Abstract
P-type ATPases are ubiquitously abundant primary ion pumps, which are capable of transporting cations across the cell membrane at the expense of ATP. Since these ions comprise a large variety of vital biochemical functions, nature has developed rather sophisticated transport machineries in all kingdoms of life. Due to the importance of these enzymes, representatives of both eu- and prokaryotic as well as archaeal P-type ATPases have been studied intensively, resulting in detailed structural and functional information on their mode of action. During catalysis, P-type ATPases cycle between the so-called E1 and E2 states, each of which comprising different structural properties together with different binding affinities for both ATP and the transport substrate. Crucial for catalysis is the reversible phosphorylation of a conserved aspartate, which is the main trigger for the conformational changes within the protein. In contrast to the well-studied and closely related eukaryotic P-type ATPases, much less is known about their homologues in Bacteria. Whereas in Eukarya there is predominantly only one subunit, which builds up the transport system, in Bacteria there are multiple polypeptides involved in the formation of the active enzyme. Such a rather unusal prokaryotic P-type ATPase is the KdpFABC complex of the enterobacterium Escherichia coli, which serves as a highly specific K+ transporter. A unique feature of this member of P-type ATPases is that catalytic activity and substrate transport are located on two different polypeptides. This review compares generic features of P-type ATPases with the rather unique KdpFABC complex and gives a comprehensive overview of common principles of catalysis as well as of special aspects connected to distinct enzyme functions.
Introduction
P-type ATPases are grouped as a superfamily of primary ion pumps, which transport cations across lipid bilayers at the expense of ATP. Since they utilize the Gibbs free energy of ATP hydrolysis, they are capable of establishing and maintaining large electrochemical gradients across biological membranes. A key intermediate step in the catalysis of P-type ATPases is the formation of a transient phosphointermediate by the autophosphorylation of a conserved aspartic acid residue, which separates this class of enzymes from, e.g., F-type ATPases. Ion translocation is generally thought to occur by subsequent alterations in the affinity and accessibility of ion binding sites exposed to either side of the membrane. The transition of ion transfer between high- and low-affinity binding sites is accomplished by alterations in the conformational state of the enzyme, leading to the widely accepted model of the so-called E1/E2 catalytic cycle (Møller et al. Citation1996). Whereas P-type ATPases from Eukarya have been comprehensively investigated in great detail, like for example the Ca2 + -ATPase of the sarcoplasmic reticulum, the Na+, K+-ATPase of animal cells, and the H+, K+-ATPase of the gastric mucosa, much less information is available for their prokaryotic counterparts, like the CopA copper-transporting ATPase or the KdpFABC K+-translocating ATPase of the enterobacterium Escherichia coli. In case of the latter, remarkable features came to light, which clearly point to a unique status of at least this bacterial P-type ATPase with respect to its eukaryotic counterparts. Whereas also the KdpFABC complex is supposed to undergo the typical E1/E2 catalytic cycle of P-type ATPases (compare ), only one of the subunits of the complex, namely the 72 kDa KdpB, comprises all typical signature motifs of P-type ATPases and, thus, performs ATP hydrolysis.
Figure 1. Proposed reaction cycle for the bacterial KdpFABC complex. Binding of a potassium ion to the E2 state enzyme promotes high-affinity ATP binding to the N-domain of KdpB. Subsequently, the KdpFABC complex is transformed to its E1 state, and the potassium ion is able to enter the now energetically favorable binding site in the center of the selectivity filter of KdpA. Upon reaching the transition state (TSI) followed by phosphorylation, the potassium ion becomes occluded. The subsequent E1 to E2 transition, accompanied by major conformational changes in KdpB, alters the dipole formed by D583 and K586 in the transmembrane domain of KdpB. This makes the position of the potassium ion in the center of the selectivity filter of KdpA unfavorable and, consequently, pushes the ion toward the cytoplasmic side of the membrane, where it is finally released. Dephosphorylation of the complex regenerates the E2 state.
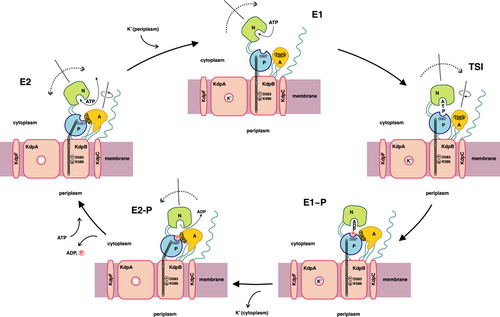
In contrast to all other P-type ATPases known so far, the translocation of the substrate (i.e., K+) is mediated by another subunit, the 59 kDa KdpA polypeptide (Altendorf et al. Citation1998). The unique status of this P-type ATPase is further underlined by the presence of two additional subunits, namely KdpF (3 kDa) and KdpC (20 kDa). Whereas the former is thought to be of structural importance for the integrity of the enzyme complex, the latter is most likely involved in catalysis. In this review, these rather unique features of the KdpFABC P-type ATPase will be put into the generic context of P-type ATPases, thereby underlining common priciples of catalysis as well as comparing special aspects, which are connected to distinct enzymatic functions.
Distribution of the KdpFABC complex and other P-type ATPases in prokaryotes
The E. coli KdpFABC system is probably the best-studied bacterial P-type ATPase. Due to the high degree of conservation, Kdp-ATPases can be easily identified in bacterial genomes. Despite the evolutionary distance between Bacteria and Archaea, organisms from both kingdoms share the same fate: the necessity of potassium uptake. Prokaryotes, both bacteria and archaea, have to respond immediately to potassium limitation in the medium due to the vital function of K+ in the cell, acting for example as enzyme co-factor, osmoprotectant or being involved in pH regulation, which explains why bacteria employ the Kdp-ATPase as a high-affinity K+-scavenging system. On the other hand, several organisms do not contain a kdp operon, some prokaryotes indeed completely lack members of the P-type ATPase family, such as Borrelia burgdorferi, Buchnera sp. APS, Rickettsia prowazekii, Xylella fastidiosa, and the archaeon Pyrococcus horikoshii. To date, 224 KdpB sequences have been found in bacterial genomes (651 finished and unfinished genomes were included in the search). Genes for KdpABC were identified in six archaeal genomes (note that KdpF is not present in all archaeal operons), namely Halobacterium sp. NRC-1, Thermoplasmaacidophilum, Ferroplasma acidarmanus Fer1, Thermoplasma volcanium GSS1, Picrophilus torridus DSM 9790, and Thermofilum pendens Hrk5. By far the most widespread P-type ATPases in bacteria (as well as in archaea) are the heavy metal-transporting ATPases (type IB). Type IB ATPases can be further subdivided into monovalent (Cu+, Ag+) and divalent (Cu2 + , Zn2 + , Pb2 + , Cd2 + , and Co2 + ) cation transporters (Lutsenko & Kaplan Citation1995, Axelsen & Palmgreen Citation1998, Rensing et al. Citation1999). Examples of bacterial type IB ATPases are the zinc-transporting ZntA (Rensing et al. Citation1997), the copper-transporting CopA (Odermatt et al. Citation1993, Citation1994), and the cadmium transporter CadA (Nucifora et al. Citation1989). Type IIA ATPases are also found in bacteria, although these Ca2 + -transporting enzymes are not very abundant in bacterial species. The Bacillus subtilis YloB gene product was shown to be a Ca2 + -transporting ATPase, which is expressed during sporulation (Raeymaekers et al. Citation2002). Another example for a Ca2 + -transporting ATPase is the PacL protein from Synechococcus sp. PCC 7942 (Berkelman et al. Citation1994). The bacterial MgtA (and MgtB) proteins belong to the type IIIB ATPases and catalyze the influx of Mg2 + (Maguire Citation1992). Interestingly, H+-ATPases, which are found in both archaea and eukarya (De Hertogh et al. Citation2004), seem to be absent in bacteria.
