid uptake to the phosphatidylinositol 3-kinase inhibitor wortmannin and the general protein kinase C inhibitor Ro31-8220. Both human and murine SR-BI were overexpressed in an LDL receptor deficient mutant CHO cell line, ldlA7, allowing us to monitor SR-BI-mediated lipid uptake in the absence of LDL receptor function Citation[22], Citation[29]. We monitored the transfer of the fluorescent lipid DiI from labeled lipoproteins into cells using a well-characterized lipid transfer assay Citation[22]. LdlA[hSR-BI] or ldlA[mSR-BI] cells cultured in the presence of serum were treated with increasing concentrations of wortmannin for 10 min prior to and during incubation with DiI-HDL (A and B). DiI uptake was measured using a fluorescence plate reader and normalized to cellular protein content. Wortmannin treatment of ldlA[hSR-BI] cells resulted in a dose dependent decrease in DiI uptake from DiI-HDL, to ∼50% at the highest concentrations (1 and 5 µM) tested (A). In contrast, DiI uptake in ldlA[mSR-BI] cells was relatively insensitive to wortmannin, exhibiting only a 20% reduction at the highest wortmannin concentration (B). Treatment of ldlA[mSR-BI] cells with increasing concentrations (0.5–10 µM) of Ro31-8220 resulted in a dose dependent inhibition of DiI uptake from DiI-HDL by ∼60% at the highest Ro31-8220 concentrations (D). HDL-lipid uptake in ldlA[hSR-BI] cells, on the other hand, was not affected by Ro31-8220 at any concentration tested (C). Neither wortmannin nor Ro31-8220 affected the levels of expression of hSR-BI in transfected cells (data not shown). Thus, HDL lipid uptake mediated by human and murine SR-BI in ldlA7 cells differ in their sensitivity to PI3K and PKC inhibitors. This suggests that, despite their similar ligand binding properties and primary sequences (80% identity in predicted amino acid sequences Citation[21], Citation[22]), human and murine SR-BI mediate HDL lipid uptake by pathways that are subject to different regulation in CHO-derived cells.
Figure 1. Effects of PI3K or PKC inhibition on HDL-lipid uptake mediated by human or mouse SR-BI. LdlA[hSR-BI] cells (Panels A and C) and ldlA[mSR-BI] cells (Panels B and D), were treated without (black bars) or with (grey bars) increasing concentrations of either wortmannin (Panels A and B) or Ro31-8220 (Panels C and D) for 10 min prior to and during incubation for 2 h at 37°C with 5 µg/ml DiI-HDL. Untransfected ldlA7 cells (white bars) were incubated with DiI-HDL in parallel. Cells were washed and fluorescence in each well was determined using a cytofluor 3000 fluorescence plate reader as described in the Methods section. DiI-uptake is expressed as relative fluorescence units normalized to the amount of protein in each well. Results are averages±standard deviations of three determinations.
![Figure 1. Effects of PI3K or PKC inhibition on HDL-lipid uptake mediated by human or mouse SR-BI. LdlA[hSR-BI] cells (Panels A and C) and ldlA[mSR-BI] cells (Panels B and D), were treated without (black bars) or with (grey bars) increasing concentrations of either wortmannin (Panels A and B) or Ro31-8220 (Panels C and D) for 10 min prior to and during incubation for 2 h at 37°C with 5 µg/ml DiI-HDL. Untransfected ldlA7 cells (white bars) were incubated with DiI-HDL in parallel. Cells were washed and fluorescence in each well was determined using a cytofluor 3000 fluorescence plate reader as described in the Methods section. DiI-uptake is expressed as relative fluorescence units normalized to the amount of protein in each well. Results are averages±standard deviations of three determinations.](/cms/asset/409d47a0-a81c-42a5-9ded-0310a269d908/imbc_a_229945_f0001_b.gif)
The PI3K and PKC signaling pathways participate in the regulation of a variety of endocytic, phagocytic and membrane transport processes, including SR-BI dependent phagocytosis of apoptotic cells Citation[30], endocytosis of the β-adrenergic receptor Citation[31] and receptor recycling/recruitment to the cell surface Citation[32], Citation[33]. Recently, wortmannin has been shown to block insulin/serum stimulated recruitment of SR-BI to the cell surface in serum starved human hepatoma HepG2 cells and mouse 3T3-L1 adipocytes Citation[10], Citation[11]. Murine SR-BI mediates lipid uptake from HDL and LDL via a process that does not require endocytosis Citation[16], Citation[17]. However endocytosis and re-secretion may be necessary for SR-BI mediated lipid efflux Citation[15]. We therefore compared the effects of inhibitors of endocytosis on human and murine SR-BI-mediated lipid uptake from HDL.
Hypertonic shock or potassium depletion both prevent the interaction of clathrin with adaptor proteins and reduce the formation of clathrin coated pits on the cell surface, blocking clathrin-dependent endocytosis Citation[27], Citation[28], Citation[34], Citation[35]. Both treatments also block clathrin-independent endocytosis, involving lipid rafts, although the mechanisms are not clear Citation[28]. The cellular uptake of alexa 594-transferrin by control or treated ldlA[hSR-BI] cells was monitored as a control for the effects of these treatments on endocytosis (A–C). Transferrin, taken up by clathrin-mediated endocytosis Citation[35], Citation[36], accumulates in the perinuclear endosomal recycling compartment (A). As previously observed by others Citation[16], Citation[35], potassium depletion (B) and hypertonic shock (C) each substantially decreased perinuclear fluorescence and therefore cellular internalization of transferrin compared to control untreated cells (A). Potassium depletion and hypertonic shock also significantly decreased hSR-BI-mediated DiI-uptake from HDL by 70 and 60%, respectively, almost to the low level of DiI-uptake in control ldlA7 cells which do not overexpress SR-BI (F and G). In contrast, potassium depletion and hypertonic shock only slightly (and non-significantly) reduced HDL-lipid uptake by ldlA[mSR-BI] cells overexpressing murine SR-BI (H and I), consistent with the findings of others using either DiI or 3H cholesterol to follow HDL-lipid uptake. These findings suggests that unlike murine SR-BI Citation[16], Citation[17], human SR-BI mediates lipid uptake from HDL by an endocytosis-sensitive pathway.
