Abstract
Cholesterol in the plasma membrane of eukaryotic cells contributes to modulating the functions and signalling pathways of numerous transmembrane proteins, including G protein Coupled Receptors (GPCRs). We have previously shown that the function of the human µ-opioid receptor (hMOR) expressed in Saccharomyces cerevisiae is modulated by sterols including cholesterol. Here, we investigated the effects of cholesterol content on hMOR pharmacology and on hMOR partitioning in cholesterol-poor and -rich domains in eukaryotic mammalian cells (CHO). We show that cholesterol is required for the stabilization of a receptor conformation with high agonist affinity and for triggering G-protein activation after agonist binding to the receptor. Biochemical analysis of untreated and cholesterol-depleted membranes in cells expressing hMOR indicated that the receptor is only present in cholesterol poor domains, in the basal state. After agonist binding to untreated CHO membranes, two distinct populations of receptor were found in cholesterol-rich and -poor domains. Cholesterol depletion or treatment of CHO membranes with the G-protein-decoupling agent GppNHp prevented the redistribution, indicating that receptor activated states localized into cholesterol-rich domains. Pharmacological data and biochemical analysis indicate that distinct activated conformations of hMOR exist in CHO plasma membrane and correspond to microdomains differing by thickness and proportions of lipid components, including cholesterol.
| Abbreviations | ||
| Chol | = | Cholesterol |
| SC | = | Saccharomyces cerevisiae |
| PL | = | phospholipids, Prot, proteins |
| MβCD | = | Methyl-β-cyclodextrin |
| PC | = | phosphocholine |
| PE | = | phosphatidyl ethanolamine |
| PS | = | phosphatidyl serine |
| SM | = | sphingophospholipids |
| GL | = | glyco-lipids or sphingoglycolipids |
| U | = | untreated membranes |
| D | = | Cholesterol depleted membranes |
| C+ | = | Cholesterol complemented D membranes |
| C+ + | = | Cholesterol complemented U membranes |
| E+ | = | Ergosterol complemented D membranes |
| DRM | = | Detergent resistant membranes |
| N-DRM | = | Detergent solubilized membranes |
Introduction
Plasma membranes are organized into specialized micro- and nano-domains differing in composition, biological function and physical properties Citation[1]. Unfortunately, these biologically active membrane domains cannot be studied directly in living cell plasma membranes, because available methods for their isolation and detection, and in particular selective methods of detergent extraction of resistant membrane domains (DRM), are too invasive. However, various model membranes with particular lipid compositions mimic DRM, possibly corresponding to raft domains, from natural membranes Citation[2–4]. Lipids in biological membranes are typically in a physically fluid state, called the liquid-disordered (ld) phase: the lipids are subjected to rotational, conformational and translational diffusion processes. However, work with model membranes indicates that some lipids tend to associate with each other, promoting dynamic lipid segregation. In this way, mixtures of cholesterol, sphingolipids and saturated glycophospholipids Citation[5] generate liquid-ordered (lo) phases that have more tightly structured hydrophobic cores and are thicker than ld phases that would have the same lipid composition Citation[6]. The properties of the lo-phase and that of raft microdomains in natural membranes should be similar Citation[1].
Cholesterol in the plasma membrane is involved in the modulation of the function and the signalling pathways of numerous transmembrane protein systems Citation[7–11]. Rhodopsin, a G protein-coupled receptor (GPCR), is one of the most extensively studied GPCRs in particular as concerns the effect of cholesterol, fatty acid chain length and degree of unsaturation on its conformation Citation[12] and function Citation[13], Citation[14]. Other GPCRs are difficult to purify and studies of the effect of surrounding lipids have mostly used native membranes allowing functional receptor studies (galanin Citation[15], cholecystokinin (CCK-R) Citation[16], µ-opioid (MOR) Citation[17–19], serotonin1A Citation[20], oxytocin receptors Citation[21], Citation[22]. Recently, a structure of the human β2-adrenergic receptor (hβ2-AR), which crystallized in an active form only in cholesterol-rich environments, has been reported Citation[23]. Cholesterol has been described to act on GPCR functions in two ways. The first, direct or specific interactions of cholesterol and GPCR Citation[15], Citation[21], Citation[22] can modulate receptor function positively or negatively (ligand binding and/or effector activity). The second involves the effect of cholesterol on the biophysical properties of the membrane, as described for the oxytocin receptor Citation[24].