The KdpB subunit – the P-type ATPase subunit
Comparative topology of P-type ATPases
P-type ATPases share a conserved core topology of six transmembrane helices (TMH) and two extended cytoplasmic loop regions (compare ).
Figure 2. Schematic topology of P-type ATPases. The proposed topology of KdpB (type IA) with its seven transmembrane helices (TMH) is shown in dark gray. Heavy metal-transporting ATPases (type IB) comprise an N-terminal extension (depicted in yellow) with various metal-binding clusters and eight TMH. P-type ATPases of the type II-IV class comprise 10 TMH. The putative type V of P-type ATPases (with unknown substrate specificity) might have 12 TMH. Beside differences in the number of transmembrane helices all P-type ATPases have two large cytoplasmic domains. The first loop between TMH 2 and TMH 3 is called actuator (A-) domain. The second cytoplasmic loop is composed of two modules: The phosphorylation (P-) domain contains the conserved aspartate residue that becomes reversibly phosphorylated (circle). The nucleotide-binding (N-) domain is inserted into the P-domain and contains the ATP binding site (illustrated by ATP) (modified according to Figure 1 of Haupt et al. Citation2004).
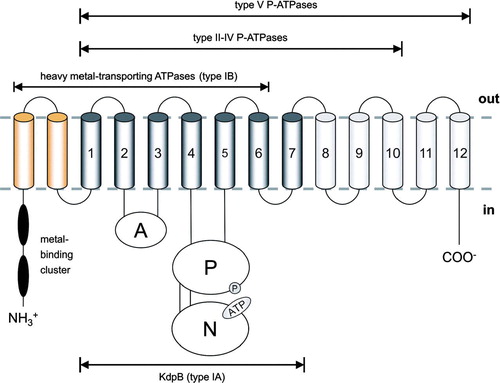
The smaller cytoplasmic loop is located between TMH 2 and 3 and is commonly named actuator (A-) domain. The larger loop, which is found between TMH 4 and TMH 5, is composed of two separate modules, which are also functionally distinct. The phosphorylation (P-) domain harbors the invariant aspartate residue and includes residues important for the hydrolysis of Mg2 + -ATP. The nucleotide-binding (N-) domain is inserted into the P-domain and contains the residues involved in the selective binding of adenosine nucleotides. These domains of various P-type ATPases have been independently expressed and purified in a functional state, suggesting that functional modules can fold autonomously (Capieaux et al. Citation1993, Moutin et al. Citation1994, Gatto et al. Citation1998, Tsivkovskii et al. Citation2001, Bramkamp & Altendorf Citation2004). Different P-type ATPases contain various insertions/extensions to the core structure, with the number of transmembrane helices ranging from seven in case of the prokaryotic KdpB polypeptide (type IA), eight for the heavy metal-transporting ATPases (type IB) together with a cytoplasmic N-terminal tail containing one ore more metal-binding clusters, ten helices for type II-IV ATPases, and, finally, twelve transmembrane helices in case of type V ATPases (which comprise no assigned substrate specificity).
The actuator (A-) domain
The first large cytoplasmic domain common to P-type ATPases is the A-domain, which is connected to the transmembrane domain by short linker sequences. In the Ca2 + -ATPase this domain is located between TMH 2 and TMH 3 and mainly consists of ten β-strands together with two α-helices (Toyoshima et al. Citation2000). The overall fold of the isolated CopA (type IB) A-domain is similar to the Ca2 + -ATPase structure (Sazinsky et al. Citation2006a). Characteristic of the A-domain is the existence of a highly conserved TGE motif, which is thought to play a role in the dephosphorylation of the E2-P form of the ATPase (Møller et al. Citation1996, Anthonisen et al. Citation2006) and which can be found in type I-V P-type ATPases and is, thus, considered a signature motif of P-type ATPases.
The phosphorylation (P-) domain
The P-domain is the most conserved module among P-type ATPases. It is composed of two parts that are separated by the insertion of the N-domain. The P-domain includes the signature motif DKTGT of P-type ATPases. The invariant Asp residue within this motif undergoes reversible phosphorylation upon ATP hydrolysis and forms the transient acylphosphate, from which the name P-type ATPase is derived. The overall secondary structure of the P-domain constitutes a classical Rossman-fold with a central six- or seven-stranded β-sheet that is flanked by α-helices (Toyoshima et al. Citation2000, Sazinsky et al. Citation2006b). The domain structure is similar to the haloacid dehalogenase fold, and, consequently, the P-type ATPases were grouped into the HAD superfamily (named after typifying L2-haloacid-dehalogenase) together with dehalogenases, phosphatases, response regulators, and epioxide hydrolases (Ridder & Dijkstra Citation1999). Because of the high structural similarity and the conservation of crucial amino acids required for hydrolysis, a common mechanism of hydrolysis among these enzymes has been proposed (Aravind et al. Citation1998). The bacterial P-type ATPases share the P-domain structure with their eukaryotic counterparts, with no large insertions or deletions within the P-domain of various enzymes. Conserved residues within the P-domain are involved in the coordination of Mg2 + , which, in turn, is needed to stabilize the γ-phosphate group of the ATP molecule during hydrolysis. During the (de)phosphorylation cycle, P-type ATPases undergo several large domain movements, which finally lead to ion transport. The ion binding sites, which are buried in the middle of the lipid bilayer, are structurally linked directly to the cytoplasmic domains performing ATP hydrolysis: one of the transmembrane helices (TMH 5 in the Ca2 + -ATPase) extends through the membrane into the P-domain and ends right underneath the phosphorylation site. In case of the Ca2 + -ATPase, the P-domain changes its internal structure and orientation with respect to TMH 5 upon binding of the γ-phosphate of ATP together with Mg2 + (Toyoshima & Mizutani Citation2004). Changes in the stranded β-sheet structure of the Rossman-fold moves the N-domain closer to the P-domain and, thus, enables the γ-phosphate to reach the reactive Asp residue in the P-domain (Toyoshima & Mizutani Citation2004).
