Abstract
Residues Tyr-110 through Gly-115 of serotonin transporter were replaced, one at a time, with cysteine. Of these mutants, only G113C retained full activity for transport, Q111C and N112C retained partial activity, but Y110C, G114C and G115C were inactive. Poor surface expression was at least partly responsible for the lack of transport by G114C and G115C. In membrane preparations, Y110C through G113C all bound a high affinity cocaine analog similarly to the wild type. Treatment with methanethiosulfonate reagents increased the transport activity of Q111C and N112C to essentially wild-type levels but had no measurable effect on the other mutants. The decreased activity of Q111C and N112C resulted from an increase in the KM for serotonin that was not accompanied by a decrease in serotonin binding affinity. Superfusion experiments indicated a defect in 5-HT exchange. Modification of the inserted cysteine residues reversed the increase in KM and the poor exchange, also with no effect on serotonin affinity. The results suggest that Gln-111 and Asn-112 are not required for substrate binding but participate in subsequent steps in the transport cycle.
Introduction
Serotonin transporter (SERT) is responsible for transporting serotonin (5-hydroxyryptamine, 5-HT) back into serotonergic neurons after its release. It is the target for many antidepressant medications, whose action is to increase the extracellular concentration of this neurotransmitter. It is also one of the targets for psychostimulants such as cocaine and amphetamine derivatives, notably 3,4-methylenedioxymethamphetamine (MDMA, also known as ‘ecstasy’).
SERT belongs to a family of neurotransmitter and amino acid transporters (the Neurotransmitter Sodium Symporter or NSS family) found in animals and prokaryoties organisms. All members of this family that have been characterized catalyze symport (coupled transport) of their substrate neurotransmitter or amino acid with one or more sodium ions. Many mammalian NSS transporters also couple chloride symport to substrate translocation, and SERT also couples potassium antiport (coupled exchange) to the movement of 5-HT.
Within the NSS family, certain regions of the sequence have particularly high incidence of identity at the amino acid level. Perhaps the strongest region of identity is in the first transmembrane domain (TM1), the first extracellular loop (EL1), and TM2. TM1 and TM2 have been subjected to analysis by cysteine scanning mutagenesis in SERT and the closely related NSS transporters for dopamine (DAT) and γ-aminobutyric acid (GAT-1) (Chen et al. [Citation1997], Henry et al. [Citation2003], Ni et al. [Citation2001], Sato et al. [Citation2004], Sen et al. [Citation2005], Zhou et al. [Citation2004]) and the EL1 region between the two TMs have been examined in GAT-1 in the context of a specific glycine mutation (Zhou & Kanner [Citation2005]) but had not been studied in SERT.
The extracellularly accessible region of TM1, as estimated from studies of SERT and GAT-1, begins around Tyr-107 of SERT (Tyr-72 in GAT-1) (Henry et al. [Citation2003], Zhou et al. [Citation2004]). Preceding TM2, the accessible region of SERT EL1 ended at Ala-116. The accessibility of Cys-109 and Ala-116 in SERT was shown to vary depending on the binding of substrates and inhibitors (Ni et al. [Citation2001], Sato et al. [Citation2004]). Between these two positions, the only available functional data comes from studies of GAT-1.
In GAT-1, residues between Asn-77 and Ala-81 (corresponding to Asn-112 through Ala-116 in SERT) were replaced with cysteine or alanine. The N77C mutant had low activity relative to the parental construct, but was stimulated by treatment with [2-(trimethylammonium) ethyl]methanethiosulfonate (MTSET), a covalent cysteine modifying reagent. Of the following three glycine to cysteine replacement mutants in GAT-1, only G80C was inactive. Both G78C and G79C were functional and were inhibited by MTSET treatment. The sensitivity to MTSET was influenced slightly by Na+ in N77C, G79C and A81C (Zhou & Kanner [Citation2005]).
Attention has recently focused on prokaryotic members of the NSS family. The demonstration that the TnaT protein of Symbiobacter thermophilum was a functional tryptophan transporter (Androutsellis-Theotokis et al. [Citation2003]) led to the crystallization of another bacterial NSS transporter, the LeuT protein of Aquifex aeolicus (Yamashita et al. [Citation2005]). The resulting structure has provided many insights into the structure and function of other NSS transporters. With respect to EL1, the amino acid sequence is highly conserved, and the structure of LeuT is likely to provide a good model for SERT ().
Figure 1. Predicted structure of SERT by analogy with LeuT. A model of SERT was constructed by Drs Harel Weinstein, Lei Shi, and Thijs Beuming of Cornell University based on the structure of LeuT (Yamashita et al. [Citation2005]). Residues 110 through 116 are shown as stick diagrams with the adjacent portions of TM1 and TM2 shown as ribbons.
![Figure 1. Predicted structure of SERT by analogy with LeuT. A model of SERT was constructed by Drs Harel Weinstein, Lei Shi, and Thijs Beuming of Cornell University based on the structure of LeuT (Yamashita et al. [Citation2005]). Residues 110 through 116 are shown as stick diagrams with the adjacent portions of TM1 and TM2 shown as ribbons.](/cms/asset/6abe68bd-a9d8-4980-a0dc-4c9a7ea1975b/imbc_a_263171_f0001_b.gif)
The structure of LeuT contains a leucine molecule bound together with two Na+ ions near the center of the molecule. There is a large aqueous opening in the extracellular face of the protein that reaches almost to the bound substrate, suggesting that the structure approximates the conformation of the transporter that binds extracellular ligands (Rudnick [Citation2006], Yamashita et al. [Citation2005]). In this conformation, some residues of EL1 are relatively well exposed to solvent (). However, considerable conformational change is expected for bound substrate to be released to the cytoplasm, since there is almost 20Å of packed protein structure separating leucine from the cytoplasmic face of the protein (Yamashita et al. [Citation2005]). Recent experiments suggest that a pathway adjacent to TM5 becomes accessible to the cytoplasm in the form of SERT that releases bound substrates (Zhang & Rudnick [Citation2005], Zhang & Rudnick [Citation2006]).
Two other extracellular loops of SERT have been examined using cysteine scanning mutagenesis. EL5 was shown to encompass a larger range of amino acid residues than predicted by hydropathy analysis (Keller et al. [Citation2004]). In light of the LeuT structure, this exposed region includes EL5 and approximately half of TM10. EL4 also has been examined and was found to be conformationally active (Mitchell et al. [Citation2004]). Highly reactive residues in EL4 became unreactive in the presence of substrates, suggesting that conformational changes associated with transport converted SERT to a form in which those residues were occluded. The occluded residues correspond to a highly exposed region of EL4 in the LeuT structure. Thus, dynamic information about loop conformation can provide insight into the transport mechanism. In this report, we describe an examination of EL1 by cysteine scanning mutagenesis.
