Abstract
We investigated the effect of receptor mobility on HIV-1 envelope glycoprotein (Env)-triggered fusion using B16 mouse melanoma cells that are engineered to express CD4 and CXCR4 or CCR5. These engineered cells are resistant to fusion mediated CD4-dependent HIV-1 envelope glycoprotein. Receptor mobility was measured by fluorescence recovery after photobleaching (FRAP) using either fluorescently-labeled antibodies or transient expression of GFP-tagged receptors in the cells. No significant differences between B16 and NIH3T3 (fusion-permissive) cells were seen in lateral mobility of CCR5 or lipid probes. By contrast CD4 mobility in B16 cells was about seven-fold reduced compared to its mobility in fusion-permissive NIH3T3 cells. However, a CD4 mutant (RA5) that localizes to non-raft membrane microdomains exhibited a three-fold increased mobility in B16 cells as compared with WT-CD4. Interestingly, the B16 cells expressing the RA5 mutant (but not the wild type CD4) and coreceptors supported HIV-1 Env-mediated fusion. Our data demonstrate that the lateral mobility of CD4 is an important determinant of HIV-1 fusion/entry.
| Abbreviations | ||
| HIV-1 | = | Human Immunodeficiency Virus type 1 |
| HIV-2 | = | Human Immunodeficiency Virus type 2 |
| CMTMR | = | (5-(and-6) (((4chloromethyl)benzoyl)amino) tetramethylrhodamine) |
| CMFDA | = | 5-cholormethyl fluorescein diacetate) |
| GSL | = | glycosphingolipid(s) |
| DiO | = | 3,3′-dioctadecyloxacarbocyanine perchlorate ('DiO', DiOC18(3)) |
| FAST DiO™ | = | 3,3'-dilinoleyloxacarbocyanine perchlorate (DiOΔ9,12-C18(3), ClO4) |
| GM3 | = | Monosialoganglioside, (NeuNAc α2→3Galβ1→4Glcβ1→1Cer) |
| PPMP | = | 1-phenyl-2-hexadecanoylamino-3-morpholino-1-propanol |
| SM | = | sphingomyelin |
Introduction
HIV-1 infects permissive cells via sequential interactions of the viral envelope glycoprotein (gp120-gp41) with the cellular receptors (CD4, CXCR4 and/or CCR5) resulting in fusion between viral and target membrane Citation[1], Citation[3], Citation[8], Citation[11], Citation[19], Citation[24]. It has been hypothesized that recruitment of receptors at the plasma membrane microdomains enriched in sphingolipids and cholesterol (rafts) regulates initial interactions necessary for productive infection Citation[20], Citation[21], Citation[31], Citation[32]. However, the notion that plasma membrane rafts serve as portals for HIV-1 entry has been challenged recently Citation[23], Citation[25], Citation[36], and, therefore, an understanding of plasma membrane micro-environment at the site of virus-receptor interactions warrants further investigation.
We have reported earlier that a mouse Citation[30] melanoma cell line (B16) was resistant to HIV-1 Env-mediated membrane fusion, although the HIV-1 receptors (CD4, CXCR4, or CCR5) were functionally expressed in B16 cells. Interestingly, this fusion restriction was only observed for viral envelopes that were dependent in CD4 for fusion. Our observations that pretreatment of B16 cells with a GSL biosynthesis inhibitor (PPMP) completely restored fusion activity Citation[30], raised several possible mechanisms for this block mediated via direct or indirect interaction(s) of CD4 receptor with the plasma membrane components. Since GM3 has been reported to interact with HIV-1 receptors Citation[12], we postulated that either (i) elevated expression of GM3 in B16 cells immobilized CD4 and/or chemokine receptors within the plasma membrane enriched microdomains, or (ii) GM3 clustering into microdomains resulted in a shift in relative distribution of the receptor(s) from raft to non-raft domains or vice versa. This study was designed to investigate a correlation of lateral mobility of CD4 with the fusion résistance in B16 cells.
We monitored expression, lateral mobility and CD4-mediated HIV-1 Env triggered fusion susceptibility of B16 cells expressing a CD4 mutant, RA5 Citation[25]. The mutant RA5 bears cytoplasmic tail region substitution of the positively charged RHRRR motif with alanine residues, without any change in palmitoylation and the presence of the Lck binding motif Citation[25]. RA5, when expressed in CD4+ lymphocytes, has been shown to promote efficient HIV-1 entry, despite its preferential localization into non-raft fraction Citation[25]. Here we report that B16 cells expressing a GFP-construct of RA5 (but not the wt-CD4), and the coreceptors supported HIV-1 Env-mediated fusion. Efficiency of fusion was similar when compared with the GM95 or NIH3T3 target cells. Although, a correlation between CD4 mobility and HIV-1 entry-fusion has been reported previously Citation[9], Citation[34], Citation[35], the modulation of CD4 receptor mobility in those studies was achieved by pretreatment of target cells with reagents that interfere with raft-formation or by chemical cross-linking of CD4. Both approaches are likely to introduce additional defects/artifacts in the plasma membrane and distribution of their components. Our observations that the lateral diffusion of wt-CD4 is restricted in B16 cells despite its preferential localization in a GSL-rich plasma membrane microenvironment, suggest a novel mechanism of interplay between membrane lipids and receptors by which host cells can escape viral infections. Our findings that receptor mobility is an important determinant in viral entry may have broad implications in understanding the viral fusion process and virus infection mechanisms.