The nucleotide-binding (N-) domain
In contrast to the collection of crystal structures of the Ca2 + -ATPase in different conformational states (Toyoshima et al. Citation2000, Citation2004, Toyoshima & Nomura Citation2002, Olesen et al. Citation2004, Sørensen et al. Citation2004, Toyoshima & Mizutani Citation2004), there is only little structural information on other P-type ATPases. However, advances have been made recently in the elucidation of structures of soluble modules like the nucleotide binding domain. The solution structures of the Wilson disease protein (WDP) (Dmitriev et al. Citation2006) and the E. coli KdpB-ATPase (Haupt et al. Citation2004, Citation2006) nucleotide binding domains were solved using NMR spectroscopy. The catalytic loop comprising P- and N-domain of the archaeal Archeoglobus fulgidus Cu+-ATPase CopA was solved by X-ray crystallography (Sazinsky et al. Citation2006b). The structure of the Na+,K+-ATPase N-domain was solved by both NMR (Hilge et al. Citation2003) and crystallography (Håkansson Citation2003). Hence, structural data for the nucleotide binding module of type IA (KdpB), type IB (WDP, CopA), and type II (Ca2 + -ATPase, Na+, K+-ATPase) P-type ATPases are now available, thus enabeling a detailed inspection of the nucleotide binding mode.
Within the catalytic loop, the N-domain is linked to the P-domain via flexible linkers. The N-domain of KdpB (KdpBN) is the smallest N-domain of all P-type ATPases and is supposed to represent the minimal core structure of this domain ().
Figure 3. Comparison of the nucleotide-binding domain structures of various P-type ATPases. A comparative view on the structures of different P-type ATPases displays the similar core structures, but also reveals the insertions found in the eukaryotic members of the P-type ATPase family. Structures of nucleotide-binding domains of KdpB [KdpBN, 2A29] (Haupt et al. Citation2006), CopA [CopA_N, 2B8E] (Sazinsky et al. Citation2006b), Wilson disease protein [WDP_N, 2ARF] (Dmitriev et al. Citation2006), Na+,K+-ATPase [NaK_N, 1Q31] (Håkansson Citation2003), and Ca2 + -ATPase [SERCA_N, 1VFP] (Toyoshima & Mizutani Citation2004) are shown side by side (panel A). KdpBN exhibits a minimal scheme of a nucleotide binding domain with a central six-stranded, anti-parallel β-sheet, flanked by two α-helices on either side. This core motif is found in all N-domains. The comparison between prokaryotic (KdpBN, CopA_N) and eukaryotic (WDP-N, NaK_N, and SERCA_N) N-domains shows that additional amino acid residues are inserted in a way that leaves the central ATP binding pocket unaffected. The different sets of amino acids that are employed by type IB heavy metal-transporting ATPases (WDP_N) on the one hand and KdpB (type IA) as well as type II ATPases (Ca2 + -ATPase) on the other hand are highlighted in panel B. Amino acid residues that interact with the nucleotide are shown in green, and the nucleotidesare depicted in red.
![Figure 3. Comparison of the nucleotide-binding domain structures of various P-type ATPases. A comparative view on the structures of different P-type ATPases displays the similar core structures, but also reveals the insertions found in the eukaryotic members of the P-type ATPase family. Structures of nucleotide-binding domains of KdpB [KdpBN, 2A29] (Haupt et al. Citation2006), CopA [CopA_N, 2B8E] (Sazinsky et al. Citation2006b), Wilson disease protein [WDP_N, 2ARF] (Dmitriev et al. Citation2006), Na+,K+-ATPase [NaK_N, 1Q31] (Håkansson Citation2003), and Ca2 + -ATPase [SERCA_N, 1VFP] (Toyoshima & Mizutani Citation2004) are shown side by side (panel A). KdpBN exhibits a minimal scheme of a nucleotide binding domain with a central six-stranded, anti-parallel β-sheet, flanked by two α-helices on either side. This core motif is found in all N-domains. The comparison between prokaryotic (KdpBN, CopA_N) and eukaryotic (WDP-N, NaK_N, and SERCA_N) N-domains shows that additional amino acid residues are inserted in a way that leaves the central ATP binding pocket unaffected. The different sets of amino acids that are employed by type IB heavy metal-transporting ATPases (WDP_N) on the one hand and KdpB (type IA) as well as type II ATPases (Ca2 + -ATPase) on the other hand are highlighted in panel B. Amino acid residues that interact with the nucleotide are shown in green, and the nucleotidesare depicted in red.](/cms/asset/94c1c9ed-53ef-4c42-af46-46e4bccd538d/imbc_a_241777_f0003_b.jpg)
In particular, the eukaryotic proteins contain larger insertions. KdpBN consists of a curved, six-stranded anti-parallel β-sheet domain flanked by two α-helices on either side (Haupt et al. Citation2004, Citation2006). The overall structures of the different N-domains (Ca2 + -ATPase, Na+, K+-ATPase, WDP, and CopA) are surprisingly similar to KdpBN, given the low sequence homology between the different P-type ATPases. However, there are some variations among these structures. For example, the eukaryotic type IB WDP N-domain, although representing a closer relative, contains a flexible loop (Dmitriev et al. Citation2006), which matches the sequence insert that is present in mammalian copper ATPases, but which is absent in the bacterial (and lower eukaryotic) homologues (for example CopA; compare ). The insertion of additional sequences into the minimal scheme of the nucleotide-binding domain present in bacterial P-type ATPases is, therefore, likely to reflect an evolutionary development. In higher organisms, transport proteins may underlie more rigid regulatory circuits compared to their prokaryotic counterparts and, hence, may have developed some extra sequences in order to enable accessory proteins to interact.
The nucleotide binding mechanism
Binding of ATP to the nucleotide-binding domain of P-type ATPases is a crucial step in the reaction cycle of these proteins. The elucidation of N-domain structures of the Ca2 + -ATPase, the Na+, K+-ATPase, WDP, CopA, and KdpB made the comparison of nucleotide-binding to various type IA, type IB, and type II ATPases possible. It became apparent that the actual nucleotide binding is accomplished by distinct sets of amino acid residues, although the overall structure of the various N-domains is highly conserved.