Materials and methods
Mutagenesis
EL1 cysteine mutant transporters were generated by site-directed mutagenesis using the QuikChangeTM kit (Stratagene, La Jolla, CA). The mutated region was excised by digestion with appropriate restriction enzymes and subcloned into the X5C mutant of rat SERT, (C15A/C21A/C109A/C357I/C622A) which contains sequences encoding a c-Myc epitope tag at the N-terminus and a FLAG epitope tag at the C-terminus (Sato et al. [Citation2004]) and is lacking all of the endogenous cysteine residues known to react with MTS reagents. In some experiments, SERT C109A (Chen et al. [Citation1997]), which is unreactive toward extracellular MTS reagents, was used as a negative control. All mutations were confirmed by DNA sequencing.
Expression
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 units/ml penicillin, and 100 µg/ml streptomycin at 37°C in a humidified 5% CO2 incubator. Cells were plated in 96-well culture plates and allowed to grow for 36–40 h. The confluent cells were infected with recombinant VTF7-3 virus and transfected with a plasmid containing rat SERT cDNA under the control of the T7 promoter as described previously (Blakely et al. [Citation1991]). Transfected cells were incubated for 20–22 h at 37°C before assaying for transport.
Transient expression of rSERT cDNAs (as described above) in HEK-293 cells was achieved by transfection using the CaP04-precipitation method. Cells were maintained essentially as described; for stable expression, cells were selected by addition of geneticin (Sitte et al. [Citation2001]).
Transport assay
Transfected HeLa cells in 96-well plates were washed once with 100 µl of of phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.3) containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM). Transport of 5-HT was measured by adding 100 µl of PBS/CM containing 20 nM [3H]5-HT (PerkinElmer, Boston, MA) to each well and incubating for 10 min at room temperature. For determination of KM and Vmax in the mutants, 5-HT concentrations were varied by using 20 nM [3H]5-HT with unlabeled 5-HT to the final concentration. The assays were terminated by three rapid washes with ice-cold phosphate-buffered saline. The cells were then lysed in 30 µl of 0.1 M NaOH for 30 min, 120 µl/well of Optifluor (PerkinElmer, Boston, MA) added, and the plates were counted in a Wallac MicroBeta plate counter. Protein concentration was determined with the Micro BCA protein assay reagent kit (Pierce).
Treatment with MTS reagents
EL1 mutants were tested for their sensitivity to the MTS reagents (2-(trimethylammonium)ethyl)methanethiosulfonate bromide (MTSET), 2-(aminoethyl)methanethiosulfonate hydrobromide (MTSEA) and (2-sulfonatoethyl)methanethiosulfonate (MTSES) (Anatrace, Maumee, OH). The transfected Hela cells were washed with PBS/CM or a modified medium as indicated, and incubated with the indicated MTS reagent in 100 µl of medium for 10 min at room temperature. The cells were then washed three times with 100 µl of PBS/CM and assayed for transport. In some cases, a second incubation with a different MTS reagent was added prior to the transport assay.
The modified PBS/CM media were as follows: NaCl; 9.5 mM K3PO4, 138 mM NaCl, 2.7 mM K-acetate, 1 mM MgCl2, 0.1 mM Ca(acetate)2 pH 7.1, NMDG (N-methyl-D-glucamine)-Cl; 9.5 mM K3PO4, 138 mM NMDG-Cl, 2.7 mM K-acetate, 1 mM MgCl2, 0.1 mM Ca(acetate)2, pH 7.1, Na-Isethionate; 9.5 mM K3PO4, 138 mM Na-isethionate, 2.7 mM K-acetate, 1 mM Mg(acetate)2, 0.1 mM Ca(acetate)2, pH 7.1. When 5-HT or cocaine was present during the incubation with MTS-reagents, they were at 2 µM final concentration.
Cell surface biotinylation
HeLa cells expressing SERT EL1 mutants were biotinylated twice with NHS-SS-biotin (Pierce, Rockford, IL) for 20 min on ice as described (Chen et al. [Citation1998]). Following the biotinylation, the cells were rinsed with glycine to quench excess reagent. The cells were then lysed, and the biotinylated proteins were recovered using streptavidin-agarose beads (Pierce) in an overnight incubation at 4°C with gentle agitation. The beads were washed, and the biotinylated proteins were eluted with 100 µl of SDS-PAGE sample buffer (Laemmli [Citation1970]) containing 100 mM DTT. The eluted samples were applied to a 10% SDS-polyacrylamide gel and detected with monoclonal M2 anti-Flag-tag antibody (Sigma, St. Louis, MO) against the FLAG epitope tag at the COOH terminus of rat SERT (Tate & Blakely [Citation1994]). A horseradish peroxidase-conjugated second anti-rabbit IgG (1:10,000) was used to visualize the signal by Super Signal West Femto (Pierce). The signals from the Western blot were quantitated with luminescence imaging using a UVP (Upland, CA) Biochemi imaging system.
Membrane preparation and binding assay
HeLa cells grown in 75-cm2 cell culture flasks were transfected with SERT EL1 mutant cDNA as described above. After overnight incubation, the cells were rinsed once with room temperature 10 mM lithium-HEPES buffer (10 mM HEPES-free acid brought to pH 8.0 with LiOH) and scraped into 10 ml of homogenization buffer (10 mM HEPES, pH 8.0, containing 0.5% of a protease inhibitor mixture (Sigma) and 100 µM PMSF). The cells were lysed by two cycles of freeze-thawing and sonication, and the resulting crude membrane fraction was collected by centrifugation at 15,000 g for 20 min at 4°C. The membranes were resuspended in 1 ml of homogenization buffer and stored at −80°C in 0.1-ml aliquots until used.