We demonstrated previously that the ligand-binding properties of the human µ-opioid receptor (hMOR) expressed on native membranes of Saccharomyces cerevisiae, was present in a low-affinity conformation at a basal state and recovered a receptor high-affinity state in cholesterol-loaded, ergosterol-depleted yeast membranes Citation[19].
Here, we investigate ligand binding (agonist and antagonist, peptide and alkaloid) and G-protein coupling according to the cholesterol content of CHO cell membranes, to elucidate the effect of cholesterol on the function of hMOR. We assessed the distribution of various pharmacological states of hMOR in plasma membranes according to the lateral distribution of cholesterol, lipids and membrane proteins in the cholesterol-rich membrane fractions (DRMs) and cholesterol-poor membrane fractions (N-DRMs). We found that hMOR function in CHO membranes may thus be dependent on the level of cholesterol and we suggested that various hMOR conformations would be confined to distinct domains of CHO cell membranes.
Materials and methods
Methyl-β-cyclodextrin (MβCD), diprenorphine (DPN), cholesterol and ergosterol were purchased from Sigma-Aldrich RBI, [D-Ala2, N-MePhe4, Gly-ol5] enkephalin (DAMGO) and [D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2] (CTAP) from Bachem. 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate (TMA-DPH) was purchased from Molecular Probes (Eugene, Oregon, USA). The purity of TMA-DPH and sterols was checked by thin-layer chromatography. Salts and solvents were of analytical grade.
Cell culture
CHO-K1 cells stably transfected with a construct encoding the hMOR (3×105 receptors/cell) labeled at the N-terminus (N-term) with a T7 tag sequence (called CHO/T7hMOR cells), were grown on Ham-F12 medium plates supplemented with 10% fetal calf serum and antibiotics (50 U/ml penicillin, 50 µg/ml streptomycin, Invitrogen) and selected with 400 µg/ml geneticin (Invitrogen).
Preparation of CHO cell membranes enriched in plasma membranes
Plasma membrane of CHO cells was prepared as previously described Citation[25], under pressure of nitrogen, in a Kontes pressure homogenizer. Plated cells were incubated in phosphate-buffered saline-EDTA (PBS-1.5 mM EDTA, pH 7.4) for 15 min at 37°C to release them from the culture dish. The resulting suspension was centrifuged at 500 g for 5 min and resuspended in Tris buffer (TE: 50 mM Tris-HCl, 1 mM EDTA, pH 7.4, containing a protease inhibitor cocktail tablet (Complete Mini, Roche, Germany) at an optical density of 0.7 at λ = 650 nm. Cells were then equilibrated at 4°C for 10 min at a pressure of 10 standard atmospheres (atm) in a Kontes pressure homogenizer (Avantec, France). Lysates were obtained by releasing the cell suspension dropwise from the homogenizer. The homogenate was centrifuged at 1500 g for 10 min to remove intact cells, nuclei and mitochondria Citation[25]. The supernatant was then centrifuged at 110,000 g for 35 min in a Beckman centrifuge with a 60Ti rotor. The membrane pellet named untreated plasma membranes of CHO cells (U membranes) was resuspended in TE and the proteins or lipids were quantified. This protocol led to a cell lysis rate of around 100%, with no disruption of subcellular organelles, but a partial contamination by endoplasmic reticulum and lysosomes Citation[25]. To decrease the rate of contamination of the plasma membrane by these elements, a Percoll gradient could be used Citation[25], but it is not possible in our case, because of the loss of hMOR activity (data not shown).
Preparation of MβCD solution and sterol-MβCD complexes
First, a solution of 40 mM MβCD was prepared in TE. Cholesterol-MβCD complexes (1:18 molar ratio) were prepared by adding a solution of 8.6 mg cholesterol in 100 µl DMF dropwise to MβCD solution at a temperature of approximately 80°C under agitation. Typically, 100 µl cholesterol solution was added to 10 ml of 40 mM MβCD solution. Ergosterol/MβCD complexes were prepared by adding 100 µl ergosterol (4.4 mg in 100 µl CHCl3:CH3OH [9/1]) solution to 10 ml of 40 mM MβCD solution (1:36 molar ratio of ergosterol to MβCD).