Figure 2. Effects of inhibition of endocytosis on HDL-lipid uptake by human or mouse SR-BI. LdlA[hSR-BI] or ldlA[mSR-BI] were either depleted of intracellular potassium, exposed to hypertonic sucrose, 10 µM cytochalasin D or 5 mM NaN3 and 50 mM 2-deoxyglucose (Energy Depletion) as described in the Methods section. A-E: Alexa 594 transferrin uptake (red fluorescence) by ldlA[hSR-BI] cells was monitored as a control for endocytosis. Cell nuclei were stained with DAPI (blue fluorescence). Representative images are shown. Equivalent results were obtained for ldlA[mSR-BI] cells (not shown). Scale bars = 10 µm. Panels F–I: Cells were either depleted of potassium (panels F and H) or exposed to hypertonic sucrose (panels G and I) prior to and during incubation with 5 µg/ml of DiI-HDL for 2 h at 37°C in the absence or presence of 200 µg/ml of maleyl-BSA (a general competitor for SR-BI-mediated lipoprotein lipid uptake). Cellular uptake of DiI was measured by flow cytometry. Specific uptake of DiI was determined as the difference in the mean fluorescence values of the population of cells incubated without and with maleyl-BSA (equivalent results were obtained when unlabeled lipoproteins were used instead of maleyl-BSA). Values were normalized to control, untreated ldlA[hSR-BI] (panels F and G; black bars, set at 100% for ease of comparison) or ldlA[mSR-BI] cells (panels H and I; black bars, set at 100% for ease of comparison). Grey bars correspond to treated cells and white bars correspond to untreated control untrasnfected ldlA7 cells. Data are averages±standard deviations of triplicates. Stars (*) indicate p<0.05 for comparisons with untreated ldlA[hSR-BI] or ldlA[mSR-BI] cells by Student's t-test (Microsoft Excel).
![Figure 2. Effects of inhibition of endocytosis on HDL-lipid uptake by human or mouse SR-BI. LdlA[hSR-BI] or ldlA[mSR-BI] were either depleted of intracellular potassium, exposed to hypertonic sucrose, 10 µM cytochalasin D or 5 mM NaN3 and 50 mM 2-deoxyglucose (Energy Depletion) as described in the Methods section. A-E: Alexa 594 transferrin uptake (red fluorescence) by ldlA[hSR-BI] cells was monitored as a control for endocytosis. Cell nuclei were stained with DAPI (blue fluorescence). Representative images are shown. Equivalent results were obtained for ldlA[mSR-BI] cells (not shown). Scale bars = 10 µm. Panels F–I: Cells were either depleted of potassium (panels F and H) or exposed to hypertonic sucrose (panels G and I) prior to and during incubation with 5 µg/ml of DiI-HDL for 2 h at 37°C in the absence or presence of 200 µg/ml of maleyl-BSA (a general competitor for SR-BI-mediated lipoprotein lipid uptake). Cellular uptake of DiI was measured by flow cytometry. Specific uptake of DiI was determined as the difference in the mean fluorescence values of the population of cells incubated without and with maleyl-BSA (equivalent results were obtained when unlabeled lipoproteins were used instead of maleyl-BSA). Values were normalized to control, untreated ldlA[hSR-BI] (panels F and G; black bars, set at 100% for ease of comparison) or ldlA[mSR-BI] cells (panels H and I; black bars, set at 100% for ease of comparison). Grey bars correspond to treated cells and white bars correspond to untreated control untrasnfected ldlA7 cells. Data are averages±standard deviations of triplicates. Stars (*) indicate p<0.05 for comparisons with untreated ldlA[hSR-BI] or ldlA[mSR-BI] cells by Student's t-test (Microsoft Excel).](/cms/asset/8475c019-7968-4bdc-bfc8-de021ccce369/imbc_a_229945_f0002_b.jpg)
Inhibition of endocytosis increases the efficiency of selective uptake by human SR-BI
We tested the ATP requirement of hSR-BI mediated lipid uptake from HDL by pretreatment of cells for 30 min with 0.5 mM NaN3 and 50 mM 2-deoxyglucose (‘energy depletion’) Citation[17]. This reduced the perinuclear accumulation of alexa 594-transferrin (E) confirming that it reduced endocytosis. Energy depletion resulted in a slight increase in the biotinylation of hSR-BI in ldlA[hSR-BI] cells and a slight decrease in the biotinylation of mSR-BI in ldlA[mSR-BI] cells (A, top panel) suggesting that it had opposite effects on the cell surface localization of human and murine SR-BI. Total human or murine SR-BI levels in cell lysates were not affected (A, bottom panel). In contrast, energy depletion did not significantly affect the level of DiI-HDL binding to either ldlA[hSR-BI] or ldlA[mSR-BI] cells, measured at 4°C (B). We measured the degree of selective lipid uptake in control and energy depleted ldlA[hSR-BI] cells by monitoring the uptake of both the protein and lipid portions of HDL simultaneously using HDL doubly labeled with alexa 488 and DiI, and 2-colour flow cytometric analysis (). Untreated control ldlA[hSR-BI] cells were incubated in the absence or presence of excess unlabeled HDL competitor (200 µg/ml). Untreated ldlA[hSR-BI] cells exhibited high levels of DiI lipid and alexa 488 protein uptake, which were each reduced by a similar amount (3–4 fold) when a 40-fold mass excess of unlabeled HDL was included (, panels A and B) such that the ratio of cell association of HDL-lipid/HDL-protein ( panel C) was not affected. Cells subjected to energy depletion exhibited an approximately 60% reduction in DiI uptake ( panel A) and a greater than 90% reduction in alexa 488 uptake ( panel B). Thus, the ratio of DiI/alexa 488 uptake ( panel C) increased by almost 5-fold compared to untreated control cells or to cells incubated with double labeled HDL in the presence of unlabeled HDL competitor. These data suggest that energy depletion decreased HDL-lipid uptake by human SR-BI by decreasing the internalization of HDL particles rather than by decreasing the transfer of lipids from associated HDL particles to cells. In contrast, energy depletion did not alter the efficiency of murine SR-BI-mediated HDL lipid uptake Citation[17]. Thus, endocytosis appears to play an important role in human SR-BI-mediated HDL lipid uptake normally, and inhibition of endocytosis can result in an increase in the degree of selective HDL lipid uptake mediated by human SR-BI.