For membrane binding assays, aliquots of membranes from cells expressing SERT EL1 mutants were thawed on ice and diluted with 1 ml of binding buffer (10 mM HEPES, adjusted to pH 8.0 with NaOH, 150 mM NaCl, 0.1 mM CaCl2, and 1 mM MgCl2). Binding was measured in Multiscreen-FB 96-well filtration plates (Millipore, Bedford, MA), which were pretreated overnight at 4°C with 200 µl of 0.1% polyethyleneimine in each well. The wells were rinsed three times with 100 µl of room temperature binding buffer, and then 100 µl of the diluted membrane solution was added per well. The membranes were washed twice by filtration with 200 µl of binding buffer, and binding was initiated by the addition of 100 µl of binding buffer containing 0.1 nM [125-I]β-CIT (PerkinElmer Life Sciences, Wellesley, MA) and 0.1–40 µM unlabeled 5-HT. After 1.5 h incubation at room temperature with gentle rocking, the reaction was stopped by washing all wells three times with 100 µl of ice-cold binding buffer. The filters were removed from the plate and counted after soaking in 150 µl of Optifluor (PerkinElmer). In some experiments the membranes were incubated with 1 mM MTSEA or MTSET for 15 min at room temperature prior to measuring β-CIT binding.
5-HT exchange assays
Superfusion experiments have been described previously in detail (Sitte et al. [Citation2001]). In brief, cells were grown overnight on poly-D-lysine coated round glass coverslips and preincubated in the presence of 0.4 µM [3H]5-HT for 20 min at 37°C in a final volume of 0.1 ml Krebs-HEPES buffer (120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgSO4, 20 mM dextrose, final pH 7.35, 10 mM HEPES, pH adjusted to 7.4 with NaOH) and then in the presence or absence of MTSEA, for 10 min at 37°C; final concentration 2.5 mM. After two additional washes, coverslips were then transferred to superfusion chambers (0.2 ml) and superfused with buffer at 25°C and a perfusion rate of 0.7 ml/min. Technical difficulties with MTSET precluded testing N112C-mediated efflux with this reagent. However, to ensure consistency, and since MTSEA activated influx and efflux by both Q111C and N112C, all efflux studies were performed with MTSEA.
After a washout period of 45 min to establish stable efflux of radioactivity, the experiment was started with collection of 2-minutes-fractions. After 6 min (baseline values), 5-HT (10 µM) was added to buffer for additional 10 min. At the end of the experiment, cells were lysed in 1% SDS and the amount of remaining tritium in the cells was determined. The release of [3H] is expressed as fractional rate; i.e. the radioactivity released during a fraction was expressed as percentage of the total radioactivity present in the cells at the beginning of that fraction.
Reagents
Tissue culture reagents were from Invitrogen Life Technologies. [3H]5-HT and [125I]β-CIT was from Perkin-Elmer (Waltham, MA) and New England Nuclear (Vienna, Austria). Cold 5-HT was from Sigma-Aldrich (St. Louis, MO and Handels GmbH Vienna, Austria). MMTS, MTSEA and MTSET were from Anatrace (Maumee, OH) and MTSCE was from Biotium (Hayward, CA). All other chemicals were from commercial sources.
Data analysis
Nonlinear regression analysis of experimental data were performed with Origin (OriginLab, Northampton, MA), which uses the Marquardt-Levenberg nonlinear least squares curve fitting algorithm. Data with error bars represent the mean and standard deviation for triplicate measurements.
Results
Activity of EL1 mutants
We constructed serotonin transporter (SERT) mutants in which a cysteine replaced the endogenous residue at six positions in external loop 1 (EL1) of SERT. These residues include some of the most highly conserved positions within the NSS transporter family. shows a model of this sequence in SERT, based on the published structure of LeuT, a bacterial homologue of SERT (Yamashita et al. [Citation2005]). Also shown is an alignment between LeuT and SERT in which seven of the 12 residues are identical. Residues preceding and following this sequence had been previously mutated (Chen et al. [Citation1997], Henry et al. [Citation2003], Sato et al. [Citation2004]). The mutations were generated in a background (X5C) lacking five endogenous cysteine residues. These five positions were previously shown to react with methanethiosulfonate (MTS) reagents, whereas X5C was relatively resistant to these reagents (Androutsellis-Theotokis & Rudnick [Citation2002], Ni et al. [Citation2001], Sato et al. [Citation2004]).
When tested for their ability to transport 5-HT, only G113C was active at levels approaching that of X5C (A). Q111C and N112C had less than 30% the activity of X5C and the remaining mutants (Y110C, G114C and G115C) retained negligible transport activity. Despite this low level of transport activity, most of the mutants were capable of binding the high-affinity cocaine analog 2β-carbomethoxy-3β-(4-[125I]iodophenyl)tropane (β-CIT) at levels close to those of X5C. B shows β-CIT binding to membranes prepared from cells expressing each of the mutants. Mutants Y110C, Q111C, N112C and G113C all bound approximately 70% the level of X5C, although much less binding was detected in G114C and G115C. Thus, the transport defect in mutants Y110C, Q111C and N112C was not due to gross misfolding of the protein, since an intact binding site was still present. The lack of binding activity in G114C and G115C was likely to be due to poor expression. C shows a Western blot (quantified in D) of the SERT mutants from cells surface labeled with NHS-biotin. Although there is some variability, most of the mutants were expressed on the cell surface at levels close to those of X5C. However, the expression of G114C was markedly reduced and G115C was undetectable. The poor expression of G114C and G115C is likely to be responsible for the lack of binding or transport activity in these mutants.
Figure 2. 5-HT transport activity and cell surface expression of EL1 cysteine mutants. (A) 5-HT transport activity. HeLa cells expressing each EL1 cysteine mutant were assayed for [3H]5-HT uptake for 10 min as described under ‘Materials and methods’. The activity of each mutant transporter is represented as a percentage of parental mutant X5C activity (0.16±0.03 pmol/mg per min). Each value represents the mean and SD of three experiments, each of which was performed in triplicates. (B) Binding activity of EL1 cysteine mutants. Membranes prepared from HeLa cells expressing each of the EL1 cysteine mutants and the control X5C were incubated with [125I]β-CIT as described (Androutsellis-Theotokis & Rudnick [Citation2002]). The binding to each mutant transporter is represented as a percentage of the amount bound to parental mutant X5C (1.52±0.11 pmol/mg). The data represent means from three experiments. (C) Surface expression of EL1 cysteine mutants. HeLa cells expressing the EL1 mutants were treated with NHS-SS-biotin to label cell surface proteins. The cells were lysed in detergent, and surface proteins were extracted with streptavidin beads. SERT mutants were detected by immunoblotting with a monoclonal antibody to the FLAG tag attached to the SERT COOH-terminus. The 96-kDa form of SERT, which represents mature and fully glycosylated protein is shown here. (D) Quantitation of surface expression. The surface expression levels of EL1 mutants were measured by densitometry and are shown as a percentage of X5C. The data represent averaged results from three experiments.