Materials and methods
Materials
Fluorescent probes were obtained from Molecular Probes (Eugene, OR), and tissue culture media were obtained from Invitrogen Corporation (Carlsbad, CA). Other reagents were from Sigma-Aldrich (St Louis, MO). 1-Phenyl-2-hexadecanoylamino-3-morpholino-1-propanol (PPMP) and GM3 were from Matreya, Inc. (Pleasant Gap, PA). Anti-GM3 mouse IgM Ab (GMR6, cat# 370695-1) was bought from SEIKAGAKU AMERICA (Falmouth, MA) Citation[15]. T4-4 CD4 antiserum was obtained through AIDS Research Reference and Reagent Program, NIAID and was initially contributed by R. Sweet Citation[5]. Anti-CD4 mouse mAb, IgG1,(R-PE conjugated RPA-T4,) was from BD Pharmingen (San Jose, CA). The source and culture conditions of B16, GM95, and HeLa cells stably expressing HIV-1 receptors are described elsewhere (Rawat et al, 2004). CD4 expression in B16CD4CXCR4 and NIH3T3CD4CXCR4 cells was determined by immuno-staining with R-PE conjugated RPA-T4 Mab (Rawat et al. 2004). The vaccinia constructs expressing either the full length processed HIV-1IIIB gp160 (gp120-gp41) (vPE16) Citation[6] or the mutant with a 104 amino acid deletion in the cytoplasmic tail of gp41 (vPE17) Citation[7] were obtained through the AIDS reagent and Reference Program (NIAID, NIH). Wild type CD4-GFP and the mutant RA5-GFP constructs were kindly provided by Dr Popik Citation[25]. CCR5-gfp was a gift from Dr Santos-Manes Citation[18].
HIV-1 envelope glycoprotein expression
CD4-requiring envelope proteins were transiently expressed on the surface of HeLa cells using the recombinant vaccinia constructs vPE16 (HIV-1IIIB, X4-utilizing) Citation[6], or vPE17, (Truncated HIV-1IIIB, X4-utilizing) Citation[7] following the protocol described Citation[30].
Fusion assay
Cell-cell fusion between CD4+ targets and gp120-gp41-expressing HeLa cells was assayed using a fluorescent dye transfer method Citation[28]. We used the fluorescent dyes CMTMR (green emission, ex/em 540/566 nm), CMFDA (blue emission, ex/em 492/516 nm) or Calcein (blue emission, ex/em 496/517 nm) to label the cells for fusion assay.
Typically target cells were plated on 12 well clusters at a density of 25–30,000 cells/well one day prior to fusion experiments and were labeled with 10 µM CMTMR following manufacturer's instructions. For PPMP treatment, target cells were plated at low density two days prior to fusion assay and treated with 10 µM PPMP for 24 hours at 37°C. Envelope-expressing cells were labeled with CMFDA (10 µM) according to Manufacturer's instructions. Fluorescently labeled effector and target cells were co-cultured under various experimental conditions. Details of the culture conditions relevant to each particular experiment are described in the figure legends. Dye redistribution was monitored microscopically as described Citation[14], and the extent of fusion was calculated as Percent Fusion = 100×number of bound cells positive for both dyes/ number of bound cells positive for target cells.
Transient expression of wild type CD4 and mutant RA5
B16CXCR4 or GM95-CXCR4 cells (1.5-2×107) were plated on T225 culture flasks over night and were transfected with CD4 constructs using SuperFect (transfection agent) following Manufacturer's instructions. Briefly, DNA constructs (100 µg) were suspended in 0.5 ml serum free medium and incubated with 0.15 ml SuperFect for 30–45 min to allow complex formation. At the end of incubation, volume was raised to 6.0 ml using complete medium and the DNA-complexes were gently layered onto the cells (pre-rinsed with PBS once following removal of culture medium). Incubations were continued at 37°C for 3–4 h, with intermittent rocking of the flasks to ensure homogeneous DNA distribution. Additional culture medium (20 ml per flask) was added and incubations were further continued for 24 hours at 37°C. To determine, surface expression of wt-CD4 and RA5, transected cells were harvested using the cell dissociation buffer, immuno-stained with PE-conjugated RPA-T4 Mab and fluorescence was monitored by FACS as described Citation[29]. GFP analysis showed that the transfection efficiency of wt-CD4 and RA5 in B16 and GM95 cells was up to 20%.
Sucrose density gradient analysis of CD4
Cells were harvested using cell-dissociation buffer and were washed with cold PBS (x3). The cell pellets were then treated with 1 ml Tris-HCl buffer (pH 7.6) containing 1% Triton X100 and protease inhibitors for 20–30 min at 4°C to solubilize plasma membrane fraction. The suspension was subjected to homogenization using a Dounce Homogenizer (10–15 strokes), transferred to an eppendorf tube and centrifuged at 1,000 rpm for 5 min at 4°C to remove nuclei. The supernatant was fractionated on sucrose gradients as follows: 0.8 ml supernatant was placed in a 4 ml ultra clear tube and mixed with an equal volume of 80% sucrose. Then, 1.6 ml of 30% sucrose and 0.8 ml 10% sucrose were layered on the tope sequentially. The samples were centrifuged at 100,000g (39,000 rpm) using a SW60 rotor for 16–18 h at 4°C. 0.4 ml fractions were carefully collected from the top and analyzed for CD4 and GM3 contents (see below).
CD4 distribution in sucrose-density fractions was determined by electrophoresis using 10% Tris-Glycine Gels (Invitrogen Corp. Carlsbad, CA)) under reducing conditions. Proteins were transferred to nitrocellulose membranes and probed with rabbit anti CD4 (T4-4) Ab. Bound antibodies were detected using anti-rabbit goat secondary Ab using Odyssey GelDoc System (LI-COR Inc, Lincoln, NE). GM3 analysis in sucrose-density fractions was performed using a Bio-DOT apparatus by immuno-staining with anti-GM3 mouse IgM Mab (GMR6) followed by detection with an anti-mouse secondary Ab.
Relative CD4 levels in various fractions were quantified using the Odyssey GelDoc software. Briefly, the regions corresponding to the CD4 in various lanes were selected using a region of interest (ROI) tool. The total grey levels within the individual ROI were normalized to a constant value corresponding to the 60 kD standard marker band. The CD4 intensity (relative grey levels) was plotted for individual fractions. GM3 was used as an internal raft marker for B16 cells, and relative GM3 intensity in various fractions was calculated as described above.