Figure 3. Effects of energy depletion on human or mouse SR-BI cell surface localization and HDL binding. LdlA[hSR-BI] and ldlA[mSR-BI] cells were subjected to energy depletion or control treatment as described in the Methods section. Cells were chilled to 4°C, and either released from dishes for cell surface biotinylation (panel A) or incubated with 5 µg/ml DiI-HDL for 1 h at 4°C to monitor HDL binding (panel B). (A) Cell surface biotinylation, protein solubilization and streptavidin pull-down were performed as described in the Methods section. One half of the pull-down sample (top panel) and total cell lysate equivalent to 1/70 of the pull down sample (bottom panel) were analyzed by SDS-PAGE (10% acrylamide) and immunoblotting with an antibody that recognizes the C-terminus of SR-BI and a donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase. Detection was by enhanced chemiluminescence. (B) Control (black bars) or energy depleted (grey bars) ldlA[hSR-BI] or ldlA[mSR-BI] or untreated control ldlA7 cells (white bars) were incubated with 5 µg/ml DiI-HDL for 1 h at 4°C, prior to washing and analysis of DiI-fluorescence using a fluorescence plate reader. Values are the means±standard deviations of triplicate samples and are expressed as the percent binding relative to untreated ldlA[hSR-BI] or ldlA[mSR-BI] cells.
![Figure 3. Effects of energy depletion on human or mouse SR-BI cell surface localization and HDL binding. LdlA[hSR-BI] and ldlA[mSR-BI] cells were subjected to energy depletion or control treatment as described in the Methods section. Cells were chilled to 4°C, and either released from dishes for cell surface biotinylation (panel A) or incubated with 5 µg/ml DiI-HDL for 1 h at 4°C to monitor HDL binding (panel B). (A) Cell surface biotinylation, protein solubilization and streptavidin pull-down were performed as described in the Methods section. One half of the pull-down sample (top panel) and total cell lysate equivalent to 1/70 of the pull down sample (bottom panel) were analyzed by SDS-PAGE (10% acrylamide) and immunoblotting with an antibody that recognizes the C-terminus of SR-BI and a donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase. Detection was by enhanced chemiluminescence. (B) Control (black bars) or energy depleted (grey bars) ldlA[hSR-BI] or ldlA[mSR-BI] or untreated control ldlA7 cells (white bars) were incubated with 5 µg/ml DiI-HDL for 1 h at 4°C, prior to washing and analysis of DiI-fluorescence using a fluorescence plate reader. Values are the means±standard deviations of triplicate samples and are expressed as the percent binding relative to untreated ldlA[hSR-BI] or ldlA[mSR-BI] cells.](/cms/asset/f471a1d2-f898-427c-984f-3cf924db0a29/imbc_a_229945_f0003_b.gif)
Figure 4. Inhibition of endocytosis increases the efficiency of selective HDL-lipid uptake mediated by hSR-BI. LdlA[hSR-BI] cells were either not treated (‘Control’ and ‘ + HDL’) or subjected to energy depletion (ED), potassium depletion, or treatment with cytochalasin D or 1 µM wortmannin (as indicated). DiI/alexa 488-double labeled HDL (5 µg/ml) was added without or with 200 µg/ml unlabeled HDL (‘ + HDL’). After a 2 h incubation at 37°C, two-color flow cytometric analysis was used to measure uptake of both DiI (Panel A – ‘Lipid Uptake’) and alexa 488 (Panel B – ‘Protein Uptake’). (C) For each sample, the ratio of DiI /alexa 488 fluorescence of the cell population was determined as a measure of the ratio of HDL lipid/protein uptake. Data are the averages±standard deviations of three independent samples.
![Figure 4. Inhibition of endocytosis increases the efficiency of selective HDL-lipid uptake mediated by hSR-BI. LdlA[hSR-BI] cells were either not treated (‘Control’ and ‘ + HDL’) or subjected to energy depletion (ED), potassium depletion, or treatment with cytochalasin D or 1 µM wortmannin (as indicated). DiI/alexa 488-double labeled HDL (5 µg/ml) was added without or with 200 µg/ml unlabeled HDL (‘ + HDL’). After a 2 h incubation at 37°C, two-color flow cytometric analysis was used to measure uptake of both DiI (Panel A – ‘Lipid Uptake’) and alexa 488 (Panel B – ‘Protein Uptake’). (C) For each sample, the ratio of DiI /alexa 488 fluorescence of the cell population was determined as a measure of the ratio of HDL lipid/protein uptake. Data are the averages±standard deviations of three independent samples.](/cms/asset/fe89e238-d3eb-4ded-b09d-e24793cdf195/imbc_a_229945_f0004_b.gif)
We explored this further by testing the effects of potassium depletion or treatment with cytochalasin D or wortmannin on the level of HDL-lipid and protein uptake in ldlA[hSR-BI] cells incubated with double labeled HDL. Actin filaments are involved in multiple steps in endocytic pathways, participating in both ligand internalization and in vesicular trafficking to and from the degradative and/or recycling compartments. Cytochalasin D disrupts actin polymerization and impairs both ligand uptake and trafficking, as revealed by a reduction in perinuclear alexa594-transferrin fluorescence in treated versus control cells (A and D). Cytochalasin D treatment of ldlA[hSR-BI] cells did not substantially affect HDL-lipid uptake (A) but did inhibit the cell association of HDL-protein (B), resulting in a greater than 2-fold increase in the ratio of uptake of HDL lipid/protein (C). On the other hand, potassium depletion decreased cell association of both HDL protein (84%, B) and to a lesser extent lipid (55% A), and resulted in an approximately 2.5-fold increase in the ratio of uptake of HDL lipid/HDL protein (C). Therefore, depletion of intracellular potassium or disruption of the actin cytoskeleton each resulted in an increased degree of selective lipid uptake mediated by hSR-BI, although they differed in their effects on HDL protein and lipid uptake. Treatment of cells with wortmannin resulted in similar reductions in both HDL-lipid and HDL-protein uptake (A and B), without altering the degree of selective lipid uptake from HDL (C). This suggests that wortmannin-mediated the inhibition of both endocytic and non-endocytic pathways.