![Figure 2. 5-HT transport activity and cell surface expression of EL1 cysteine mutants. (A) 5-HT transport activity. HeLa cells expressing each EL1 cysteine mutant were assayed for [3H]5-HT uptake for 10 min as described under ‘Materials and methods’. The activity of each mutant transporter is represented as a percentage of parental mutant X5C activity (0.16±0.03 pmol/mg per min). Each value represents the mean and SD of three experiments, each of which was performed in triplicates. (B) Binding activity of EL1 cysteine mutants. Membranes prepared from HeLa cells expressing each of the EL1 cysteine mutants and the control X5C were incubated with [125I]β-CIT as described (Androutsellis-Theotokis & Rudnick [Citation2002]). The binding to each mutant transporter is represented as a percentage of the amount bound to parental mutant X5C (1.52±0.11 pmol/mg). The data represent means from three experiments. (C) Surface expression of EL1 cysteine mutants. HeLa cells expressing the EL1 mutants were treated with NHS-SS-biotin to label cell surface proteins. The cells were lysed in detergent, and surface proteins were extracted with streptavidin beads. SERT mutants were detected by immunoblotting with a monoclonal antibody to the FLAG tag attached to the SERT COOH-terminus. The 96-kDa form of SERT, which represents mature and fully glycosylated protein is shown here. (D) Quantitation of surface expression. The surface expression levels of EL1 mutants were measured by densitometry and are shown as a percentage of X5C. The data represent averaged results from three experiments.](/cms/asset/a175453b-593e-4fe1-9ca0-151607141f64/imbc_a_263171_f0002_b.gif)
Response to modification with MTS reagents
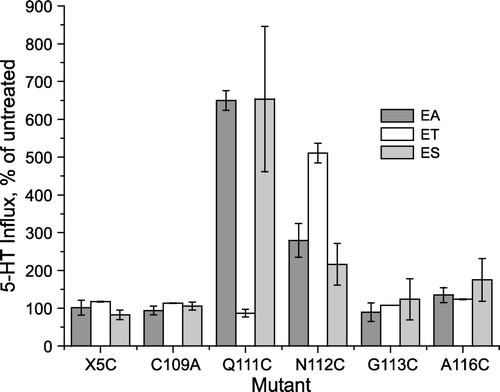
Replacement of EL1 residues with cysteine allows modification of the resulting mutants with cysteine reagents such as methanethiosulfonate (MTS) reagents. shows that two of the mutants, Q111C and N112C respond to such modification with an increase in transport activity. Three MTS reagents were tested initially, (2-aminoethyl) methanethiosulfonate (MTSEA), [2-(trimethylammonium) ethyl] methanethiosulfonate (MTSET) and (2-sulfonatoethyl)methanethiosulfonate (MTSES). We found that MTSEA and MTSES stimulated the transport activity of Q111C approximately 7-fold, while MTSET had no obvious effect. In contrast, MTSET was the most effective reagent for stimulating N112C, which increased approximately 5-fold in transport rate, although MTSEA and MTSES also stimulated, but only 2- to 3-fold. A116C, as previously reported (Sato et al. [Citation2004]), was slightly stimulated under these conditions, but no reproducible effect on transport was observed for X5C, C109A, or G113C () or for Y110C, G114C or G115C (not shown) after treatment with any of the above reagents.
Figure 3. Effect of methanethiosulfonate reagents on EL1 cysteine mutants. Hela Cells expressing SERT EL1 mutants were treated with 1 mM MTSEA (EA), MTSET (ET) or MTSES (ES) for 10 min and then assayed for 5-HT influx, as described under ‘Experimental procedures’. The results represent data combined from three experiments and are expressed as a percentage of the untreated activity of each mutant.
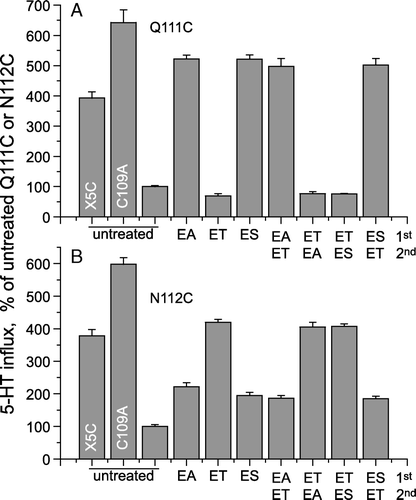
The differences in Q111C and N112C transport stimulation by MTSEA, MTSET and MTSES could result from differences in reactivity of the MTS reagents toward each mutant, or they could be due to the chemical nature of the modified cysteine in each case. (Reaction of a cysteine residue with MTSEA, MTSET and MTSES results in the formation of a mixed disulfide between the cysteine residue and 2-amino ethanethiol, 2-trimethylammoniumethanethiol, or 2-sulfonatoethyl ethanethiol, respectively.) To distinguish between these two possibilities, we treated cells expressing Q111C or N112C sequentially with one MTS reagent followed by another. If the lack of stimulation of Q111C by MTSET, for example, was due to sluggish reactivity toward this reagent, then subsequent modification by MTSEA or MTSET should still have activated. However, if MTSET reacted completely with Q111C with no effect on activity, then subsequent modification by MTSEA or MTSES would have been precluded.
The results in indicate that treatment with each of the MTS reagents resulted in complete modification of the reactive cysteine residue. Treatment of Q111C with MTSET prevented subsequent activation by MTSEA or MTSES, and treatment of N112C with MTSEA or MTSES prevented additional activation by MTSET. In each case, the final level of activity was determined by the first MTS treatment and was not affected by subsequent treatments with another reagent. This experiment also demonstrates that the MTS-stimulated activity of Q111C and N112C was similar to that of X5C (for N112C) or between that of X5C and C109A, which has only one endogenous cysteine replaced (for Q111C).
Figure 4. Extent of activation was dependent on the modifying reagent. Hela Cells expressing Q111C or N112C mutants were first treated with 1 mM MTSEA (EA), MTSET (ET) or MTSES (ES) for 10 min and the cells were washed to remove the unreacted MTS-reagents. The washed cells were further treated with a second MTS-reagent as indicated and then assayed for 5-HT influx. Data are expressed as a percentage of the untreated activity of Q111C (A) or N112C (B). (The figure shows results of a typical experiment that was replicated three times with similar results.)