ATP binding to the N-domain of KdpB was analyzed by NMR in both the absence (apo-form) and presence (holo-form) of nucleotide and, thus, provided a detailed picture of the binding mechanism in solution. The structures of apo- and holo-KdpBN were almost identical with notably exceptions in only two regions (Haupt et al. Citation2005, Citation2006). Helix α1 extents by three residues upon nucleotide-binding, and the loop connecting β2 and β3 closes down by 15° on the ligand as the curved β-sheet stretches. P276 and S384 represent the pivotal points in β2 and β3, respectively. The nucleotide is positioned by side chains of β3 and β5 and is capped by helices α1 and α2. Contacts between the enzyme and the nucleotide were surprisingly limited in case of KdpBN. F377 interacts via an aromatic Π-stacking with the purine ring, and the ξ-amino group of K395 approaches the purine ring from the other side, thereby resulting in a cation/Π-stacking (). The observed low binding constants of around 1.4 mM for AMP-PNP and ATP (Bramkamp & Altendorf Citation2004, Haupt et al. Citation2006) were explained by the low binding energy of -2 to -5 kcal/mol for these kind of interactions (Haupt et al. Citation2005). D344 forms a hydrogen bond with the amino group N6 of the purine in the holo-form, while it stabilizes the charge of K395 in the apo-form of KdpBN. In addition to K395, only M383 and G396 showed NOE cross-peaks to the H2 and H1′ hydrogens of AMP-PNP. No NOEs were described between the enzyme and the ribose moiety (possible consequences thereof are discussed in the KdpC part of this review). The phosphate residues are protruding into the solvent away from the binding site. This orientation may be stabilized by two arginine residues, R317 and R382, which are located at the outer rim of the binding pocket. E348 and D399 are possibly involved in the stabilization of these two arginines.
Nucleotide binding to the N-domain of the eukaryotic Ca2 + -ATPase is similar when compared to KdpBN. The Ca2 + -ATPase structure with bound AMP-PCP revealed that F487 interacts via Π-Π-stacking with the purine ring, and M494 and E442 may contribute side chain hydrogen bonds. The role of K515 (which corresponds to K395 in KdpB) is not yet clear in comparison to the KdpBN structure. The ξ-amino group of K515 is not positioned correctly in order to contribute to a possible cation-Π-stacking, but rather forms a salt bridge with E442 in the apo- and holo-form of the enzyme, while the corresponding salt bridge is opened in the holo-KdpBN. The phosphate groups are also positioned by two arginine residues in case of the Ca2 + -ATPase, namely R489 and R560. These residues are, thus, functionally homologous to R317 and R382 in case of KdpBN. Despite the slight differences it seems plausible that P-type ATPases sharing the conserved KGxxD/E motif (with the lysine residue being K395 in KdpBN and K515 in Ca2 + -ATPase) have a similar nucleotide binding mechanism. When comparing the bacterial type IA and the eukaryotic type II ATPases, the fundamental contacts between the ATPases and the substrate are essentially the same, with only some differences in the fine-tuning of the binding mechanisms.
Nucleotide binding in type IB ATPases is performed by a complete different set of amino acid residues and, hence, resembles a significant difference between the heavy metal-transporting type IB ATPases and the potassium-transporting type IA ATPases.
The Archeoglobus Cu+-transporting ATPase CopA resembles a prokaryotic version of the heavy metal-transporting ATPases and may serve as a structural model for bacterial type IB P-type ATPases. The overall structure of the nucloetide binding domain of CopA is almost identical to that of KdpB with a root mean square deviation of 1.54 Å. Although the crystal structure was in the apo-form, an overlay with the KdpBN structure revealed that the nucleotide is essentially located in a corresponding position between the central β-strands and the capping α-helices. Five amino acids, E457, H462, G490, G492, and G501 (Archeoglobus CopA numbering) are conserved in type IB ATPases and are predicted to interact with the nucleotide. E457, H462, and G492 seem to orient and/or contact the purine ring through hydrogen bonding, and G490 and G501 interact with the ribose moiety and the α-phosphate. Comprehensive mutagenesis of the corresponding residues in the Zn2 + -transporting ATPase ZntA of E. coli (Bissig et al. Citation2001, Okkeri et al. Citation2002, Citation2004) underlined the importance of these residues for ATP binding in this class of ATPases. It is interesting to note that the same structural elements are conserved in the N-domains of all P-type ATPases, while the amino acid sequences are highly variable. However, only the type IB ATPases have a distinct mode of nucleotide binding, which more closely resembles the nucleotide-binding mechanism in protein kinases such as cAPK protein kinase with their GxGxxG motif (Zheng et al. Citation1993, Okkeri et al. Citation2004).
The solution structure of the human Wilson disease protein was solved in its holo-form () and confirmed the contribution of residues E1064, H1069, G1099, and G1101 to nucleotide binding (the corresponding residues in CopA are E457, H462, G490, G492). Furthermore, NOEs between H1′ of the ribose and G1149 (G501 in CopA) and 1Hα, 1Hβ, 1HN, and N1150 were described. I1102 and I1180 coordinate the purine ring, whereas G1149 and N1150 bind the ribose moiety. The glycine residue 1149 is analogous to G396 in KdpB, although in this enzyme the glycine residues are not involved in ribose binding. These differences may explain the 10-fold higher affinity of ATP for the WDP N-domain compared to KdpB.
The transmembrane domain
As already mentioned, the transmembrane domain of P-type ATPase comprises various numbers of α-helices, ranging from 7–12 depending on the transported substrate and, hence, their individual subclass (type I – type V). The prokaryotic KdpB protein most likely has the rather unique number of seven TM helices (Bramkamp & Altendorf Citation2005). Within the Ca2 + -ATPase, a clear segregation between TMH 1-6 and TMH 7-10 is observed (Toyoshima et al. Citation2000), which is consistent with the lack of TMH 7-10 in the type IB ATPases.
The fact that KdpB might have seven instead of six TMHs separates the KdpB subunit clearly from the related heavy metal-transporting P-type ATPases. The hypothesis that the KdpFABC complex represents an ancestor of the later-evolved P-type ATPases might, therefore, not be true. More likely, the KdpB subunit might have lost TMH 8–10 (which are found in the larger eukaryotic P-type ATPases) when it acquired the channel-like subunit KdpA.