Fluorescence recovery after photobleaching (FRAP) acquisition and analysis
FRAP was performed using a Zeiss LSM 510 (Carl Zeiss, Jena, Germany) confocal laser scanning microscope. Transfected cells were plated on 35 mm glass bottom dishes (MatTek, Ashland, MA) and kept at physiological conditions of 37°C and 5% CO2 in a stage incubation system (Incubator S; PeCon GmbH, Erbach, Germany). A 488 nm Ar+ laser line was used for GFP excitation and emission light was collected with a 500–550 bandpass filter. A 40X/1.3 NA oil immersion objective lens was used with a zoom factor of 4. By increasing the zoom to 4, the pixel size was reduced to 0.1 um2 and a 20 pixel wide rectangle was used for the bleach region. This made the total size of the bleach region 2 um tangential to the direction of diffusion within the membrane, which is, needed for the one-dimensional (1D) FRAP analysis. Laser output was controlled by an acousto-optic tunable filter (AOTF) and kept as minimal as possible during image acquisition to prevent inadvertent photobleaching. The detector pinhole was opened slightly to acquire an optical section of 2 um thickness. This allowed more light to be collected for better quantification. Three pre-bleach images were acquired to determine the rate of inadvertent photobleaching. Photobleaching was performed by increasing the transmission of the AOTF up to 100% for 20–50 iterations to get as complete of bleach as possible. After photobleaching, images were acquired at one second apart for 8–10 images and then time resolution was changed to ten seconds to follow the recovery to completion. A total of 20–40 data points were acquired for image analysis. FRAP analysis was performed using the Medical Imaging Processing, Analysis, and Visualization (MIPAV; CIT/NIH, Bethesda, MD, McAuliffe MJ 2005)) software package. Data were corrected with background subtraction, as well as normalization for the inadvertent photobleaching rate calculated from the whole cell. Bleach regions for analysis were either read automatically by the software from the image header file or manually created. Data was analyzed using the 1D FRAP model Citation[18].
Results
B16CD4CXCR4 cells do not support fusion with cells expressing the cytoplasmic tail deletion mutant HIV-1 Gp120-gp41
Since the restriction in fusion was only observed for B16 target membrane and CD4-requiring HIV-1 envelope, we first investigated fusion activity of a cytoplasmic deletion mutant HIV-1 envelope that is dependent on CD4 but promotes fusion with extremely high efficiency as compared to the wild-type envelope Citation[2], Citation[7], Citation[37]. HeLa cells were infected with a vaccinia construct, vPE17, to express this mutant of HIV-1 envelope and fusion activity with B16CD4CXCR4 was examined using the fluorescence dye-transfer assay (see Methods sections). Interestingly, B16CD4CXCR4 cells failed to promote fusion with the cytoplasmic tail deletion HIV-1 Env mutant (), whereas, NIH3T3CD4CXCR4 cells fused efficiently with this mutant HIV-1 envelope under identical conditions ().
Figure 1. B16CD4CXCR4 cells do not support fusion with cells expressing cytoplasmic tail deletion mutant HIV-1 Gp120-gp41. CD4-requiring envelope proteins were transiently expressed on the surface of HeLa cells using the recombinant vaccinia constructs vPE16 (HIV-1IIIB, X4-utilizing) and vPE17 (Cytoplasmic Tail deletion mutant HIV-1IIIB, X4-utilizing). B16CD4CXCR4 cells (2.5 or 5.0×104 per well) were plated on 12 well clusters two day prior to fusion experiments. The cells at high density were treated with 10 µM PPMP for 24 h at 37°C. The cells were labeled with CMTMR and were co-cultured with calcein-labeled envelope-expressing cells for 4–8 h at 37°C in the absence or presence of C34 as indicated. Fusion was monitored using the fluorescent dye transfer and by formation of multinucleated cells (syncytia) upon fusion (see Methods section). Phase images are shown for the cocultures of HeLa cells expressing cytoplasmic tail deletion mutant HIV-1IIIB with B16CD4CXCR4 cells without (A) or with PPMP pretreatment. B16CD4CXCR4 cells did not support fusion with HeLa cells expressing HIV-1IIIB envelope (Not shown) (C) Extent of fusion. The values represent an average (±SD) from 3–5 samples with in a single experiment. The results were reproducible from at least three independent experiments. NIH3T3CD4CXCR4 cells were used as positive controls in these experiments.
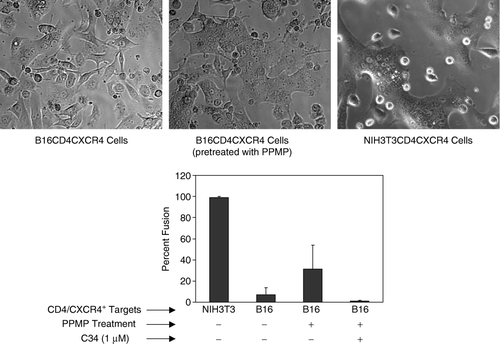
PPMP treatment of the B16 target cells restored fusion activity with the mutant Env similar to that for wt HIV-1 Env as reported earlier. Restoration of fusion was specific to viral envelope-receptor interactions as the fusion was inhibited in the presence of C34, a reagent that interferes with the gp41 six-helix bundle formation (). These data show that enhancing the fusion efficiency of HIV-1 Env is not sufficient to overcome the barriers imposed by the B16 cell membrane components. Therefore, we examined membrane disposition/localization of CD4 in B16 cells by employing the classical approach of fractionation of detergent resistant membranes on sucrose-density gradients (see below).