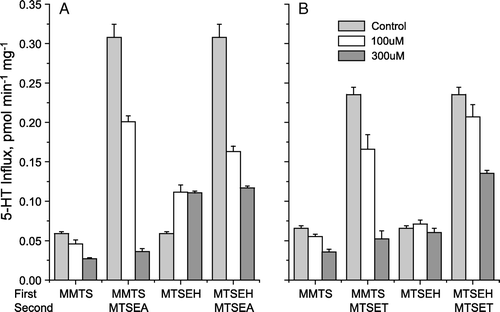
The effect of MTS substituent was further explored by testing the sensitivity of Q111C and N112C to methyl methanethiosulfonate (MMTS) and (2-hydroxyethyl methanethiosulfonate) (MTSEH). As shown in , MMTS, which adds a methanethiol moiety to the reactive cysteine, further inhibited both Q111C (A) and N112C (B). Reaction with MMTS also blocked the ability of a subsequent incubation with MTSEA or MTSET, respectively, to stimulate the activity of Q111C (A) and N112C (B). MTSEH stimulated the activity of Q111C, although to a much smaller extent than MTSEA, and pretreatment with MTSEH prevented further stimulation by MTSEA. MTSEH had no effect on the activity of N112C, up to concentrations that caused cells to detach from the plate. At testable concentrations, MTSEH partially blocked activation of N112C by MTSET, but the blockade was not complete, probably due to lower reactivity of MTSEH with this mutant.
Figure 5. Modification with MMTS and MTSEH. Hela Cells expressing Q111C (A) or N112C (B) mutants were treated with MMTS or MTSEH (0, 100 or 300 µM) in a first (10 min) incubation and then, where indicated, either 10 µM MTSEA or 500 µM MTSET as described in the legend to . Absolute transport rates are shown.
Ion and ligand effects on reactivity
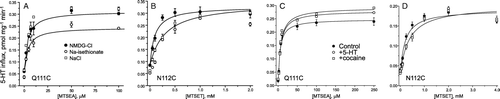
The reactivity of many endogenous or inserted cysteine residues in SERT was found to be modulated in the presence of various ions, substrates or inhibitors. In the context of EL1, Cys-109 reactivity was increased by Li+ and decreased by cocaine (Ni et al. [Citation2001]) and the reactivity of A116C was increased by 5-HT (Sato et al. [Citation2004]). To measure the effect of ions and ligands on the reactivity of Q111C and N112C, we treated cells expressing these mutants with a range of MTSEA or MTSET concentrations for 10 min, and determined the concentration leading to half-maximal stimulation. This EC50 concentration of MTSEA or MTSET was used to calculate a rate constant with the assumption of bimolecular kinetics and a first-order time course of activity loss (Rudnick [Citation2002]). In the 10 min treatment, half-maximal activation gives a t½ of 10 min and a pseudo first order rate constant of 0.069 min−1. From this value and the concentrations of MTS reagent required for half-maximal stimulation, individual rate constants were calculated.
Analysis of the data shown in indicates that there was little effect of replacing either Na+ or Cl− (with N-methyl-D-glucamine (NMDG) or isethionate, respectively) on the activation of Q111C by MTSEA (A). However, approximately twice the concentration of MTSET was required to half-maximally activate N112C in the presence of Na+, suggesting that the reactivity of N112C toward MTSET decreased by about 2-fold in the presence of Na+ (B). Additional experiments (not shown) in which Na+ was replaced by NMDG in the absence of Cl− (replacement with diatrizoate) also demonstrated a decreased reaction rate in the presence of Na+. Addition of 2 µM cocaine or 2 µM 5-HT had little effect on the activation of Q111C by MTSEA (C) or of N112C by MTSET (D). We noted a small but reproducible decrease in the maximum stimulation of Q111C by MTSEA when Cl− was absent during the MTSEA treatment. We currently do not understand the reason for this effect, but it may be due to some instability of Q111C when Cl− was removed.
Figure 6. Ionic and ligand effects on activation. HeLa cells expressing the Q111C mutant (A and C) or the N112C mutant (B and D) were treated with MTSEA or MTSET, respectively, at the indicated concentrations for 10 min in the presence of NaCl (control), NMDG-Cl or Na-isethionate, or in the presence of 2 µM 5-HT or cocaine (C and D), and then washed and assayed for 5-HT influx. The figure shows results of a typical experiment that was replicated at least six times with similar results.
The nature of Q111C and N112C activation by MTS reagents
To determine the nature of MTS-dependent activation, we measured steady-state kinetics of 5-HT influx in cells expressing Q111C and N112C before and after treatment with MTSEA and MTSET, respectively. clearly shows that treatment with MTS reagents, had little effect on the Vmax for transport, but decreased KM dramatically for both mutants. Analysis of the data in A showed that the KM for Q111C decreased from approximately 8 µM before treatment to about 0.4 µM after treatment with MTSEA. Similarly, the KM for N112C decreased from about 5.5 µM before treatment to approximately 0.8 µM after treatment with MTSET (B). The KM values for the MTS-treated mutants is close to the value obtained for the parental construct X5C (0.67 µM), while the untreated mutants had significantly higher KM values. Thus, the increase in activity observed with MTS treatment derives primarily from a decrease in KM.
Figure 7. Activation resulted from a decrease in KM. The rate of 5-HT transport by the Q111C mutant (A) or of the N112C mutant (B) before and after modification with MTSEA or MTSET, respectively, was determined over the indicated range of 5-HT concentrations using 20 nM [3H]5-HT plus added unlabeled 5-HT to achieve the indicated concentrations. The figure shows results of one experiment that was replicated at least twice with similar results. The Km value of the parental control stain X5C is 0.67±0.10.
![Figure 7. Activation resulted from a decrease in KM. The rate of 5-HT transport by the Q111C mutant (A) or of the N112C mutant (B) before and after modification with MTSEA or MTSET, respectively, was determined over the indicated range of 5-HT concentrations using 20 nM [3H]5-HT plus added unlabeled 5-HT to achieve the indicated concentrations. The figure shows results of one experiment that was replicated at least twice with similar results. The Km value of the parental control stain X5C is 0.67±0.10.](/cms/asset/0c94f1f9-3c21-4726-92fb-b9b247ceae30/imbc_a_263171_f0007_b.gif)
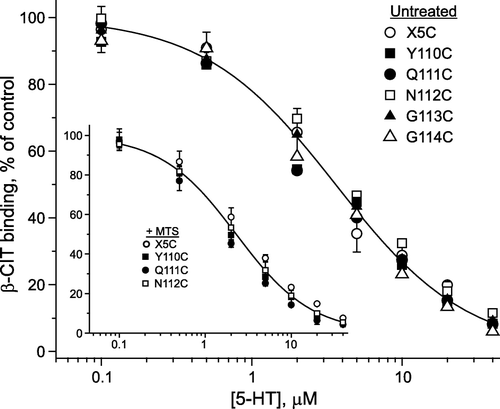
The KM decrease could result from an increase in 5-HT affinity, but it could also result from a change in the rates of individual steps in the transport cycle. To distinguish between these two possibilities, we determined the equilibrium binding affinity of 5-HT in membranes from cells expressing Q111C and N112C before and after treatment with MTSEA and MTSET, respectively. 5-HT binding was measured by its ability to displace β-CIT. Comparison of X5C with Y110C, Q111C, N112C, G113C, and G114C indicated very similar ability of 5-HT to displace β-CIT for both untreated () and MTS-treated (inset) conditions. Thus, the dramatic change in KM observed upon treatment of Q111C or N112C with MTS reagents was not due to a change in 5-HT affinity and thus was likely to result from a change in the rate of a step subsequent to 5-HT binding in the transport cycle.