MβCD manipulation of membrane sterol content
Membranes were depleted of cholesterol and loaded with cholesterol. Cholesterol was depleted from CHO native membranes (U for untreated membranes), prepared as described above at a protein concentration of approximately 2 mg/ml, by incubation with equal volume of 10 mM MβCD solution for 30 min at 20°C under agitation. Depleted membranes (D membranes) were pelleted as described above and resuspended at 2 mg/ml in TE. U and D membranes were incubated with cholesterol or ergosterol with an equal volume of 10 mM appropriate sterol-MβCD complexes for 30 min at 20°C under agitation. Native membranes loaded with cholesterol (C+ + ) and depleted membranes loaded with cholesterol (C+) or ergosterol (E+), were centrifuged as described above in a Beckman centrifuge with a 60Ti rotor to remove MβCD and then resuspended in TE.
Lipid analysis
Lipids from CHO membranes or from DRMs and N-DRMs were extracted according to the classical Bligh and Dyer procedure. Sterols were quantified by colorimetric methods based on the oxidation of the hydroxyl group (Roche Molecular Materials) or on the formation of a cholestatetraenylic cation by the Zak reaction Citation[26].
Phospholipids (PL) were titrated according to the Eaton and Dennis procedure. Phospholipids and sphingolipids where characterized by thin-layer chromatography (TLC): the total lipid extract from one experiment was spotted at one edge of TLC channeled plates (Kieselgel 60 CF254, Merck) and allowed to migrate with chloroform/methanol/water (65:25:4 v/v/v). The plate was sprayed with sulfuric acid, heated at 250°C for 15 min and analyzed. Quantity One 1-D software (Biorad) was used to quantify the spots by densitometry.
Fatty acids were titrated according to gas chromatography (GC) analysis. Two internal standards (15:0–15:0 PC and 17:0–17:0 PC) in a 2:1 molar ratio were added to the lipid extract before fatty acid transmethylation. Organic solvents were evaporated under a stream of nitrogen, and the samples were dissolved in 250 µl boron trifluoride/methanol (14% w/v). The tubes were flushed with N2, capped with a Teflon-lined cap and heated at 90°C for 1 h. They were then cooled to room temperature, 1 ml of water was added, and methyl esters were extracted twice with 0.5 ml petroleum ether and vigorous shaking. The pooled organic phases were dried with anhydrous sodium sulfate and concentrated under N2. GC analysis was carried out on a semi-capillary column (15 m×0.25 mm) coated with 5% diphenyl, 95% dimethylsiloxane (HP-5MS) with argon as the carrier gas. The oven temperature was programmed to rise from 160–200°C at a rate of 4°C/min, remain at 200°C for 8 min, then rise to 260°C at a rate of 6°C/min and remain at this temperature for 15 min to elute free sterols and other contaminants. The response factor for each fatty acid was calculated relative to that of internal standards.
Steady-state fluorescence polarization experiments
Steady-state fluorescence polarization experiments were carried out with a homemade automatic T-format apparatus connected to a microcomputer, as described previously Citation[27]. The suspension of labeled cells was placed in a closed, thermostatically controlled housing system equipped with temperature monitoring devices (Peltier elements). The temperature increased from 4 to 37±0.3°C at a rate of 1°C/3 min. Polarized fluorescence intensities (IV and IH) were measured at each degree. Wavelengths of 348 nm for excitation and 426 nm for emission were used. Polarization rate values (pTMA-DPH=(IV-IH)/(IV+IH)) were determined to within±1% (relative values).
Membranes (U, D, C+, C+ + ) were prepared as described above and diluted in PBS buffer to obtain an OD348<0.08. TMA-DPH (1 µM) was incubated with the membranes for 5 min before the experiment.