The transmembrane domain of P-type ATPases usually harbors the ion binding sites. A detailed picture of the ion binding sites in their various states is so far only available for the Ca2 + -ATPase (Toyoshima et al. Citation2000, Toyoshima & Nomura Citation2002, Sørensen et al. Citation2004 . TMH 4 and TMH 6 of the Ca2 + -ATPase are unwound within the membrane, and TMH 10 is kinked. Especially the unwound areas of TMH 4 and TMH 6 contribute to the Ca2 + binding sites. In cooperation with TMH 8, the helices TMH 4-TMH 6 provide side chain and backbone oxygen atoms to coordinate the calcium ions. The channel, by which the ions gain access to the cavity (occluded state), is located between TMH 2, TMH 4, and TMH 6. The dissociation (or binding) of Ca2 + is mediated by large rearrangements of six (TMH 1-TMH 6) out of the ten transmembrane helices. The reason for the unwinding of TMH 4 is a proline residue in the conserved PEGL motif (Møller et al. Citation1996). In fact, the Pro residue is conserved throughout the P-type ATPases, thereby suggesting a universal role for the correct function of the transport process. In case of the type IB heavy metal transporting ATPases, the corresponding residue is within the typifying CPx-motif in TMH 6 (which is analogous to TMH 4 in other P-type ATPases). The essential role of the cysteine residue of the CPx motif has been shown for bacterial P-type ATPases (Bissig et al. Citation2001, Fan & Rosen Citation2002, Liu et al. Citation2006, Dutta et al. Citation2007). For the zinc-transporting ATPase ZntA it was shown recently that additional charged residues in TMH 7 and TMH 8 may contribute to the metal-binding site together with the CPC motif (Okkeri & Haltia Citation2006). Thus, it is clear that all P-type ATPases from type IB to type V seem to use a similar ion binding region within the TM domain of the central subunit with only little adjustments in the amino acid composition of the distinct binding sites.
In contrast to all other P-type ATPases, the prokaryotic KdpFABC complex exhibits a unique transport mechanism. Within the KdpFABC complex the KdpA subunit is the ion-translocating subunit, while KdpB energizes the process by the classical ATP hydrolysis-driven reaction cycle of P-type ATPases. Electrophysiological experiments with the KdpFABC complex suggest that no counterion is transported and, hence, that the K+ influx is electrogenic (Fendler et al. Citation1996, Citation1999). Since KdpB is not transporting any ion via its TM domain, it may have reduced its number of transmembrane helices during evolution. However, the polypeptide also contains a specific set of amino acids that are most likely involved in the coupling process between subunits KdpA and KdpB. Sequence comparison between KdpB and other P-type ATPases identified two charged residues (D583 and K586) within TMH 5 that are highly conserved in KdpB proteins, but not in other P-type ATPases. A mutational analysis of these residues revealed that the loss of these charges results in an uncoupling between ion transport and ATP hydrolysis (Bramkamp & Altendorf Citation2005). The two residues, which are located in the middle of the lipid bilayer, are probably forming a strong dipole. Movement of such a dipole might, however, have rather indirect than direct effects on the potassium ions bound within the selectivity filter region of KdpA.
The unique subunits
KdpA – the K+ conduit
As already mentioned, the KdpFABC complex comprises a unique spatial separation between energy conversion (i.e., ATP hydrolysis in KdpB) and substrate translocation (K+ transport mediated by KdpA) on two different polypeptides. Hence, no corresponding counterpart or domain element of KdpA can be found in other P-type ATPases nor does KdpA comprise any significant homologies to transmembrane domains of other P-type ATPases. Based on sequence alignments it has been concluded that KdpA is evolutionary derived from a homotetrameric MPM-type (membrane/P-loop/membrane) K+ channel by gene duplication and fusion events, thus assigning KdpA to the superfamily of prokaryotic MPM-type K+ channel proteins (Durell et al. Citation2000). In contrast to the well-characterized Streptomyces lividans K+ channel KcsA featuring a homotetrameric structure with each subunit comprising a single MPM motif (Doyle et al. Citation1998), KdpA was shown to possess four putative MPM motifs within a single polypeptide sequence, flanked by two additional transmembrane helices one on each side. Although primarily based on sequence similarity patterns, this hypothesis proves to be in good accord with experimental data relying on both the analysis of mutants affecting K+ selectivity and the determination of transmembrane topology (Buurman et al. Citation1995, Durell et al. Citation2000, van der Laan et al. Citation2002, Bertrand et al. Citation2004). In addition to the high selectivity for potassium – only rubidium is also tolerated – mutations affecting ion binding and selectivity of the transport complex cluster in defined regions within the KdpA polypeptide, thereby arguing in favor of these clusters to establish corresponding selectivity filter regions within the assembled K+ conduit. Corresponding sequence alignments of the P-loop regions of KdpA can be found in Durell et al. (Citation2000). The eight central helices containing the four putative MPM motifs can easily be modeled pairwise together with the connecting loop regions against the coordinates of the S. lividans KcsA K+ channel (Doyle et al. Citation1998) ().
Figure 4. MPM-type K+ channel model of KdpA. The four MPM motifs of KdpA were modeled pairwise together with the connecting loop regions against the coordinates of the S. lividans KcsA K+ channel [1K4C] (Zhou et al. Citation2001). (A) Residues effecting ion selectivity upon mutagenesis are indicated in red (significant residues) and blue (essential residues). (B) Tetrameric assembly of A, with the color code ranging from yellow to blue with increasing effect on ion selectivity upon mutagenesis.
![Figure 4. MPM-type K+ channel model of KdpA. The four MPM motifs of KdpA were modeled pairwise together with the connecting loop regions against the coordinates of the S. lividans KcsA K+ channel [1K4C] (Zhou et al. Citation2001). (A) Residues effecting ion selectivity upon mutagenesis are indicated in red (significant residues) and blue (essential residues). (B) Tetrameric assembly of A, with the color code ranging from yellow to blue with increasing effect on ion selectivity upon mutagenesis.](/cms/asset/98acac83-a284-4a81-970c-39bcde6d029d/imbc_a_241777_f0004_b.jpg)
In KdpA, each of the four KcsA-like MPM protein domains is composed of two transmembrane helices with an intervening P segment (Durell et al. Citation2000). These segments comprise a reentrant structural pattern, which is made up of a short pore helix followed by a loop. In KcsA, the four P segments are oriented in a symmetric tetrameric assembly, thereby forming the selectivity filter region of the transmembrane pore. For KdpA, a similar quaternary arrangement of the MPM domains can be assumed, since the assembly of structural elements strongly follows the KcsA pattern. In a corresponding tetrameric array of the four MPM elements, the P segments of KdpA also form a 45° tilted, descending pore helix, which in KcsA is followed by a subsequently ascending filter region, which contains the well-conserved central glycine residue. Sites of K+ binding are formed via backbone carbonyl oxygens of neighboring and opposing P segments. E. coli KdpA also features central P segment glycines within the three C-terminal P motifs, whereas in the N-terminal P segment there is an asparagine at the corresponding position. This distribution of glycines within the P segments was reported to be conserved among members of the KtrB, TrkH, and KdpA K+ transporter family (Durell et al. Citation2000), and the corresponding residue is also conserved among K+ channels (Durell et al. Citation1999). Thus, the structural model of KdpA also reflects the physiological importance of the selectivity filter glycine residue. However, it should be noted that despite the high homology of KdpA and KcsA, K+ gating in KdpA should differ from the mechanics of KcsA, since at least KdpB is also involved in ion translocation by providing energy to the site of substrate transport. Furthermore, the two additional transmembrane helices flanking the four MPM domains of KdpA could either mediate subunit interactions or could also be directly involved in the ion translocation pathway. In addition, the channel has to function as a unidirectional valve in order to prevent passive K+ backflow, which would be due to an active, ATP-driven internal accumulation of K+. This aspect renders a different gating mechanism involving accessory protein factors other than the four MPM domains even more likely.