Figure 8. 5-HT binding affinity was unchanged by mutation of Tyr-110, Gln-111, Asn-112 or Gly-113. Membranes prepared from HeLa cells expressing each of the EL1 cysteine mutants and the control X5C were incubated with β-CIT in the presence of the indicated concentrations of 5-HT for 1 h, washed, and assayed for the residual β-CIT binding as described under ‘Experimental procedures’. The concentration of β-CIT used was approximately 10% of the KD value. Inset: Membranes from three EL1 mutant stains and X5C were modified with MTS-reagents prior to measuring 5-HT displacement of β-CIT. Y110C and Q111C were treated with 1 mM MTSEA and N112C was treated with 1 mM MTSET for 15 min. Data are from a typical experiment which was repeated twice with similar results. The results are expressed as a percentage of β-CIT binding activity in the absence of 5-HT.
To further define the defect caused by mutation at Gln-111 or Asn-112, we tested the ability of MTSEA to influence the exchange of intracellular [3H]5-HT with extracellular unlabeled 5-HT (). This reaction requires only the steps in the transport reaction that involve 5-HT binding and translocation, but not the K+ stimulated return steps that occur after release of 5-HT to the cytoplasm, converting SERT to a form that binds another 5-HT from the extracellular medium. HEK-293 cells expressing X5C, Q111C and N112C SERT mutants were incubated with [3H]5-HT and then superfused with medium, which was collected and counted. After efflux had stabilized, 5-HT was added to the superfusion medium and the subsequent increase in efflux was considered 5-HT exchange (see Materials and Methods). As shown by the filled symbols in , panels A-C, the exchange was much faster for X5C relative to Q111C or N112C, reflecting the difference in their influx rates (). When the cells were treated with MTSEA (open symbols, ), there was an enhanced efflux in response to subsequent 5-HT addition, which was most dramatic for Q111C (B), robust for N112C (C) and barely detectable for X5C (A). Combined results from 3 to 7 experiments (D) show that the effect on X5C was not significant but that MTSEA significantly increased 5-HT exchange for Q111C and N112C. MTSEA activation of N112C was not as dramatic as that of Q111C, in keeping with the relative transport response of these two mutants to MTSEA (). Consequently, the time course for efflux following 5-HT addition was slower (C, D).
Figure 9. Effect of MTSEA on [3H]5-HT efflux induced by extracellular 5-HT. Representative efflux experiments of X5C-SERT (panel A; n=6–7), Q111C-SERT (panel B; n=5–13, all experiments performed in triplicate) and N112C-SERT (panel C; n=10–11) stably expressed in HEK293 cells. Cells were grown on glass coverslips and preloaded with tritiated serotonin for 20 min in the presence (open symbols) or absence of MTSEA (2.5 mM; filled symbols). After a subsequent wash step, coverslips were transferred to small superfusion chambers and superfused (for details, see ‘Materials and methods’). The experiment was started after a 45 min washout period at t = 0 with the collection of superfusate in 2-min fractions. 5-HT (10 µM) was added to the superfusion buffer after 6 min. The collected radioactivity was counted and is expressed as a fractional efflux rate, i.e., as a percentage per 2 min of the cellular [3-H]5-HT content at that very time point. Baseline efflux was adjusted for more accurate comparison between control and MTSEA-treated cells. Panel D summarizes the integrated effects of 5-HT treatment in the presence or absence of MTSEA (as indicated) with subtraction of baseline efflux levels.
![Figure 9. Effect of MTSEA on [3H]5-HT efflux induced by extracellular 5-HT. Representative efflux experiments of X5C-SERT (panel A; n=6–7), Q111C-SERT (panel B; n=5–13, all experiments performed in triplicate) and N112C-SERT (panel C; n=10–11) stably expressed in HEK293 cells. Cells were grown on glass coverslips and preloaded with tritiated serotonin for 20 min in the presence (open symbols) or absence of MTSEA (2.5 mM; filled symbols). After a subsequent wash step, coverslips were transferred to small superfusion chambers and superfused (for details, see ‘Materials and methods’). The experiment was started after a 45 min washout period at t = 0 with the collection of superfusate in 2-min fractions. 5-HT (10 µM) was added to the superfusion buffer after 6 min. The collected radioactivity was counted and is expressed as a fractional efflux rate, i.e., as a percentage per 2 min of the cellular [3-H]5-HT content at that very time point. Baseline efflux was adjusted for more accurate comparison between control and MTSEA-treated cells. Panel D summarizes the integrated effects of 5-HT treatment in the presence or absence of MTSEA (as indicated) with subtraction of baseline efflux levels.](/cms/asset/56fc6250-7586-4500-bb35-a45ee7292555/imbc_a_263171_f0009_b.gif)
Discussion
Among mammalian transporters in the NSS family the region from Cys-109 to Phe-117 of SERT contains the highest concentration of conserved residues. Within this region, the identity between transporters varies from a low of 56% for Gln-111 to a high of 100% for Gly-113 through Gly-115 with an average value of 85%. Indeed, the apparently ‘low’ value for Gln-111 is misleading because, aside from a methionine at this position in the system IMINO transporter SIT1 (SLC6A20), all mammalian NSS transporters have a polar residue (K, Q, R, T, or S) at this position. Even among prokaryotic members of the NSS family, the degree of conservation in this region is high, as exemplified in for the comparison between SERT and LeuT from Aquifex aolicus. This study is the first attempt to systematically examine this region of SERT by cysteine scanning mutagenesis.