Preparation of DRMs
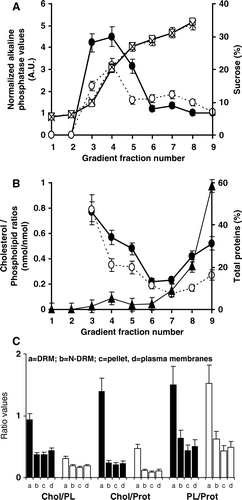
Preparations of total native or depleted membranes (1 ml) with a protein concentration of 5–7.5 mg/ml in TE were deposited at the bottom of 15 ml ultracentrifuge tubes (Ultraclear Beckman). These samples were incubated in the presence and absence of hMOR agonists (1 µM) and antagonists (1 µM) for 1 h at 25°C in a water bath and then cooled on ice. TX100 was used at a final concentration of 0.25% (higher concentrations of TX100 (up to 0.5%) completely solubilized T7hMOR, limiting its presence in DRMs) for 5 mg of proteins, to extract more than 95% of T7hMOR from the pellet of sucrose gradients of U or D CHO membranes. In contrast more than 50% of the proteins were still present in the pellet. The mixed solution was kept on ice for 20 min. A discontinuous sucrose gradient was prepared: the above preparation was mixed with 1 ml 70% sucrose (w/v) in TE (without TX100 and antiproteases), overlaid with 4 ml 30% sucrose (w/v) and then 4 ml 5% sucrose (w/v). The gradient was centrifuged at 200,000 g for 18 h at 4°C in a Beckman SW41 rotor. Fractions of 1.20 ml were harvested from the top (fraction 1) to the bottom (fraction 8) of the tube. We identified an opaque annulus (fraction 4) corresponding to DRMs of U (DRM-U) or D (DRM-D) CHO membranes in the 20% sucrose region. Fractions 6–8 corresponded to N-DRMs of U (N-DRMs-U) or of D (N-DRMs-D) CHO membranes. Turbidity at 620 nm, sucrose percentage, quantity of proteins, and alkaline phosphatase specific activity (Sigma Fast Enzymatic kit, Sigma), used as raft marker, was determined. For each fraction, the alkaline phosphatase activity was converted as the normalized alkaline phosphatase activities (AP) by correction following their relative total protein contents. DRMs were recovered mainly in fractions three to five. DRMs were washed twice by ultracentrifugation at 200,000 g for 1 h at 4°C to prepare them for binding experiments. The pellet (fraction 9) was suspended in Tris-HCl 50 mM, pH 7.4.
Western blotting
Aliquots of gradient fractions identified as DRM and N-DRM fractions were suspended in Laemmli sample buffer (50 mM Tris-HCl, 0.8% SDS (w/v), 4% glycerol (v/v), 0.2% β-mercaptoethanol (v/v), bromophenol blue, pH 6.8) supplemented with 4 M urea, boiled for 5 min, separated by 10% SDS-polyacrylamide gel electrophoresis and electrotransferred to a nitrocellulose membrane (Hybond ECL, Amersham). Equal amounts of proteins (15 µg) were loaded in each lane. The nitrocellulose membrane was blocked for 60 min at room temperature in 20 mM Tris-HCl/125 mM NaCl buffer, pH 7.6 with 0.1% Tween-20 (v/v), 5% milk powder (w/v) and 10% FCS (v/v). The membranes were incubated for 1 h incubation with anti-T7 Tag antibody-HRP conjugate (dilution 1/5000, Novagen) at 20°C, and Western Lightning™ Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences, Inc.) was used to detect bound antibody. ImageJ 1.32J software (Wayne Rasband, NIH USA) was used to quantify and analyze band intensities. The western analysis was normalized according to the true mass of proteins contained in the gradient fractions by determining and applying correction factors to the corresponding lane of the western blot.
Saturation binding experiments
CHO/T7hMOR membranes, DRM and N-DRM aliquots (5–20 µg of protein) were used for saturation experiments in 0.5 ml of 50 mM Tris-HCl, pH 7.4 with [3H]-DPN (Perkin Elmer Life Sciences), a non-selective opioid antagonist, and [3H]-DAMGO (NEN Life Sciences Products), a MOR-selective agonist. Competitive inhibition of [3H]-DPN (1 nM) was assessed with 12 concentrations of competitors (4–20 pM morphine or 4 pM to 200 µM CTAP, according to the selectivity of the inhibitor used). Saturation and competitive experiments were done in the presence or in the absence of the GTP analogue, Gpp(NH)p (100 µM). Non-specific binding was determined in the presence of 1 µM unlabeled DPN or DAMGO. Following incubation for 1 h at 25°C, free ligand was removed by filtration through a Whatman GF/B filter. Filters were then washed three times with 10 mM ice-cold Tris-HCl buffer. Scintillation fluid (Ready protein+, Beckman Coulter, USA) was added and bound radioactivity measured with a Packard Tri-carb 2100 TR scintillation counter. Data were analyzed with GraphPad Prism software using the one-site binding equation.