KdpC – the ‘catalytical chaperone’
Although the KdpC subunit is reported to be an essential part of the transport complex (Gaßel & Altendorf Citation2001), only little is known so far about its specific structure and function. Based on sequence analysis, KdpC is supposed to contain just one transmembrane helix in the N-terminal region, ranging from residues 9–28, with the rest of the polypeptide facing the cytoplasm. Together with secondary structure prediction, hydropathy plot analysis, and extensive sequence alignments the polypeptide was found to comprise four domains, two of which are highly conserved among different organisms and are located within the hydrophilic portion of KdpC. Especially the highly conserved C-terminal portion of the polypeptide was found to be important, since deletion mutagenesis revealed that the truncation of 11 residues led to the inactivation of the complex (Gaßel & Altendorf Citation2001). Although the hydrophilic portion of KdpC is the only larger hydrophilic domain of the KdpFABC complex beside the P-, N-, and A-domains of the catalytic KdpB subunit, its distinct function is still unknown. An orthologous stabilizing role has been discussed with respect to the β subunit of Na+, K+- and H+, K+-ATPase in Eukarya (Axelsen & Palmgreen Citation1998). However, since the soluble extension of the β subunit faces the extracellular space, a corresponding function of KdpC seems unlikely due to an opposing inward orientation of the hydrophilic part. On the other hand, several small proteins exist within the P-type ATPase superfamily like phospholamban or calmodulin in case of the Ca2 + -ATPase (Kimura & Inui Citation2002) or the γ subunit in case of Na+, K+-ATPase (Therien et al. Citation1999), which exert a regulatory function on enzyme activity not via interactions within the membrane, but with the large cytoplasmic loop of the pump. Neurospora crassa H+-ATPase, which is also a single-subunit P-type ATPase, contains an intrinsic hydrophilic regulatory R domain at the C terminus, which is proposed to interact with the catalytic N- and P-domains upon (de-)phosphorylation by a specific kinase (Kühlbrandt et al. Citation2002). Thus, modulation of P-type ATPase activity or ATP affinity by small polypeptides comprising just one transmembrane span together with a hydrophilic extension, which interacts with the catalytic domains of the pump, is likely to be a common principle. However, in strong contrast to KdpC, these so-called FXYD proteins are not essential for enzymatic activity, thereby arguing in favor of the notion that KdpC exhibits a more unique type of regulatory function. Recently, it has been demonstrated that the isolated hydrophilic portion of KdpC (KdpCsol) specifically binds one ATP molecule at a well-defined binding site, which is in accord with former observations that upon labeling of the KdpFABC complex with the photoreactive ATP analogue 2-azido-ATP, in addition to KdpB also KdpC was labeled significantly (Ahnert et al. Citation2006). This argues in favor of also a regulatory function of KdpC either via interaction with the nucleotide binding pocket of KdpB or via ATP binding itself. Both the nucleotide affinity and specificity of KdpC was found to be quite low (Ahnert et al. Citation2006), thereby rendering a cooperative regulatory mechanism together with KdpB likely. The site of ATP binding could be located within the conserved C-terminal segment of KdpC. This stretch of residues can easily get into contact with the catalytic domains of the KdpB subunit. Such an interaction could already be abolished by C-terminal truncations of the peptide. Thus, the formation of a ternary complex of the hydrophilic domain of KdpC together with KdpBN (the nucleotide binding domain of KdpB) and the nucleotide is quite reasonable for the following aspects: Isolated KdpBN comprises a rather high binding constant for ATP of 1.4 mM (Haupt et al. Citation2006). In contrast, the other well-characterized nucleotide binding domains exhibit a much lower Kd for ATP, like 10–100 µM in case of the Ca2 + -ATPase (Abu-Abed et al. Citation2002) or 70 µM for the Wilson disease protein, which is reported to be within the typical range of simple protein/ligand systems (Dmitriev et al. Citation2006). Another special feature of KdpBN with respect to Wilson disease protein is that in the former there are no interactions between the ribose moiety of the nucleotide and the protein (Haupt et al. Citation2006), whereas in the latter the ribose is well coordinated within a proper ribose binding pocket (Dmitriev et al. Citation2006), which might explain the low ATP affinity of KdpBN. Furthermore, in KdpBN, the nucleotide has to be oriented in order to enter the binding site properly together with a precisely balanced equilibrium of nucleotide uptake and release (Haupt et al. Citation2006). Together with the low nucleotide affinity and the lack of additional coordination sites targeting the ribose moiety this renders an additional regulatory process likely. At least in case of the Wilson disease protein, there is no additional subunit or regulatory domain. Although phospholamban or calmodulin are present in the Ca2 + -ATPase, the mode of regulation most likely differs from that of KdpFABC due to an evolutionary early divergence of mammalian and bacterial P-type ion pumps. Since the Wilson disease protein is more closely related to KdpB than to the Ca2 + -ATPase, these two mechanisms of nucleotide interactions can directly be compared (Dmitriev et al. Citation2006). Interestingly, in the ATP binding studies with KdpCsol, alterations in neither the phosphate moiety (i.e., the comparison of ATP, ADP, and AMP), nor the base (ATP vs. GTP and CTP) had any influence on the overall nucleotide binding ratio. This would leave the ribose moiety of the nucleotide to be bound to KdpC, whereas the base moiety is embedded in the nucleotide binding pocket of KdpB with the phosphates protruding toward the site of phosphorylation, i.e., the P-domain of the KdpB subunit. Taken together, KdpC might orient and/or lock the nucleotide into the low-affinity catalytic binding site of KdpB via cooperative ribose interactions.