Localization of CD4 and mutant RA5 in plasma membrane of B16 cells
CD4 is considered a raft protein Citation[13], and efforts have been made to correlate localization of CD4 in membrane raft microdomains with susceptibility to HIV-1 Env-mediated Citation[4], Citation[16], Citation[17], Citation[20], Citation[22], Citation[23], Citation[26] fusion. In contrast, studies with cell lines engineered to express mutant and/or wt-CD4 demonstrate that CD4 localization into rafts is not an essential determinant for HIV-1 envelope mediated fusion. Along these lines, studies also show that membrane modulation by treatments such as cholesterol depletion Citation[36] and enzyme treatments Citation[9] results in inhibition of gp120-gp41 mediated fusion. However, the effect of these modulations was dependent on the levels of receptor expression. Taken together, these studies indicate that CD4 plasma membrane microenvironment is vital for HIV-1 fusion. Therefore, we examined whether wt-CD4 distribution was altered in B16 cells.
B16CD4CXCR4 cells were treated with TX100 (see Methods section) and detergent insoluble fractions were separated on sucrose density gradient analysis. Presence (or absence) of CD4 in various fractions was determined using an anti CD4 Ab (T4-4) as described (see Methods section). The results were compared with NIH3T3CD4CXCR4 cells that efficiently support HIV-1 fusion. Although surface-expressed CD4 in the B16 cells was significantly lower as compared to NIH3T3 cells (grey bars, wt-CD4, open bars, Isotype control, A), the lack of fusion in these cells is not due to differences in CD4 levels. We have previously established that PPMP treatment fully restores fusion in B16 cells without having an effect on CD4 receptor expression Citation[30].
Figure 2. Raft localization of wt-CD4 in B16CD4CXCR4 cells. Relative CD4 distribution in detergent insoluble and soluble fractions was determined by sucrose-density gradient analysis. B16 or NIH3T3 cells stably expressing CD4 and CXCR4 (B16CD4CXCR4 and NIH3T3CD4CXCR4) were subjected to TX100 solubilization, followed by sucrose density gradient fractionation as described in Methods section. Various fractions were analyzed for CD4 by Western blot analysis. A shows cell-surface expression of CD4 in cell lines used in this experiment. Solid bars, CD4 expression by PE-OKT4 staining; Open bars, Isotype control. B shows relative CD4 in various sucrose density gradient fractions (S = M.W. standards).The lanes corresponding to CD4 only are shown in the figure. The CD4 present in each fraction was quantified using the Odyssey Software relative to the 60 kD standard as described in Methods section (C).
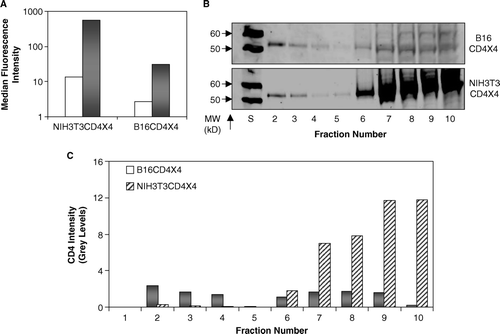
Sucrose density gradient analysis (B) shows that a major fraction of CD4 was associated with detergent insoluble fraction in B16CD4CXCR4 cells (fraction 2–4). Interestingly, in NIH3T3CD4CXCR4 cells, a most of CD4 was associated with fractions 6–10, and only a very small fraction was associated with fractions 2–4 (B). Quantitation of CD4 (expressed as relative grey levels, see Methods section) in various fractions is shown in C. Hence, in spite of CD4 distribution in raft fraction in B16 cells, HIV-1 fusion was blocked. GM3 (that selectively partitions into detergent insoluble fractions) was used as an internal ‘raft’ marker in these experiments (see B&C below).
Figure 3. Expression, and raft distribution of CD4 mutant RA5 in B16 cells. RA5 mutant (RA5-GFP) was expressed in B16-CXCR4 cells by transient transfection, examined for cell surface expression, analyzed for raft location by detergent insolubility and for HIV-1 envelope glycoprotein-mediated fusion. The results were compared with wt-CD4 (wt-CD4-GFP) under identical conditions, using GM95-CXCR4 cells as controls. A shows the site of mutation in RA5 in the membrane proximal amino acid region (from RHRRR to AAAAA) (adapted from [25]). B shows cell surface expression of RA5 and wt-CD4 in B16-CXCR4 or GM95-CXCR4 cells by immuno-staining with RPA-T4 Mab. C–E show results of CD4 and RA5 distribution in sucrose density gradient fractions of TX100-solubilized membranes of B16 cells. C and D show quantification of relative grey levels in various fractions normalized to the 60 kD M.W. standards (see Methods section). We used GM3 as and internal raft marker in our gradient analysis that was quantified by dot-blot analysis using the GMR6 Mab. C and D, solid bars, wt-CD4 and RA5, respectively. C and D, diagonal bars, GM3 levels. The protein content for wt-CD4 and RA5 in various cell fractions is shown in Figure E.
![Figure 3. Expression, and raft distribution of CD4 mutant RA5 in B16 cells. RA5 mutant (RA5-GFP) was expressed in B16-CXCR4 cells by transient transfection, examined for cell surface expression, analyzed for raft location by detergent insolubility and for HIV-1 envelope glycoprotein-mediated fusion. The results were compared with wt-CD4 (wt-CD4-GFP) under identical conditions, using GM95-CXCR4 cells as controls. Figure 3A shows the site of mutation in RA5 in the membrane proximal amino acid region (from RHRRR to AAAAA) (adapted from [25]). Figure 3B shows cell surface expression of RA5 and wt-CD4 in B16-CXCR4 or GM95-CXCR4 cells by immuno-staining with RPA-T4 Mab. Figure 3C–E show results of CD4 and RA5 distribution in sucrose density gradient fractions of TX100-solubilized membranes of B16 cells. Figure 3C and D show quantification of relative grey levels in various fractions normalized to the 60 kD M.W. standards (see Methods section). We used GM3 as and internal raft marker in our gradient analysis that was quantified by dot-blot analysis using the GMR6 Mab. Figure 3C and D, solid bars, wt-CD4 and RA5, respectively. Figure 3C and D, diagonal bars, GM3 levels. The protein content for wt-CD4 and RA5 in various cell fractions is shown in Figure E.](/cms/asset/3cb86b79-0311-40e4-bc5e-8d69e13ba28d/imbc_a_261222_f0003_b.gif)
To gain further insight into the nature of this restriction, we designed studies to using a CD4 mutant, RA5, (A) that localizes to the non-raft membrane microdomain, but supports HIV-1 Env-mediated fusion in lymphocytic cell lines Citation[25]. Receptor expression in the cells transfected with wt-CD4 and RA5 are shown in B (see Methods section). It is clear from the data that relative cell-surface expression of wt-CD4 and RA5 were similar in GM95 and B16 cells.