Replacement with cysteine had dramatically different effects on different residues in this region. In particular, of the three adjacent glycine residues, replacement with cysteine had no effect on Gly-113 but ablated transport activity at the following two positions (A). The inability of G114C and G115C to transport is most likely a result of the low surface expression of these mutants (C,D). In the structure of LeuT, the corresponding glycine residues are almost entirely buried. Gly-40 has no solvent exposed surface area and Gly-41 has only 2% of its surface area exposed to solvent. By comparison, Gly-39 of LeuT, which corresponds to Gly-113 of SERT, has 40% of its surface exposed in the structure. Thus, the bulkier cysteine side chain is likely to be more readily accommodated as a replacement for Gly-113 than for Gly-114 or Gly-115.
Tyr-110 of SERT corresponds to Ala-36, which is also buried in the LeuT structure. However, replacement of Tyr-110 with cysteine did not create a severe expression defect (C, D). The Y110C mutant retained almost the same level of β-CIT binding activity as the parental X5C, probably because the cysteine side chain is less bulky than the phenolic side chain of tyrosine and can be accommodated in the structure without difficulty. Despite relatively normal surface expression (C, D) and binding activity (B, 7), Y110C did not transport 5-HT, indicating that some step in transport subsequent to substrate binding was defective. We speculate that in another conformation of SERT (possibly the internal-facing form), Tyr-110 is required to interact with some other residues in a way that cannot be substituted by cysteine. Thus, the conformation that binds 5-HT and β-CIT can accept a cysteine in the place of Tyr-110 but cysteine cannot replace Tyr-110 in a conformation required for some other step in the transport cycle.
Gln-111 and Asn-112 of SERT correspond to Glu-37 and Asn-38 of LeuT, which were accessible in the structure, with 30% and 18%, respectively, of their surface area exposed to solvent. The transport activity of Q111C and N112C was significantly lower than that of the parental X5C construct (A), although binding and surface expression appeared normal (B–D). Of the cysteine mutants in the EL1 region, Q111C and N112C were the ones to show the largest activity change when treated with MTS reagents (). We previously showed that the nearby A116C also reacted with MTSEA to increase transport activity (Sato et al. [Citation2004]), although the effect was not as large as with Q111C or N112C. From the rate constants for modification, these two mutants appear to be relatively reactive. We believe that the characteristics of the Q111C and N112C mutants and their reactivity toward MTS reagents may shed some light on the mechanism of 5-HT transport by SERT.
Replacement of Gln-111 or Asn-112 with cysteine inhibited transport activity (A) but, in both mutants, modification of the novel cysteine with MTS reagents restored activity back to the level of the parental X5C construct (). This surprising behavior might reflect the need for polar character at positions 111 and 112. In the case of Q111C, modification by MTSEA and MTSES was equally effective, despite their widely divergent ionic properties. MTSEA modifies cysteine by adding a –S-C2H4-NH3+ moiety to the end of the side chain, but MTSES contributes instead an – S-C2H4-SO3− group. It is unlikely that simply blocking the cysteine sulfhydryl group was responsible for reactivation, because treatment with MTSET had no effect on Q111C, except to block reactivation by other MTS reagents (A). In N112C, all three MTS reagents activated transport, but only MTSET activated maximally ( and B). These results are almost the reverse of those seen with Q111C except that MTSEA and MTSES partially activated N112C (). Clearly, there are requirements in addition to increasing the polar nature of the cysteine at 111 or 112 that must be satisfied for maximal function, although our results do not allow a complete understanding of these requirements.
Treatment with MMTS and MTSEH allowed some further understanding of the process. MMTS, which adds a methanethiol moiety to the reactive cysteine, was slightly inhibitory for both Q111C and N112C, as expected if polar character is important at these positions. MTSEH, which adds an – S-C2H4-OH group, activated Q111C, although not as much as did MTSEA, but did not stimulate N112C. Thus, the level of activity seemed to reflect the extent of polar character in the side chains of these two residues. Moreover, the two positions did not respond equivalently to increased polarity since MTSEH stimulated Q111C but not N112C.
The rates of Q111C activation by MTSEA or N112C activation by MTSET were quite rapid relative to other endogenous or inserted cysteine residues in SERT. From , we calculated that MTSEA reacted with Q111C at a rate of 200-300 sec−1 M−1. Although cysteine at some positions, such as Cys-200 in EL2 (Chen et al. [Citation1997]) or Cys-357 in IL3 (Androutsellis-Theotokis et al. [Citation2001]) reacted much more slowly with MTSEA (3 and 26 sec−1 M−1, respectively), others such as A441C in TM8 reacted at comparable rates (Androutsellis-Theotokis & Rudnick [Citation2002]). The rate of Q111C modification was not significantly affected by removal of Na+ or Cl−, nor was it sensitive to the presence of 5-HT or cocaine. This result suggests that conformational changes accompanying ion and ligand binding and transport do not reduce the accessibility of Cys-111 in Q111C.
We found a different situation for N112C. The rate of about 5 sec−1 M−1 for MTSET reaction with N112C is comparable to some rates measured for MTSET modification of cysteines inserted into other extracellular loops, but was much slower than the maximum rates measured. In EL5, for example, values ranged up to 200 sec−1 M−1 (Keller et al. [Citation2004]). Furthermore, significant differences were measured in the rate of N112C modification by MTSET depending on conditions. The most dramatic change was a >2-fold increase in the rate when Na+ was replaced with NMDG (). The same difference was observed both in the presence and absence of Cl− (data not shown). In addition, a smaller decrease in rate was observed in the presence of saturating concentrations of 5-HT or cocaine. In general, we have frequently found that cysteine residues in SERT that are highly exposed and react rapidly also are less sensitive to conformational changes due to ligand binding or transport. Thus, a cysteine replacing Gln-111, which corresponds to the more highly accessible Glu-37 in LeuT was more reactive and less sensitive to Na+ or ligands but a cysteine in place of Asn-112, which corresponds to the less accessible Asn-38 of LeuT, reacted slower but was more sensitive to changes in Na+.
The most striking observation reported here is that cysteine substitution of Gln-111 or Asn-112 decreased transport rate, primarily at low 5-HT concentrations, through an increase in KM, without affecting 5-HT affinity. This KM increase did not result from an irreversible folding defect, since it could be reversed by modification of either cysteine mutant with an appropriate MTS reagent. Changes in KM are frequently attributed to changes in substrate affinity, which are certainly one determinant of KM, but our data clearly demonstrate large KM changes with no change in affinity for both Q111C and N112C. KM is also determined by the relationship between the kinetics of individual transport steps, implying that a step in transport following 5-HT binding is affected by cysteine substitution at Gln-111 and Asn-112 and is restored by modification of that cysteine by MTS reagents. Efflux experiments indicate an impairment in the steps involving 5-HT translocation () and MTSEA treatment repaired this defect, as it did for transport (). These results indicate that the mutation of Gln-111 or Asn-112 to cysteine impaired the part of the transport cycle in which 5-HT is translocated. Although we cannot exclude the possibility that the return steps (through which the empty carrier appears on the cell surface) are also affected, this result defines the effect of these mutations as influencing 5-HT translocation and rules out the possibility that only the return steps are affected.