KdpF – the molecular glue
In comparison to the rather detailed view of the catalytic subunit KdpB and the substrate-translocating KdpA polypeptide as well as the catalytic chaperone KdpC, little information is available on the fourth subunit of the KdpFABC transport complex, KdpF. Although this polypeptide comprises only one transmembrane helix with no further cytoplasmic or periplasmic extensions, it is an integral part of the KdpFABC complex (Gaßel et al. Citation1999). Although deletion mutagenesis revealed that the KdpF subunit is not essential in vivo, the isolated KdpABC complexes lacking KdpF did not exhibit ATPase activity and disintegrated quite easily (Gaßel et al. Citation1999). The activity could be restored by the addition of either purified KdpF or E. coli lipids, which suggests a stabilizing, lipid-like function of the strongly hydrophobic KdpF polypeptide. Whereas such small hydrophobic subunits are not found among other P-type ATPases, there are at least examples from other membrane protein complexes like the Paracoccus denitrificans cytochrome c-oxidase, in which subunit IV was found to delicately assemble into a groove formed by the larger subunits of the complex, thereby most likely serving as a lipid-like molecular glue, which stabilizes subunit interactions (Iwata et al. Citation2002).
Perspectives
Whereas the KdpFABC complex exhibits all classical P-type ATPase properties, it also comprises some unique features with respect to its subunit composition and mode of action. Its remarkable allocation of ATP hydrolysis and substrate transport on two different subunits opens a fascinating avenue for the investigation of coupling and energy transfer between the sites of catalysis and ion translocation. Thus, the elucidation of contact sites and dynamic interactions between the participating subunits (KdpA and KdpB) will be the focus of future work. In addition, also the role of the KdpC subunit as a potential catalytical chaperone, which probably guides the nucleotide to its corresponding binding site, has to be further elucidated in order to open new perspectives of nucleotide binding and catalysis in P-type ATPases.
Acknowledgements
We would like to thank Dr Oleg Dmitriev for sharing the pdb-file used to construct the WDP N-domain with bound nucleotide in B. Furthermore, we thank Henrik Strahl for the KdpA homology modeling used in . Work in the authors’ laboratory was supported by the DFG and the Fonds der Chemischen Industrie.
Related Research Data
References
- Abu-Abed M, Mal TK, Kainosho M, MacLennan DH, Ikura M. Characterization of the ATP-binding domain of the sarco(endo)plasmic reticulum Ca2 + -ATPase: probing nucleotide binding by multidimensional NMR. Biochemistry 2002; 41: 1156–1164
- Ahnert F, Schmid R, Altendorf K, Greie J-C. ATP binding properties of the soluble part of the KdpC subunit from the Escherichia coli K+-transporting KdpFABC P-type ATPase. Biochemistry 2006; 45: 11038–11046
- Altendorf K, Gaßel M, Puppe W, Möllenkamp T, Zeek A, Boddien C, Fendler K, Bamberg E, Dröse S. Structure and function of the Kdp-ATPase of Escherichia coli. Acta Physiol Scand Suppl 1998; 643: 173–146
- Anthonisen AN, Clausen JD, Andersen JP. Mutational analysis of the conserved TGES loop of sarcoplasmic reticulum Ca2 + -ATPase. J Biol Chem 2006; 281: 31572–31582
- Aravind L, Galperin MY, Koonin EV. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem Sci 1998; 23: 469–472
- Axelsen KB, Palmgreen MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol 1998; 46: 84–101
- Berkelman T, Garret-Engele P, Hoffman NE. The pacL gene of Synechococcus sp. strain PCC 7942 encodes a Ca2 + -transporting ATPase. J Bacteriol 1994; 176: 4430–4436
- Bertrand J, Altendorf K, Bramkamp M. Amino acid substitutions in putative filter regions III and IV in KdpA alter ion selectivity of the KdpFABC complex from E. coli. J Bacteriol 2004; 186: 5519–5522
- Bissig KD, Wunderli-Ye H, Duda PW, Solioz M. Structure-function analysis of purified Enterococcus hirae CopB copper ATPase: effect of Menkes/Wilson disease mutation homologues. Biochem J 2001; 357: 217–223
- Bramkamp M, Altendorf K. Functional modules of KdpB, the catalytic subunit of the Kdp-ATPase from Escherichia coli. Biochemistry 2004; 43: 12289–12296
- Bramkamp M, Altendorf K. Single amino acid substitution in the putative transmembrane helix V in KdpB of the KdpFABC complex of Escherichia coli uncouples ATPase activity and ion transport. Biochemistry 2005; 44: 8260–8266
- Buurman ET, Kim K-T, Epstein W. Genetic evidence for two sequentially occupied K+ binding sites in the Kdp transport ATPase. J Biol Chem 1995; 207: 6678–6685
- Capieaux E, Rapin C, Thines D, Dupont Y, Goffeau A. Overexpression in Escherichia coli and purification of an ATP-binding peptide from the yeast plasma membrane H + -ATPase. J Biol Chem 1993; 268: 21895–21900
- De Hertogh B, Lantin AC, Baret PV, Goffeau A. The archaeal P-type ATPases. J Bioenerg Biomembr 2004; 36: 135–142
- Dmitriev O, Tsivkovskii R, Abildgaard F, Morgan CT, Markley JL, Lutsenko S. Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc Natl Acad Sci USA 2006; 103: 5302–5307
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998; 280: 69–77
- Durell SR, Bakker EP, Guy HR. Does the KdpA subunit from the high affinity K + -translocating P-type Kdp-ATPase have a structure similar to that of K+ channels?. Biophys J 2000; 78: 188–199
- Durell SR, Hao Y, Nakamura T, Bakker EP, Guy HR. Evolutionary relationship between K+ channels and symporters. Biophys J 1999; 77: 775–788
- Dutta SJ, Liu J, Stemmler AJ, Mitra B. Conservative and nonconservative mutations of the transmembrane CPC motif in ZntA: effect on metal selectivity and activity. Biochemistry 2007; 46: 3692–3703
- Fan B, Rosen BP. Biochemical characterization of CopA, the Escherichia coli Cu(I)-translocating P-type ATPase. J Biol Chem 2002; 277: 46987–46992
- Fendler K, Dröse S, Altendorf K, Bamberg E. Electrogenic K+ transport by the Kdp-ATPase of Escherichia coli. Biochemistry 1996; 35: 8009–8017
- Fendler K, Dröse S, Epstein W, Altendorf K, Bamberg E. The Kdp-ATPase of Escherichia coli mediates an ATP-dependent, K+-independent electrogenic partial reaction. Biochemistry 1999; 38: 1850–1856
- Gaßel M, Altendorf K. Analysis of KdpC of the K+-transporting KdpFABC complex of Escherichia coli. Eur J Biochem 2001; 268: 1772–1781