The cells were solubilized in tx100 and fractionated on sucrose density gradients, and the CD4 content in individual fractions was determined as described (Methods section). The relative CD4 levels in various fractions are shown in C (grey bars, wt-CD4) and D (crossed-bars, RA5) in B16 cells. GM3, the only ganglioside expressed in B16 cells has been shown to predominantly localize in raft fractions Citation[38]. Therefore, we used GM3 as an internal raft marker in our gradients (diagonal bars, C and D). The total protein content in various gradient fractions is shown in E. The relative amount of total protein in various fractions used for sucrose density gradient analysis was similar for cells expressing wt-CD4 or the mutant RA5 (E). These results show that wt-CD4 and RA5 when expressed in B16 cells preferentially localize in raft and non-raft fraction respectively similar to that reported for lymphocytes Citation[25] (the results on fusion activity of wt-CD4 and RA5-expressing cells are presented later in this section).
Lateral diffusion of CD4, CCR5 and lipids in B16 cells
Immuno-electron microscopy studies on spatial distribution of CD4, CXCR4 and CCR5 in lymphocytes and macrophages show that these molecules are segregated and preferentially clustered on microvilli Citation[33]. It is apparent that lateral mobility of receptors/envelope complexes within the plasma membrane will influence their assembly and recruitment at the fusion site and, hence, influence HIV-1 entry Citation[9]. Therefore, we examined lateral diffusion of lipids and receptors in B16 cells by the fluorescence recovery after photobleaching (FRAP) measurements (see Methods section for FRAP measurements and data analysis). The results were compared with two fusion permissive cell lines constitutively expressing CD4 and coreceptors, (a) GM95 (GSL-deficient) cells derived from B16 cells, and (b) mouse fibroblast NIH3T3 cells, that express normal levels of GSLs. In our first set of FRAP experiments, we tested B16 cells constitutively expressing CD4 (B16CD4CXCR4) after immunostaining with an anti-CD4 Mab (FITC-OKT4). Lipid mobility was determined by labeling the cells with the lipid probes, DiO/FAST-DiO, and for coreceptor mobility, we used cells transiently expressing CCR5-gfp (see Methods section).
To monitor lipid mobility, we initially used a lipophilic membrane probe (DiO). Since the diffusion process appears to be accelerated by introducing unsaturation in the alkyl tails of the probes, we compared lipid mobility using the lipid probes DiO and FAST-DiO. The two probes differ in the composition of their fatty acyl chains, such that the FAST DiO has diunsaturated linoleyl (C18:2) tails in place of the saturated octadecyl tails (C18:0) of DiO.
The results on lipid diffusion are presented in . The values expressed are an average of repeat measurements (SEM, n=7–25, as indicated in the Table) within a single experiment. The results were consistent from at least two independent experiments. DiO diffusion in B16 cells (7.77±0.8×10−10cm2) was only slightly slower as compared to GM95 (9.47±2.50×10−10cm2) or NIH3T3 (10.89±3.16×10−10cm2). Similarly, we observed a slight decrease in diffusion of FAST-DiO in B16 cells (8.39±5.51×10−10cm2) as compared to GM95 (13.1±8.39×10−10cm2) and NIH3T3 cells (13.8±8.98×10−10cm2). Since the two lipid probes (DiO, FAST-DiO) that preferentially partition into ordered and disordered membrane Citation[27] resulted in similar diffusion in the three cell lines tested, we conclude that lipid diffusion in B16 cells is similar to that of GM95 or NIH3T3 cells.
Table I. Lateral mobility of plasma membrane lipids in B16 Cells.3
Next, we evaluated the diffusion coefficient(s) of CD4, and CCR5 in various cell lines. The results are presented in , and the values expressed are an average of repeat measurements (SEM, n=8–17, as indicated in the table) within a single experiment.
Table II. Lateral mobility of HIV-1 receptors in B16 cells.3
The results were consistent from at least two independent experiments. It is clear that there were no significant differences in lateral diffusion of CCR5 in B16/GM95/NIH3T3 cells (4.28/1.95/4.72×10−10cm2/sec) respectively. In contrast, we observed a significant decrease in lateral mobility of CD4 in only B16 cells (1.87×10−11cm2/sec) when compared with CD4 diffusion in GM95 (20.9±3.0×10−11cm2/sec), or NIH3T3 cells (13.5×10−11cm2/sec). Interestingly, CD4 diffusion in GM95 cells was even faster than NIH3T3 cells. These data support our previous observations that only GM95 cells (constitutively expressing HIV receptors) supported fusion at low temperatures (≤25°C) Citation[29].
The results presented in strongly suggest that the restricted mobility of CD4 is likely to confer HIV − 1 fusion restriction in B16 cells. Therefore, we examined lateral mobility of the mutant CD4, RA5, which has been previously shown to partition into non-raft fraction, but retains its ability to support HIV-1 entry in T lymphocytes (See A,). The results were compared with wt-CD4 and RA5 diffusion in GM95 cells. The results shown in are an average of multiple measurements (details in legend) within a single experiment and were at least reproducible from two independent experiments.