Transporters such as SERT are believed to function by interconversion between two forms, each of which exposes the substrate binding site to a different surface of the membrane. The orientation of EL1 shown in , in which Gln-111 and Asn-112 are exposed, may approximate the conformation in which 5-HT binds to SERT from the external medium. However, at some point in the reaction cycle Gln-111 and Asn-112 may interact with other parts of the protein in a manner that is disrupted by replacement with cysteine. This intermediate might be the form of SERT that exposes the binding site to the cytoplasm. We would expect that the relatively open pathway leading from the periplasmic side of LeuT to the bound leucine in the published structure is closer to the external-facing form of the transporter and that it substantially collapses in the cytoplasmic-facing form (Rudnick [Citation2006]). Indeed, there is data to suggest that cysteine residues in the extracellular half of TM1 and in EL4 of SERT became occluded under conditions expected to favor the cytoplasmic-facing form (5-HT + NaCl) but are exposed in the presence of the non-transported inhibitor cocaine, which should favor the external-facing form (Henry et al. [Citation2003], Mitchell et al. [Citation2004]). These treatments have the opposite effects on residues in TM5 proposed to contribute to the cytoplasmic pathway (Zhang & Rudnick [Citation2005], Zhang & Rudnick [Citation2006]). Although the cysteines that replaced Gln-111 and Asn-112 did not become dramatically occluded in the presence of 5-HT, their impact on transport but not binding suggests that these mutations might affect steps subsequent to binding, for example those involving the cytoplasmic-facing form of SERT in which the external permeation pathway is closed.
Acknowledgements
This work was supported by a Program Project Grant, DA012408, from the National Institute on Drug Abuse (GR) and the Austrian Science Foundation/FWF, grant P18706 (HHS). We thank Dr. Lei Shi and the bioinformatics core of the Program Project Grant for the structural data and solvent accessibility calculations.
References
- Androutsellis-Theotokis A, Ghassemi F, Rudnick G. A conformationally sensitive residue on the cytoplasmic surface of serotonin transporter. J Biol Chem 2001; 276: 45933–45938
- Androutsellis-Theotokis A, Goldberg NR, Ueda K, Beppu T, Beckman ML, Das S, Javitch JA, Rudnick G. Characterization of a functional bacterial homologue of sodium-dependent neurotransmitter transporters. J Biol Chem 2003; 278: 12703–12709
- Androutsellis-Theotokis A, Rudnick G. Accessibility and conformational coupling in serotonin transporter predicted internal domains. J Neurosci 2002; 22: 8370–8378
- Blakely RD, Clark JA, Rudnick G, Amara SG. Vaccinia-T7 RNA polymerase expression system: evaluation for the expression cloning of plasma membrane transporters. Anal Biochem 1991; 194: 302–308
- Chen JG, Liu-Chen S, Rudnick G. External cysteine residues in the serotonin transporter. Biochemistry 1997; 36: 1479–1486
- Chen JG, Liu-Chen S, Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. JBC 1998; 273: 12675–12681
- Henry LK, Adkins EM, Han Q, Blakely RD. Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J Biol Chem 2003; 278: 37052–37063
- Keller PC, 2nd, Stephan M, Glomska H, Rudnick G. 2004. Cysteine-scanning mutagenesis of the fifth external loop of serotonin transporter. Biochemistry 43:8510–8516.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680–685
- Mitchell SM, Lee E, Garcia ML, Stephan MM. Structure and function of extracellular loop 4 of the serotonin transporter as revealed by cysteine-scanning mutagenesis. JBC 2004; 279: 24089–24099
- Ni YG, Chen JG, Androutsellis-Theotokis A, Huang CJ, Moczydlowski E, Rudnick G. A lithium-induced conformational change in serotonin transporter alters cocaine binding, ion conductance, and reactivity of cys-109. JBC 2001; 276: 30942–30947
- Rudnick G. Chemical modification strategies for structure-function studies. Transmembrane transporters, MW Quick. Wiley-Liss, Inc, Hoboken, NJ 2002; 125–141
- Rudnick G. Structure/function relationships in serotonin transporter: new insights from the structure of a bacterial transporter. Handbook of experimental physiology, HH Sitte. Springer-Verlag, Berlin, Heidelberg 2006; 59–73
- Sato Y, Zhang Y-W, Androutsellis-Theotokis A, Rudnick G. Analysis of transmembrane domain 2 of rat serotonin transporter by cysteine scanning mutagenesis. J Biol Chem 2004; 279: 22926–22933
- Sen N, Shi L, Beuming T, Weinstein H, Javitch JA. A pincer-like configuration of TM2 in the human dopamine transporter is responsible for indirect effects on cocaine binding. Neuropharmacology 2005; 49: 780–790
- Sitte HH, Hiptmair B, Zwach J, Pifl C, Singer EA, Scholze P. Quantitative analysis of inward and outward transport rates in cells stably expressing the cloned human serotonin transporter: Inconsistencies with the hypothesis of facilitated exchange diffusion. Mol Pharmacol 2001; 59: 1129–1137
- Tate C, Blakely R. The effect of N-linked glycosylation on activity of the Na+- and Cl−-dependent serotonin transporter expressed using recombinant baculovirus in insect cells. JBC 1994; 269: 26303–26310
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005; 437: 215–223
- Zhang YW, Rudnick G. Cysteine scanning mutagenesis of serotonin transporter intracellular loop 2 suggests an alpha-helical conformation. JBC 2005; 280: 30807–30813
- Zhang YW, Rudnick G. The cytoplasmic substrate permeation pathway of serotonin transporter. JBC 2006; 281: 36213–36220
- Zhou Y, Bennett ER, Kanner BI. The aqueous accessibility in the external half of transmembrane domain I of the GABA transporter GAT-1 is modulated by its ligands. J Biol Chem 2004; 279: 13800–13808
- Zhou Y, Kanner BI. Transporter-associated currents in the γ-aminobutyric acid transporter GAT-1 are conditionally impaired by mutations of a conserved glycine residue. J Biol Chem 2005; 280: 20316–20324