- Gaßel M, Möllenkamp T, Puppe W, Altendorf K. The KdpF subunit is part of the K + -translocating Kdp complex of Escherichia coli and is responsible for the stabilization of the complex in vitro. J Biol Chem 1999; 274: 37901–37909
- Gatto C, Wang AX, Kaplan JH. The M4M5 cytoplasmic loop of the Na,K-ATPase, overexpressed in Escherichia coli, binds nucleoside triphosphates with the same selectivity as the intact native protein. J Biol Chem 1998; 273: 10578–10585
- Håkansson KO. The crystallographic structure of Na,K-ATPase N-domain at 2.6 Å resolution. J Mol Biol 2003; 332: 1175–1182
- Haupt M, Bramkamp M, Coles M, Altendorf K, Kessler H. Inter-domain motions of the N-domain of the KdpFABC complex, a P-type ATPase, are not driven by ATP-induced conformational changes. J Mol Biol 2004; 342: 1547–1558
- Haupt M, Bramkamp M, Coles M, Kessler H, Altendorf K. Prokaryotic Kdp-ATPase: recent insights into the structure and function of KdpB. J Mol Microbiol Biotechnol 2005; 10: 120–131
- Haupt M, Bramkamp M, Heller M, Coles M, Deckers-Hebestreit G, Herkenhoff-Hesselmann B, Altendorf K, Kessler H. The holo-form of the nucleotide binding domain of the KdpFABC complex from Escherichia coli reveals a new binding mode. J Biol Chem 2006; 281: 9641–9649
- Hilge M., Siegal G, Vuister GW, Guntert P, Gloor SM, Abrahams JP. ATP-induced conformational changes of the nucleotide-binding domain of Na,K-ATPase. Nat Struct Biol 2003; 10: 468–474
- Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 2002; 376: 660–669
- Kimura Y, Inui M. Reconstitution of the cytoplasmic interaction between phospholamban and Ca2 + -ATPase of cardiac sarcoplasmic reticulum. Mol Pharmacol 2002; 61: 667–673
- Kühlbrandt W, Zeelen J, Dietrich J. Structure, mechanism, and regulation of the Neurospora plasma membrane H + -ATPase. Science 2002; 297: 1692–1696
- Liu J, Dutta SJ, Stemmler AJ, Mitra B. Metal-binding affinity of the transmembrane site in ZntA: implications for metal selectivity. Biochemistry 2006; 45: 763–772
- Lutsenko S, Kaplan JH. Organization of P-type ATPases: significance of structural diversity. Biochemistry 1995; 34: 15607–15613
- Maguire ME. MgtA and MgtB: prokaryotic P-type ATPases that mediate Mg2 + influx. J Bioenerg Biomembr 1992; 24: 319–328
- Møller JV, Juul B, le Maire M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1996; 1286: 1–51
- Moutin MJ, Cuillel M, Rapin C, Miras R, Anger M, Lompre AM, Dupont Y. Measurements of ATP binding on the large cytoplasmic loop of the sarcoplasmic reticulum Ca2 + -ATPase overexpressed in Escherichia coli. J Biol Chem 1994; 269: 11147–11154
- Nucifora G, Chu L, Misra TK, Silver S. Cadmium resistance from Staphylococcus aureus plasmid pI258 cadA gene results from a cadmium-efflux ATPase. Proc Natl Acad Sci USA 1989; 86: 3544–3548
- Odermatt A, Krapf R, Solioz M. Induction of the putative copper ATPases, CopA and CopB, of Enterococcus hirae by Ag+ and Cu2 + , and Ag+ extrusion by CopB. Biochem Biophys Res Commun 1994; 202: 44–48
- Odermatt A, Suter H, Krapf R, Solioz M. Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J Biol Chem 1993; 268: 12775–12779
- Okkeri J, Bencomo E, Pietila M, Haltia T. Introducing Wilson disease mutations into the zinc-transporting P-type ATPase of Escherichia coli. The mutation P634L in the ‘hinge’ motif (GDGXNDXP) perturbs the formation of the E2P state. Eur J Biochem 2002; 269: 1579–1586
- Okkeri J, Haltia T. The metal-binding sites of the zinc-transporting P-type ATPase of Escherichia coli. Lys693 and Asp714 in the seventh and eighth transmembrane segments of ZntA contribute to the coupling of metal-binding and ATPase activity. Biochim Biophys Acta 2006; 1757: 1485–1495
- Okkeri J, Laakkonen L, Haltia T. The nucleotide-binding domain of the Zn2 + -transporting P-type ATPase from Escherichia coli carries a glycine motif that may be involved in binding of ATP. Biochem J 2004; 377: 95–105
- Olesen C, Sørensen TL, Nielsen RC, Møller JV, Nissen P. Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 2004; 306: 2251–2255
- Raeymaekers L, Wuytack E, Willems I, Michiels CW, Wuytack F. Expression of a P-type Ca2 + -transport ATPase in Bacillus subtilis during sporulation. Cell Calcium 2002; 32: 93
- Rensing C, Ghosh M, Rosen BP. Families of soft-metal-ion-transporting ATPases. J Bacteriol 1999; 181: 5891–587
- Rensing C, Mitra B, Rosen BP. The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc Natl Acad Sci USA 1997; 94: 14326–14331
- Ridder IS, Dijkstra BW. Identification of the Mg2 + -binding site in the P-type ATPase and phosphatase members of the HAD (haloacid dehalogenase) superfamily by structural similarity to the response regulator protein CheY. Biochem J 1999; 339: 223–226
- Sazinsky MH, Agarwal S, Arguello JM, Rosenzweig AC. Structure of the actuator domain from the Archaeoglobus fulgidus Cu+-ATPase. Biochemistry 2006a; 45: 9949–9955
- Sazinsky MH, Mandal AK, Arguello JM, Rosenzweig AC. Structure of the ATP binding domain from the Archaeoglobus fulgidus Cu+-ATPase. J Biol Chem 2006b; 281: 11161–11166
- Sørensen TL, Møller JV, Nissen P. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 2004; 304: 1672–1675
- Therien AG, Karlish SJD, Blostein R. Expression and functional role of the gamma subunit of the Na, K-ATPase in mammalian cells. J Biol Chem 1999; 274: 12252–12256
- Toyoshima C, Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature 2004; 430: 529–535
- Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000; 405: 647–655
- Toyoshima C, Nomura H, Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 2004; 432: 361–368
- Toyoshima C, Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 2002; 418: 605–611
- Tsivkovskii R, MacArthur BC, Lutsenko S. The Lys1010-Lys1325 fragment of the Wilson's disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem 2001; 276: 2234–2242
- Van der Laan M, Gaßel M, Altendorf K. Characterization of amino acid substitutions in KdpA, the K + -binding and -translocating subunit of the KdpFABC complex of Escherichia coli. J Bacteriol 2002; 184: 5491–5494
- Zheng J, Knighton DR, ten Eyck LF, Karlsson R, Xuong N, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 1993; 32: 2154–2161
- Zhou, Y, Morais-Cabral, JH, Kaufman, A, MacKinnon, R. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature, 414:43–48.