Fluorescent protein tagged-hSR-BI is functional
To visualize the dynamics of the distribution of hSR-BI in live cells, we generated human SR-BI tagged on its N-terminus with either eGFP or mRFP. EGFP-tagged human SR-BI exhibited lower mobility than either untagged human or mouse SR-BI upon SDS-PAGE and immunoblotting (A), consistent with increased size of the polypeptide resulting from the eGFP-fusion. After incubation with 5 µg/ml DiI-HDL, ldlA[eGFP-hSR-BI] cells (overexpressing eGFP-hSR-BI) exhibited ∼20-fold greater HDL-lipid uptake than either untransfected ldlA7 cells (B) or ldlA[eGFP-hSR-BI] cells that had not been incubated with DiI-HDL (not shown). Similar results were obtained using cells expressing mRFP-tagged hSR-BI (not shown). Therefore, fluorescent protein tagged hSR-BI was functional.
Figure 5. Expression and activity of eGFP-tagged human SR-BI in transfected ldlA7 cells. (A) LdlA[mSR-BI] (lane 1) and ldlA7 cells (lanes 2, 3 and 5) were cultured in medium containing 5% FBS. LdlA7 cells were either not transfected (lane 2) or transfected with hSR-BI (lane 3) or eGFP-hSR-BI expression plasmids (lane 5). After 3 days, cell lysates were prepared and analyzed by SDS-PAGE and immunoblotting for SR-BI. Lane 4 is empty. (B) Cells transfected with the eGFP-hSR-BI expression plasmid were subjected to G418 selection and cell sorting as described in Materials and Methods, to generate ldlA[eGFP-hSR-BI] cells that stably overexpress eGFP-hSR-BI. Untransfected ldlA7 (grey histogram) and ldlA[eGFP-hSR-BI] (black histogram) cells were incubated with 5 µg/ml DiI-HDL for 2 h at 37°C. DiI uptake was measured by flow cytometry as described in the Methods section. Representative histograms are shown.
![Figure 5. Expression and activity of eGFP-tagged human SR-BI in transfected ldlA7 cells. (A) LdlA[mSR-BI] (lane 1) and ldlA7 cells (lanes 2, 3 and 5) were cultured in medium containing 5% FBS. LdlA7 cells were either not transfected (lane 2) or transfected with hSR-BI (lane 3) or eGFP-hSR-BI expression plasmids (lane 5). After 3 days, cell lysates were prepared and analyzed by SDS-PAGE and immunoblotting for SR-BI. Lane 4 is empty. (B) Cells transfected with the eGFP-hSR-BI expression plasmid were subjected to G418 selection and cell sorting as described in Materials and Methods, to generate ldlA[eGFP-hSR-BI] cells that stably overexpress eGFP-hSR-BI. Untransfected ldlA7 (grey histogram) and ldlA[eGFP-hSR-BI] (black histogram) cells were incubated with 5 µg/ml DiI-HDL for 2 h at 37°C. DiI uptake was measured by flow cytometry as described in the Methods section. Representative histograms are shown.](/cms/asset/4a4a6614-4e0a-4e63-9b26-74c26c1ac562/imbc_a_229945_f0005_b.gif)
EGFP-hSR-BI and mRFP-hSR-BI exhibited similar distributions in stably expressing cells. They localized to the cell surface at regions of cell-cell contact (A, B and G–J) and in tether-like extensions between cells (A, G, H). EGFP- and mRFP fluorescence was also detected in punctate vesicle-like structures within cells (A, B and G–J). Similar patterns of SR-BI localization were detected by immunofluorescence in permeabilized ldlA[mSR-BI] (C) Citation[4] and ldlA[hSR-BI] cells (data not shown). The mRFP-hSR-BI-positive, punctate, vesicle-like structures observed in ldlA[mRFP-hSR-BI] cells did not co-localize with the lysosomal markers cathepsin A or saposin A or with eGFP-clathrin light chain at 37°C in ldlA7 cells stably expressing both ().
Figure 6. Localization and dynamics of eGFP- hSR-BI. LdlA[eGFP-hSR-BI], ldlA[mRFP-hSR-BI] and ldlA[mSR-BI] cells were cultured on poly-D-lysine-coated glass coverslips fixed to 35 mm culture dishes. Cells were fixed with 2.5% paraformaldehyde and either visualized directly (panels A and B) or after permeabilization and indirect immunofluorescence using anti-SR-BI495 antibody and alexa 488-anti-rabbit secondary antibody (panel C) using wide-field fluorescence microscopy with standard rhodamine and FITC filter sets. Panels D–F correspond to DIC images of the cells shown in A–C. Scale bars = 10 µm. (G–J) Live-cell confocal imaging of ldlA[eGFP-hSR-BI] cells was performed as described in the Methods section. Panel G on the left shows three adjacent ldlA[eGFP-hSR-BI] cells. Scale bar = 10 µm. The white boxes represent regions that are magnified in panels H–J. Panel H shows a time series at 50 sec intervals, demonstrating eGFP-hSR-BI dynamics associated with a tether between two adjacent cells. The first panel (time 0 sec) is cropped and magnified from panel G. The arrow serves as a reference marker to illustrate the leftward motion of eGFP-hSR-BI fluorescence along the tether. Scale bar = 5 µm. This series of images corresponds to Supplementary Video 1 – online version only. Panel I shows images at 10 sec intervals, beginning at time 115 sec relative to the image in panel G. They illustrate the motion of an eGFP-hSR-BI-positive puncta (small box) towards the center of the cell. The arrow indicates an eGFP-hSR-BI-positive structure that is static within the time-frame of the images. Scale bar = 5 µm. This series of images corresponds to Supplementary Video 2 – online version only. Panel J shows a series of images taken at 5 sec intervals and focusing on the region of cell-cell contact between two adjacent cells. The cell on the upper left in this panel corresponds to the cell in the lower right in panels H and I (see panel G). Scale bar = 2.5 µm. The arrow indicates the position at which an SR-BI positive puncta, apparently derived from the cell surface, appears. This corresponds to Supplementary Video 3 – online version only.