Figure 4. Lateral diffusion of wt-CD4 or RA5 in B16 cells. The cells transiently expressing wt-CD4-GFP or RA5-GFP were examined for lateral diffusion by FRAP by monitoring GFP fluorescence as described in Methods section. The diffusion coefficients are plotted as logarithmic scale and represent an average of 15–20 measurements. The standard deviation (± SD×10-10cm2/sec) of diffusion measurements was as follows: 0.82 (B16, WT-CD4), 2.77 (B16, RA5), 3.18 (GM95, WT-CD4) and 4.14.
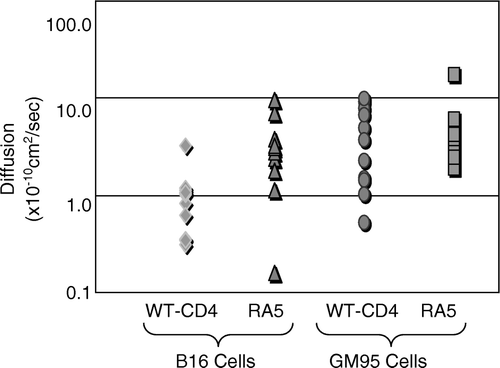
As expected, lateral diffusion of mutant RA5 (5.15±4.41×10−cm2/sec) and wt-CD4 (4.21±3.18×10−cm2/sec) when expressed in fusion-competent GM95 cells were similar. In contrast, we observed a significant statistical difference in diffusion of wt-CD4 (1.02±0.82×10−cm2/sec) and RA5 (3.23±2.77×10−cm2/sec) when expressed in B16 cells (). Therefore, our next experiments were designed to examine fusion susceptibility of RA5-expressing B16 cells (see below).
Fusion susceptibility of WT and mutant CD4 expressed in B16 cells
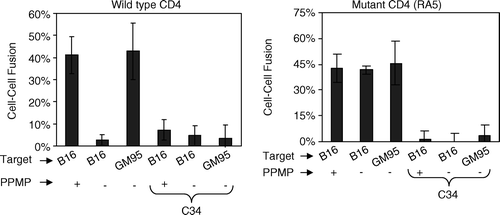
We examined the ability of RA5-expressing cells to support gp120-gp41 triggered fusion. Fusion activity was compared with GM95 cells expressing HIV-1 receptors under similar conditions. CMTMR labeled target cells were cocultured with CMFDA-labeled gp120-gp41 expressing HeLa cells for 4–6 h at 37°C, and fusion was monitored (see Methods section). Results are presented in .
We observed very low levels of fusion when B16-CXCR4 cells transiently expressing wt-CD4 were incubated with gp120-gp41 expressing cells (Left panel, ). In contrast, GM95-CXCR4 cells expressing wt-CD4 supported fusion with high efficiency. PPMP pretreatment of B16 target cells restored fusion consistent with our previous findings. Fusion activity of cells expressing the RA5 mutant is shown in the right panel (). Interestingly, B16-CXCR4 cells, transiently expressing the RA5 mutant were highly susceptible to gp120-gp41 mediated fusion. Fusion activity was similar to that of the GM95 targets. PPMP pretreatment of RA5-expressing B16 cells did not result in further enhancement in fusion activity. These observations clearly demonstrate that the HIV-1 Env-mediated fusion is only restricted to wt-CD4 but not the mutant CD4-expressing B16 cells. Specificity of gp120-gp41 triggered membrane fusion was confirmed by a block in fusion in the presence of C34, a fusion inhibitor that interferes with six-helix bundle formation ().
Figure 5. RA5-expressing B16 cells promote fusion with HIV-1 gp120-gp41 expressing cells. The cells transiently expressing wt-CD4 or RA5 were cultured with HeLa cells infected with vPE16 (see legend to ). Fusion was monitored microscopically and images were analyzed as described in the legend to .
Discussion
HIV-1 entry is a multistep process that involves sequential interactions of HIV-1 gp120-gp41 with CD4 and chemokine receptors. The process is facilitated by receptor movement, and is influenced by plasma membrane organization and/or composition. In addition to these key players, plasma membrane lipids (GSLs/cholesterol) have been implicated in modulating HIV-1 entry. However, an absolute requirement of target membrane rafts and their components in HIV-1 infection has been challenged.
Our original hypothesis that GSLs per se play a role in HIV-1 entry/fusion Citation[14] was based on our studies with PPMP, which inhibits the synthesis of glucosyl ceramide, the precursor for GSL biosynthesis. We argued that a reduction of GSL levels by blocking their synthesis would adversely affect HIV-1 fusion. PPMP has a number of different effects on sphingolipid metabolism, which include an increase of ceramide levels. However, in our follow up studies, we reported that GSL-deficient cells (GM95), that were engineered to express HIV-1 receptors, were highly susceptible to HIV-1 Env-mediated fusion Citation[29]. These results raised questions about a direct involvement of GSLs in fusion of cells expressing high levels of HIV-1 receptors. We have also reported that enhancing plasma membrane ceramide levels by other means (fenretinide, sphingomyelinase) reduces the susceptibility of certain target cells to HIV-1 fusion Citation[9], Citation[10]. Therefore, it appears that the receptor disposition with it's surrounding microenvironment (and not the specific lipid components) within the plane of the plasma membrane of a target cell is likely to dictate and/or facilitate fusion process.
Surprisingly, treatment of B16 cells with PPMP enhanced their fusion susceptibility to about the level of fusion susceptibility of NIH3T3 or GM95 cells expressing CD4 and coreceptors. We also noted that the levels of sphingomyelin in B16 cells were significantly increased following PPMP treatment Citation[30]. Since the relative amount of ceramide to sphingomyelin appears to be important for HIV receptor disposition and fusion susceptibility Citation[9], we conclude that the high levels of sphingomyelin produced in B16 cells following PPMP treatment will override the inhibitory effect (on fusion) of possible ceramide accumulation.