![Figure 6. Localization and dynamics of eGFP- hSR-BI. LdlA[eGFP-hSR-BI], ldlA[mRFP-hSR-BI] and ldlA[mSR-BI] cells were cultured on poly-D-lysine-coated glass coverslips fixed to 35 mm culture dishes. Cells were fixed with 2.5% paraformaldehyde and either visualized directly (panels A and B) or after permeabilization and indirect immunofluorescence using anti-SR-BI495 antibody and alexa 488-anti-rabbit secondary antibody (panel C) using wide-field fluorescence microscopy with standard rhodamine and FITC filter sets. Panels D–F correspond to DIC images of the cells shown in A–C. Scale bars = 10 µm. (G–J) Live-cell confocal imaging of ldlA[eGFP-hSR-BI] cells was performed as described in the Methods section. Panel G on the left shows three adjacent ldlA[eGFP-hSR-BI] cells. Scale bar = 10 µm. The white boxes represent regions that are magnified in panels H–J. Panel H shows a time series at 50 sec intervals, demonstrating eGFP-hSR-BI dynamics associated with a tether between two adjacent cells. The first panel (time 0 sec) is cropped and magnified from panel G. The arrow serves as a reference marker to illustrate the leftward motion of eGFP-hSR-BI fluorescence along the tether. Scale bar = 5 µm. This series of images corresponds to Supplementary Video 1 – online version only. Panel I shows images at 10 sec intervals, beginning at time 115 sec relative to the image in panel G. They illustrate the motion of an eGFP-hSR-BI-positive puncta (small box) towards the center of the cell. The arrow indicates an eGFP-hSR-BI-positive structure that is static within the time-frame of the images. Scale bar = 5 µm. This series of images corresponds to Supplementary Video 2 – online version only. Panel J shows a series of images taken at 5 sec intervals and focusing on the region of cell-cell contact between two adjacent cells. The cell on the upper left in this panel corresponds to the cell in the lower right in panels H and I (see panel G). Scale bar = 2.5 µm. The arrow indicates the position at which an SR-BI positive puncta, apparently derived from the cell surface, appears. This corresponds to Supplementary Video 3 – online version only.](/cms/asset/d2eb3838-a105-498a-907a-085bab518a8e/imbc_a_229945_f0006_b.gif)
Figure 7. mRFP-tagged hSR-BI does not co-localize with lysosomal markers or with clathrin. LdlA[mRFP-hSR-BI] cells (panels A–F) and ldlA7 cells stably expressing both eGFP-clathrin and mRF-hSR-BI (panels G–I) were cultured on glass cover slips and fixed. Cells were permeabilized and immunostained for lysosomal markers cathepsin A (panel A) or saposin A (panel D). Alexa 488 labeled secondary antibody was used for detection prior to wide-field epifluorescence microscopy using a Zeiss Axiovert 200 M inverted microscope. Alternatively, cells were imaged directly (panels G–I) using a Zeiss LSM510 laser scanning confocal microscope. A, D and G: Alexa 488 or eGFP fluorescence. B, E and H: mRFP fluorescence. C, F and I: merged color images in which green corresponds to cathepsin A (panel C), saposin A (panel F) or eGFP-clathrin (panel I) and red corresponds to mRFP-hSR-BI. Scale bars = 10 µm. Representative images are shown.
![Figure 7. mRFP-tagged hSR-BI does not co-localize with lysosomal markers or with clathrin. LdlA[mRFP-hSR-BI] cells (panels A–F) and ldlA7 cells stably expressing both eGFP-clathrin and mRF-hSR-BI (panels G–I) were cultured on glass cover slips and fixed. Cells were permeabilized and immunostained for lysosomal markers cathepsin A (panel A) or saposin A (panel D). Alexa 488 labeled secondary antibody was used for detection prior to wide-field epifluorescence microscopy using a Zeiss Axiovert 200 M inverted microscope. Alternatively, cells were imaged directly (panels G–I) using a Zeiss LSM510 laser scanning confocal microscope. A, D and G: Alexa 488 or eGFP fluorescence. B, E and H: mRFP fluorescence. C, F and I: merged color images in which green corresponds to cathepsin A (panel C), saposin A (panel F) or eGFP-clathrin (panel I) and red corresponds to mRFP-hSR-BI. Scale bars = 10 µm. Representative images are shown.](/cms/asset/722822c9-9770-44e8-b52a-31af156f2471/imbc_a_229945_f0007_b.jpg)
Live-cell confocal imaging revealed the dynamics of the SR-BI containing structures in ldlA[eGFP-hSR-BI] cells. For these experiments, cells were treated with cycloheximide for 1 hr to block new protein synthesis prior to imaging at 37°C. EGFP-hSR-BI in cell surface extensions between cells appeared to be most concentrated at either one or both ends of the extension and to exhibit motion towards one of the cells (G and H and Supplementary Video 1 – online version only). SR-BI at the cell surface at regions of cell-cell contact appeared to localize in smaller extensions between cells, diffusely along the cell periphery and in punctae within cells, adjacent to the periphery (G–J). EGFP-hSR-BI in the smaller extensions exhibited a similar mobility to that in the longer extensions, although it was difficult to determine direction (H and Supplementary Video 1 – online version only). SR-BI at the cell periphery in regions of cell-cell contact was also dynamic (I and J and Supplementary Videos 2 and 3 – online version only). EGFP-hSR-BI was also present in punctae within cells that appeared to represent vesicles. These were highly dynamic, moving both radially towards the center of cells as well as circumferentially (I and Supplementary Video 2 – online version only). Some of these appeared to originate at or adjacent to the cell periphery at regions of cell-cell contact (I and J and Supplementary Videos 2 and 3 – online version only). These observations are consistent with evidence that SR-BI undergoes continuous internalization from and recycling back to the cell surface Citation[9], Citation[12], Citation[14], and suggests that at least some of these punctate structures represent hSR-BI-containing vesicles either derived from or destined for the cell surface.
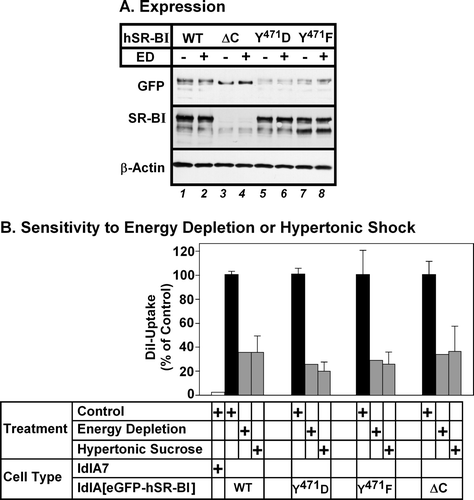
Mutations in human SR-BI's C-terminus do not alter its sensitivity to inhibition of endocytosis
To explore whether the carboxy terminal cytoplasmic domain of hSR-BI was required for its endocytosis-dependent HDL lipid uptake, we generated eGFP-tagged point or deletion mutants targeting this region and compared their expression and activity to eGFP-tagged wild type hSR-BI. The sequence Y471LFW in the cytoplasmic C-terminus of human SR-BI has the appearance of a potential endocytic motif of the form YXXφ (where X is any amino acid and φ is hydrophobic) Citation[37]. This is conserved in bovine, pig and rabbit SR-BI sequences, but those of mouse, rat and hamster contain F in place of Y471Citation[21], Citation[22], Citation[29], Citation[38–41]. To test if this contributed to the requirement for endocytosis for hSR-BI-mediated HDL lipid uptake, we generated point mutants in which Y471 was changed to either F (to mimic mouse SR-BI) or D (to mimic potential Y-phosphorylation Citation[37]). We also generated a deletion mutant lacking the entire C-terminal cytoplasmic domain (hSR-BI-ΔC) as described by others Citation[42]. LdlA7 cells stably expressing either eGFP-hSR-BI, eGFP-hSR-BI-Y471F, eGFP-hSR-BI-Y471D or eGFP-hSR-BI-ΔC were treated with or without energy depletion. Immunoblotting with antibodies specific for either GFP or the C-terminus of SR-BI (A) and eGFP fluorescence levels (not shown) revealed that eGFP-hSR-BI and the Y471D and Y471F mutants were expressed at similar levels, whereas the eGFP-hSR-BI-ΔC mutant appeared to be expressed at a higher level. Energy depletion did not affect the level of eGFP-hSR-BI expression (A). Cells were incubated with DiI-HDL for 2 h, prior to analysis of both DiI and GFP fluorescence by flow cytometry. B shows the effects of either energy depletion or hypertonic shock on HDL-lipid uptake expressed as a percentage of the level of DiI uptake in control untreated cells. Both energy depletion and exposure to hypertonic sucrose reduced DiI-uptake from DiI-HDL to similar extents in cells expressing either wild type, Y471F/D-mutant hSR-BI, or hSR-BI lacking its C-terminal cytoplasmic tail. Therefore, neither mutation of Y471 to F or D, nor deletion of the entire C-terminal cytoplasmic region of hSR-BI affected the dependence of HDL lipid uptake on endocytosis.
Figure 8. Mutations in the C-terminal cytoplasmic domain do not affect the dependence of hSR-BI mediated lipid uptake on endocytosis. LdlA7 cells were transfected with eGFP-hSR-BI (wild type, WT), eGFP-hSR-BI-Y471F, eGFP-hSR-BI-Y471D and eGFP-hSR-BI-ΔC expression plasmids and subjected to G418 selection. (A) Detergent extracts of control and energy depleted cells were prepared and proteins were subjected to SDS-PAGE and immunoblotting using antibodies specific for GFP (top panel), SR-BI-C terminal peptide (middle panel) and β-actin (loading control, bottom panel). Equal amounts of protein were applied to each lane. (B) Cells subjected to energy depletion or exposed to hypertonic sucrose (grey bars), control untreated cells (black bars) or untransfected ldlA7 cells (white bar) were incubated with DiI-HDL and DiI uptake and GFP-fluorescence were measured as described in the Methods section. The level of DiI uptake is expressed as the percentage of control, untreated cells and data represent the average±range of two independent experiments.
Discussion
Our data suggests that human SR-BI can mediate both the endocytic uptake and selective uptake of HDL lipids in a manner that is sensitive to the PI3K inhibitor wortmannin but not the PKC inhibitor Ro31-8220. SR-BI mediates the internalization of a variety of its ligands including serum amyloid A, bacterial lipopolysaccharide, bacteria and apoptotic cells Citation[30], Citation[43–46]. It also mediates the internalization and re-secretion of HDL Citation[12], Citation[14–16]. We have found that endocytosis is necessary for human SR-BI-mediated HDL-lipid uptake in CHO-derived ldlA7 cells overexpressing human SR-BI. In contrast, murine SR-BI mediates HDL-lipid uptake via a pathway that is not affected by inhibition of endocytosis Citation[16], Citation[17] but is sensitive to the PKC inhibitor Ro31-8220 Citation[23].
We have used the fluorescent lipid, DiI, for these studies as a convenient marker of HDL lipid uptake Citation[22]. The endocytosis step is not a characteristic of DiI uptake, nor does it appear to be a characteristic of the cell type used in these studies since murine SR-BI expressed in the same cell type mediated uptake of the same lipid (DiI) from HDL in an endocytosis-independent manner ( and Citation[16]). Instead, our findings suggest that the pathway by which HDL lipids are taken up is determined by the characteristics of the receptor (human versus mouse SR-BI). Disruption of endocytosis by depletion of ATP or potassium, or by disruption of actin polymerization resulted in more pronounced decreases in the cell association of HDL protein than lipid, resulting in an apparent increase in the degree of selective lipid uptake mediated by hSR-BI. These results suggest that human SR-BI can mediate HDL lipid uptake by both endocytic and selective lipid uptake pathways, and that endocytosis contributes substantially to total lipid uptake mediated by this receptor. The increased degree of selective lipid uptake (ratio of HDL lipid/protein taken up) resulting from inhibition of endocytosis suggests that endocytosis is a dynamic regulator of SR-BI's selective lipid uptake activity.