In this study we have probed the mechanism(s) underlying the block of fusion in B16 cells expressing HIV-1 receptors. First we examined a possible role of transmembrane/cytoplasmic anchor of HIV-1 Env, in observed restriction to fusion. The deletion of transmembrane anchors significantly increases the fusion mediated by HIV-1 envelope glycoprotein Citation[2], Citation[7], Citation[37]. Our observations that the mutant gp120-gp41 envelope also failed to support fusion, further supported the notion that block was due to inefficient interactions of HIV-1 envelope with the target membrane.
The results on lateral diffusion of CD4, lipids, and CCR5 in B16/GM95/NIH3T3 cells ( and ), show that CD4 diffusion in B16 cells is significantly slower when compared with GM95 or NIH3T3 cells. Since we did not observe such differences in lateral diffusion of lipids or the chemokine receptor, these data confirmed that fusion was restricted at the level of CD4-envelope interactions. Slower diffusion can prolong the formation of multi-protein complexes, as is the case with HIV-1 fusion machinery comprising of HIV-1 fusion protein CD4 and co-receptor.
The ability of RA5-expressing B16-CXCR4 cells to support gp120-gp41 mediated fusion () further suggested that the restriction was at the level of wt-CD4 disposition and/or the microenvironment in its vicinity. We found that distribution of wt-CD4 and RA5 mutant into raft and non-raft regions respectively was similar in B16 and GM95 cells. These observations led us to conclude that raft-localization of CD4 is not an essential factor for HIV-1 entry, consistent with previous reports. Interestingly, we observed significant differences in lateral diffusion of wt-CD4 and RA5 in B16 cells (). In contrast, diffusion of wt-CD4 and RA5 mutant were similar when expressed in GM95 cells () or NIH3T3 cells (data not shown). These data allow us to propose () that the failure of B16 cells expressing wt-CD4 result from restriction of CD4 lateral diffusion presumably as a result of clustering Citation[9].
Figure 6. CD4 clustering in B16 cells and susceptibility to HIV-1 fusion. HIV-1 delivers its genetic material into the cell by direct fusion of the viral membrane with the plasma membrane of the host cells mediated by trimeric gp120/gp41 molecules (blue). Interactions between gp120 and a cluster of CD4 molecules (yellow) result in conformational changes in gp120/gp41 (red). In B16 cells the WT-CD4 cluster does not readily disperse (left). However, the cluster of mutant (RA5) CD4 molecules readily disperses allowing the intermingling between those CD4 and CCR5/CXCR4 molecules (green) and enabling interactions gp120 with co-receptors. A further barrage of conformational changes then leads to membrane fusion.
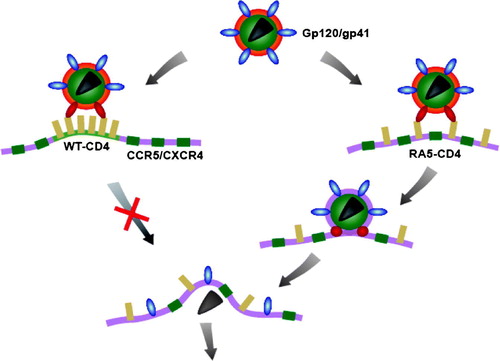
Taken together, our results suggest that over-expression of GM3 in B16 cells enhances clustering of CD4. This restricts the formation of trimolecular complex formation between CD4-corecptor (CCR5/CXCR4)-viral Envelope glycoprotein and hence block in fusion, consistent with our earlier observations Citation[30]. In our previous report we achieved the fusion rescue by decreasing the levels of GM3. In the current report we have bypassed the restriction by moving CD4 away from the GM3 annulus. Further, when CD4 is allowed to leave the GM3 annulus as in CD4-RA5 construct, trimolecular complex is formed and fusion proceeds as normal. Hence restricting components of multi-protein reaction as is the case for HIV envelope mediated fusion can achieve results akin of inhibitor for coreceptor -CD4 interaction.
Acknowledgements
Vaccinia recombinants were obtained through the AIDS reagent and Reference Program (NIAID, NIH) by Dr P. Earl and B. Moss. The CD4-GFP constructs were kindly provided by Dr Waldemar Popik (JHU, School of Medicine, Baltimore). CCR5-gfp construct was a kind gift from Dr Santos Manes (Centro Nacional de Biotechnologia/CSIC, Madrid, Spain). We appreciate members of Blumenthal lab for their sustained support during this study and Ryan Campbell-Massa for assistance in data analysis. This research was supported [in part] by the Intramural Program of the NIH, National Cancer Institute, Center for Cancer Research.
This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsements by the U.S. government.
References
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996; 272: 1955–1958
- Bhattacharya J, Peters PJ, Clapham PR. Human immunodeficiency virus type 1 envelope glycoproteins that lack cytoplasmic domain cysteines: impact on association with membrane lipid rafts and incorporation onto budding virus particles. J Virol 2004; 78: 5500–5506
- Broder CC, Dimitrov DS. HIV and the 7-transmembrane domain receptors. Pathobiology 1996; 64: 171–179
- Campbell SM, Crowe SM, Mak J. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J Clin Virol 2001; 22: 217–227
- Deen KC, McDougal JS, Inacker R, Folena-Wasserman G, Arthos J, Rosenberg J, Maddon PJ, Axel R, Sweet RW. A soluble form of CD4 (T4) protein inhibits AIDS virus infection. Nature 1988; 331: 82–84
- Earl PL, Doms RW, Moss B. Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc Natl Acad Sci USA 1990; 87: 648–652
- Earl PL, Koenig S, Moss B. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J Virol 1991; 65: 31–41
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996; 272: 872–877
- Finnegan CM, Rawat SS, Cho EH, Guiffre DL, Lockett S, Merrill AH, Jr, Blumenthal R. Sphingomyelinase restricts the lateral diffusion of CD4 and inhibits human immunodeficiency virus fusion. J Virol 2007; 81: 5294–5304
- Finnegan CM, Rawat SS, Puri A, Wang JM, Ruscetti FW, Blumenthal R. Ceramide, a target for antiretroviral therapy. Proc Natl Acad Sci USA 2004; 101: 15452–15457
- Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R. The HIV Env-mediated fusion reaction. Biochim Biophys Acta 2003; 1614: 36–50
- Hammache D, Yahi N, Maresca M, Pieroni G, Fantini J. Human erythrocyte glycosphingolipids as alternative cofactors for human immunodeficiency virus type 1 (HIV-1) entry: evidence for CD4-induced interactions between HIV-1 gp120 and reconstituted membrane microdomains of glycosphingolipids (Gb3 and GM3). J Virol 1999; 73: 5244–5248
- Horejsi V. The roles of membrane microdomains (rafts) in T cell activation. Immunol Rev 2003; 191: 148–164
- Hug P, Lin HM, Korte T, Xiao X, Dimitrov DS, Wang JM, Puri A, Blumenthal R. Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J Virol 2000; 74: 6377–6385
- Kotani M, Ozawa H, Kawashima I, Ando S, Tai T. Generation of one set of monoclonal antibodies specific for a-pathway ganglio-series gangliosides. Biochim Biophys Acta 1992; 1117: 97–103
- Kozak SL, Heard JM, Kabat D. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J Virol 2002; 76: 1802–1815
- Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses 2001; 17: 1009–1019
- Lippincott-Schwartz J, Presley JF, Zaal KJ, Hirschberg K, Miller CD, Ellenberg J. Monitoring the dynamics and mobility of membrane proteins tagged with green fluorescent protein. Methods Cell Biol 1999; 58: 261–281
- Markovic I, Clouse KA. Recent advances in understanding the molecular mechanisms of HIV-1 entry and fusion: revisiting current targets and considering new options for therapeutic intervention. Curr HIV Res 2004; 2: 223–234
- Nayak DP, Hui EK. The role of lipid microdomains in virus biology. Subcell Biochem 2004; 37: 443–491
- Nguyen DH, Giri B, Collins G, Taub DD. Dynamic reorganization of chemokine receptors, cholesterol, lipid rafts, and adhesion molecules to sites of CD4 engagement. Exp Cell Res 2005; 304: 559–569
- Pal R, Nair BC, Hoke GM, Sarngadharan MG, Edidin M. Lateral diffusion of CD4 on the surface of a human neoplastic T-cell line probed with a fluorescent derivative of the envelope glycoprotein (gp120) of human immunodeficiency virus type 1 (HIV-1). J Cell Physiol 1991; 147: 326–332
- Percherancier Y, Lagane B, Planchenault T, Staropoli I, Altmeyer R, Virelizier JL, Arenzana-Seisdedos F, Hoessli DC, Bachelerie F. HIV-1 entry into T-cells is not dependent on CD4 and CCR5 localization to sphingolipid-enriched, detergent-resistant, raft membrane domains. J Biol Chem 2003; 278: 3153–3161
- Pohlmann S, Reeves JD. Cellular entry of HIV: evaluation of therapeutic targets. Curr Pharm Des 2006; 12: 1963–1973
- Popik W, Alce TM. CD4 receptor localized to non-raft membrane microdomains supports HIV-1 entry. Identification of a novel raft localization marker in CD4. J Biol Chem 2004; 279: 704–712
- Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol 2002; 76: 4709–4722
- Pucadyil TJ, Chattopadhyay A. Effect of cholesterol on lateral diffusion of fluorescent lipid probes in native hippocampal membranes. Chem Phys Lipids 2006; 143: 11–21
- Puri A, Paternostre M, Blumenthal R. Lipids in viral fusion. Methods Mol Biol 1906; 199: 61–81
- Rawat SS, Eaton J, Gallo SA, Martin TD, Ablan S, Ratnayake S, Viard M, KewalRamani VN, Wang JM, Blumenthal R, Puri A. Functional expression of CD4, CXCR4, and CCR5 in glycosphingolipid-deficient mouse melanoma GM95 cells and susceptibility to HIV-1 envelope glycoprotein-triggered membrane fusion. Virology 2004; 318: 55–65
- Rawat SS, Gallo SA, Eaton J, Martin TD, Ablan S, KewalRamani VN, Wang JM, Blumenthal R, Puri A. Elevated expression of GM3 in receptor-bearing targets confers resistance to human immunodeficiency virus type 1 fusion. J Virol 2004; 78: 7360–7368
- Rawat SS, Johnson BT, Puri A. Sphingolipids: modulators of HIV-1 infection and pathogenesis. Biosci Rep 2005; 25: 329–343
- Rawat SS, Viard M, Gallo SA, Blumenthal R, Puri A. Sphingolipids, cholesterol, and HIV-1: A paradigm in viral fusion. Glycoconj J 2006; 23: 189–197
- Singer II, Scott S, Kawka DW, Chin J, Daugherty BL, DeMartino JA, DiSalvo J, Gould SL, Lineberger JE, Malkowitz L, Miller MD, Mitnaul L, Siciliano SJ, Staruch MJ, Williams HR, Zweerink HJ, Springer MS. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J Virol 2001; 75: 3779–3790
- Steffens CM, Hope TJ. Localization of CD4 and CCR5 in living cells. J Virol 2003; 77: 4985–4991
- Steffens CM, Hope TJ. Mobility of the human immunodeficiency virus (HIV) receptor CD4 and coreceptor CCR5 in living cells: implications for HIV fusion and entry events. J Virol 2004; 78: 9573–9578
- Viard M, Parolini I, Sargiacomo M, Fecchi K, Ramoni C, Ablan S, Ruscetti FW, Wang JM, Blumenthal R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J Virol 2002; 76: 11584–11595
- Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J Virol 2005; 79: 12231–12241
- Yamamura S, Handa K, Hakomori S. A close association of GM3 with c-Src and Rho in GM3-enriched microdomains at the B16 melanoma cell surface membrane: a preliminary note. Biochem Biophys Res Commun 1997; 236: 218–222