It has recently been demonstrated that the reduced selective uptake activity of murine SR-BII, a splice variant of SR-BI with a different C-terminal cytoplasmic domain may be the result of its increased endocytosis Citation[47]. The insertion of a di-leucine endocytic motif derived from SR-BII's C-terminal cytoplasmic domain into the corresponding region of murine SR-BI resulted in its increased endocytosis and decreased mSR-BI-mediated selective uptake activity Citation[47]. This provides further support for the idea that SR-BI's selective uptake activity may be subject to regulation by modulating its endocytosis. SR-BI contains two short cytoplasmic regions at the extreme N and C-termini of ∼9 and 45 amino acid residues, respectively. We tested if the C-terminal cytoplasmic region was involved in the endocytosis-dependent HDL-lipid uptake mediated by hSR-BI. We analyzed hSR-BI mutants that either lacked the cytoplasmic C-terminus Citation[42] or contained Y471F or Y471D point mutations. None of these mutations affected the sensitivity of hSR-BI-mediated HDL lipid uptake to inhibition of endocytosis (B). Therefore the endocytosis-dependence of HDL lipid uptake by hSR-BI is not conferred by its C-terminal cytoplasmic tail.
These findings have potentially important implications for understanding the role of human SR-BI in selective lipid uptake and its regulation in vivo. Our data suggest that, at least in CHO-derived cells, human SR-BI mediated HDL lipid uptake involves endocytosis, possibly of HDL mediated by hSR-BI. Inhibition of hSR-BI-mediated HDL endocytosis results in increased efficiency of selective lipid uptake. This suggests that the process of endocytosis is a dynamic regulator of SR-BI's selective lipid uptake activity, possibly diverting HDL-derived lipids to different intracellular pools, although this remains to be tested. Identification of the features of the hSR-BI molecule that influence the endocytic versus selective lipid uptake pathway will allow for a better understanding of the regulation of these opposing pathways and potentially provide insight into means of regulating SR-BI's selective lipid uptake activity.
Legends to Supplementary Videos (online version only)
Supplementary Video 1. Live cell confocal imaging of two adjacent ldlA[eGFP-hSR-BI] cells with an SR-BI-positive tether-like extension between them as well as smaller extensions above an area of apparently direct cell-cell contact. This video corresponds to the sequence of images shown in H. Images were captured every 5 sec over a total of 205 sec and are played back at 2 frames/sec, representing 10×normal speed.
Supplementary Video 2. Live cell confocal imaging of a different region of the same two adjacent ldlA[eGFP-hSR-BI] cells as that shown in Supplemental Video 1. Shown is the region of direct contact between the two cells, exhibiting dynamic SR-BI localization. SR-BI localizes diffusely at the region of cell contact, to punctae that appear to be at and just below the cell surface and to punctae within the cells. An SR-BI-positive puncta adjacent to the cell surface can be seen migrating towards the interior of the cell on the left (see also the corresponding series of panels in I). Images were captured every 5 sec over a total of 205 sec and are played back at 2 frames/sec, representing 10×normal speed.
Supplementary Video 3. Live cell confocal imaging of two adjacent ldlA[eGFP-hSR-BI] cells. The cell on the upper left in this video is the same as the cell on the lower right in Supplementary Video 2. Shown is a region of direct contact between the two cells, exhibiting dynamic SR-BI localization. This video corresponds to a 205 sec sequence encompassing the series of images in J. The video shows two instances of eGFP-SR-BI fluorescence accumulating in what appear to be vesicles derived from the cell surface. Images were captured every 5 sec over a total of 205 sec and are played back at 2 frames/sec, representing 10×normal speed.
Acknowledgements
We thank E. Roediger for expert technical assistance. FACS was performed by H. Liang in the McMaster University Flow Cytometry Facility. We thank J. Daniel for helpful discussions, and M. Krieger, R. Tsien, S. Grinstein and J.H, Keen for generously providing reagents. This research was supported by funding from the Heart and Stroke Foundation of Ontario and the Natural Sciences and Engineering Research Council of Canada to BLT. BLT is a Heart and Stroke Foundation of Canada New Investigator and RT is a Canadian Institutes of Health Research New Investigator.
References
- Trigatti B. Role of the scavenger receptor class B type I in lipoprotein metabolism and atherosclerosis: Insights from genetically altered mice. Biochemistry of atherosclerosis. Advances in biochemistry in health and disease. 1, SC Kaur. Springer, New York 2006; 53–69
- Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 2002; 90: 270–276
- Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol 2003; 23: 1589–1594
- Babitt J, Trigatti B, Rigotti A, Smart EJ, Anderson RG, Xu S, Krieger M. Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J Biol Chem 1997; 272: 13242–13249
- Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L. Localization and regulation of SR-BI in membrane rafts of HepG2 cells. J Cell Sci 2004; 117: 3095–3105
- Levy E, Menard D, Suc I, Delvin E, Marcil V, Brissette L, Thibault L, Bendayan M. Ontogeny, immunolocalisation, distribution and function of SR-BI in the human intestine. J Cell Sci 2004; 117: 327–337
- Shaul PW. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol 2003; 547: 21–33
- Briand O, Lestavel S, Pilon A, Torpier G, Fruchart JC, Clavey V. SR-BI does not require raft/caveola localisation for cholesteryl ester selective uptake in the human adrenal cell line NCI-H295R. Biochim Biophys Acta 2003; 1631: 42–50
- Burgos PV, Klattenhoff C, de la Fuente E, Rigotti A, Gonzalez A. Cholesterol depletion induces PKA-mediated basolateral-to-apical transcytosis of the scavenger receptor class B type I in MDCK cells. Proc Natl Acad Sci USA 2004; 101: 3845–3850
- Tondu AL, Robichon C, Yvan-Charvet L, Donne N, Le Liepvre X, Hajduch E, Ferre P, Dugail I, Dagher G. Insulin and angiotensin II induce the translocation of scavenger receptor class B, type I from intracellular sites to the plasma membrane of adipocytes. J Biol Chem 2005; 280: 33536–33540
- Shetty S, Eckhardt ER, Post SR, van der Westhuyzen DR. Phosphatidylinositol-3-kinase regulates scavenger receptor class B type I subcellular localization and selective lipid uptake in hepatocytes. Arterioscler Thromb Vasc Biol 2006; 26: 2125–2131
- Rhode S, Breuer A, Hesse J, Sonnleitner M, Pagler TA, Doringer M, Schutz GJ, Stangl H. Visualization of the uptake of individual HDL particles in living cells via the scavenger receptor class B type I. Cell Biochem Biophys 2004; 41: 343–356
- Wustner D. Steady state analysis and experimental validation of a model for hepatic high-density lipoprotein transport. Traffic 2006; 7: 699–715
- Silver DL, Wang N, Xiao X, Tall AR. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J Biol Chem 2001; 276: 25287–25293
- Pagler TA, Rhode S, Neuhofer A, Laggner H, Strobl W, Hinterndorfer C, Volf I, Pavelka M, Eckhardt ER, van der Westhuyzen DR, Schutz GJ, Stangl H. SR-BI-mediated high density lipoprotein (HDL) endocytosis leads to HDL resecretion facilitating cholesterol efflux. J Biol Chem 2006; 281: 11193–11204
- Nieland TJ, Ehrlich M, Krieger M, Kirchhausen T. Endocytosis is not required for the selective lipid uptake mediated by murine SR-BI. Biochim Biophys Acta 2005; 1734: 1744–1751
- Harder CJ, Vassiliou G, McBride HM, McPherson R. Hepatic SR-BI-mediated cholesteryl ester selective uptake occurs with unaltered efficiency in the absence of cellular energy. J Lipid Res 2006; 47: 492–503
- Liu B, Krieger M. Highly purified scavenger receptor class B, type I reconstituted into phosphatidylcholine/cholesterol liposomes mediates high affinity high density lipoprotein binding and selective lipid uptake. J Biol Chem 2002; 277: 34125–34135
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA 2002; 99: 7877–7882
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol 1999; 1: 1–7
- Calvo D, Vega MA. Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J Biol Chem 1993; 268: 18929–18935
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996; 271: 518–520
- Zhang Y, Ahmed AM, McFarlane N, Capone C, Boreham DR, Truant R, Igdoura SA, Trigatti BL. Regulation of SR-BI-mediated selective lipid uptake in Chinese hamster ovary-derived cells by protein kinase signaling pathways. J Lipid Res 2007; 48: 405–416
- Chapman MJ, Goldstein S, Lagrange D, Laplaud PM. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J Lipid Res 1981; 22: 339–358
- Krieger M, Brown MS, Goldstein JL. Isolation of Chinese hamster cell mutants defective in the receptor-mediated endocytosis of low density lipoprotein. J Mol Biol 1981; 150: 167–184
- Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA 1979; 76: 333–337
- Larkin JM, Brown MS, Goldstein JL, Anderson RG. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 1983; 33: 273–285
- Carpentier JL, Sawano F, Geiger D, Gorden P, Perrelet A, Orci L. Potassium depletion and hypertonic medium reduce ‘non-coated’ and clathrin-coated pit formation, as well as endocytosis through these two gates. J Cell Physiol 1989; 138: 519–526
- Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem 1994; 269: 21003–21009
- Osada Y, Shiratsuchi A, Nakanishi Y. Involvement of mitogen-activated protein kinases in class B scavenger receptor type I-induced phagocytosis of apoptotic cells. Exp Cell Res 2006; 312: 1820–1830
- Naga Prasad SV, Jayatilleke A, Madamanchi A, Rockman HA. Protein kinase activity of phosphoinositide 3-kinase regulates beta-adrenergic receptor endocytosis. Nat Cell Biol 2005; 7: 785–796
- Fiory F, Oriente F, Miele C, Romano C, Trencia A, Alberobello AT, Esposito I, Valentino R, Beguinot F, Formisano P. Protein kinase C-zeta and protein kinase B regulate distinct steps of insulin endocytosis and intracellular sorting. J Biol Chem 2004; 279: 11137–11145
- van Dam EM, Ten Broeke T, Jansen K, Spijkers P, Stoorvogel W. Endocytosed transferrin receptors recycle via distinct dynamin and phosphatidylinositol 3-kinase-dependent pathways. J Biol Chem 2002; 277: 48876–48883
- Larkin JM, Donzell WC, Anderson RG. Modulation of intracellular potassium and ATP: effects on coated pit function in fibroblasts and hepatocytes. J Cell Physiol 1985; 124: 372–378
- Hansen SH, Sandvig K, van Deurs B. Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol 1993; 121: 61–72
- Durrbach A, Louvard D, Coudrier E. Actin filaments facilitate two steps of endocytosis. J Cell Sci 109 ( Pt 1996; 2): 457–465
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003; 72: 395–447
- Rajapaksha WR, McBride M, Robertson L, O'Shaughnessy PJ. Sequence of the bovine HDL-receptor (SR-BI) cDNA and changes in receptor mRNA expression during granulosa cell luteinization in vivo and in vitro. Mol Cell Endocrinol 1997; 134: 59–67
- Lopez D, McLean MP. Sterol regulatory element-binding protein-1a binds to cis elements in the promoter of the rat high density lipoprotein receptor SR-BI gene. Endocrinology 1999; 140: 5669–5681
- Kim JG, Vallet JL, Nonneman D, Christenson RK. Molecular cloning and endometrial expression of porcine high density lipoprotein receptor SR-BI during the estrous cycle and early pregnancy. Mol Cell Endocrinol 2004; 222: 105–112
- Ritsch A, Tancevski I, Schgoer W, Pfeifhofer C, Gander R, Eller P, Foeger B, Stanzl U, Patsch JR. Molecular characterization of rabbit scavenger receptor class B types I and II: portal to central vein gradient of expression in the liver. J Lipid Res 2004; 45: 214–222
- Cao WM, Murao K, Imachi H, Yu X, Abe H, Yamauchi A, Niimi M, Miyauchi A, Wong NC, Ishida T. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res 2004; 64: 1515–1521
- Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem 2005; 280: 2954–2961
- Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, Remaley AT, Csako G, Thomas F, Eggerman TL, Patterson AP. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem 2005; 280: 8031–8040
- Vishnyakova TG, Bocharov AV, Baranova IN, Chen Z, Remaley AT, Csako G, Eggerman TL, Patterson AP. Binding and internalization of lipopolysaccharide by Cla-1, a human orthologue of rodent scavenger receptor B1. J Biol Chem 2003; 278: 22771–22780
- Philips JA, Rubin EJ, Perrimon N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science 2005; 309: 1251–1253
- Eckhardt ER, Cai L, Shetty S, Zhao Z, Szanto A, Webb NR, Van der Westhuyzen DR. High density lipoprotein endocytosis by scavenger receptor SR-BII is clathrin-dependent and requires a carboxyl-terminal dileucine motif. J Biol Chem 2006; 281: 4348–4353