[35S]-GTPγS binding experiments
[35S]-GTPγS binding to hMOR-CHO cell membrane preparations was measured in 96-well microplates (NEN Basic Flashplates), with 200 µl final well volumes. Membrane preparations (10 µg protein/well) were incubated for 15 min at 30°C in assay buffer (20 mM Hepes, pH 7.4, containing 100 mM NaCl, 10 µg/ml saponin, 3 mM MgCl2, 1 µM GDP), in the presence or absence (basal [35S]-GTPγS binding) of 0.1 nM to 1 µM DAMGO or morphine. These membrane preparations were then incubated with 0.1 nM [35S]-GTPγS (Amersham Biosciences) for 30 min at 30°C, followed by centrifugation (800 g for 10 min) at 4°C and removal of the supernatants. A Wallac 1450 Microbeta Trilux was used to count scintillation over 2 min in each well in the microplates. Data were analyzed with GraphPad Prism software using the one-site binding equation.
Results
Preparation of membranes from CHO cells enriched in plasma membranes
The value of 0.44 for the Chol/PL molar ratio of U membranes () indicated that some quantity of reticulum is still present in our plasma membrane preparation. In a pure CHO plasma membrane, Chol/PL has a value of 0.54, while for a total membrane homogenate, a value of 0.34 was measured Citation[25] because no cholesterol is found in endoplasmic reticulum.
Table I. Lipid, protein contents and pTMA-DPH polarization ratio value for U (untreated), D (depleted), C+ (D complemented), C+ + (U complemented) and E+ (D ergosterol complemented) CHO/T7hMOR membranes.
Reversible alterations of sterol content of CHO plasma membranes expressing T7hMOR
We depleted the sterol content of U membranes by treatment with empty MβCD and complemented D or U membranes with sterol using 10 mM of sterol-loaded MβCD leading to cholesterol-loaded (C+) or -overloaded (C+ + ) and ergosterol-loaded (E+) membranes. Total Prot and to a lesser extent PL contents did not change significantly after depletion; also PL/Prot ratios were not significantly changed after incubation with MβCD (). That is in agreement with MβCD being specific sterol-carrying agents when their concentration does not exceed 10 mM Citation[13], Citation[28].
Treatment of U membranes with empty MβCD (giving rise to the cholesterol-depleted membranes D) depleted approximately 60% of the cholesterol content, which was in keeping with previous data described in the literature Citation[29]. Reloading cholesterol or ergosterol in D or U membranes (respectively C+, E+ and C+ + ), indicated that MβCD-mediated sterol depletion was fully reversible ().
Effect of sterol content on membrane microviscosity
The sterol content of membranes greatly influences the rotational and conformational dynamics of lipids Citation[1], Citation[3] and therefore the membrane microviscosity. We evaluated changes of lipid microviscosity in U and modified membranes by measuring the polarization rate of TMA-DPH (pTMA-DPH) at temperatures between 4 and 37°C. TMA-DPH is a fluorescent probe, which indicates changes of lipid organization at the depth where cholesterol is localized Citation[30]. As described for other cell systems Citation[27], pTMA-DPH slightly decreased in a regular manner as the temperature increased (data not shown). The values of pTMA-DPH were measured for U, D, C+, C+ + , and E+ membranes at 25°C (): pTMA-DPH was dependent on the sterol content as previously described Citation[31], showing that our treatments were efficient and reversible. The polarization values measured are consistent with membranes being in a fluid phase corresponding to a disordered liquid state Citation[30]. Values of pTMA-DPH were significantly lower in D membranes (p(D)=0.29) than in U membranes (p(U)=0.36). Loading with cholesterol (C+ and C+ + membranes) or ergosterol (E+ membranes) restored the microviscosity values of U membranes.
Effect of the sterol content on ligand binding to T7hMOR
We studied binding of [3H]DPN and [3H]DAMGO to U membranes. Affinities of these ligands (Kd) () were in the nanomolar range and similar to those reported in numerous studies Citation[32]. Furthermore, and as reported previously Citation[33], we observed that the number of high affinity binding sites for [3H]DPN was twice higher than that for [3H]DAMGO (in the concentration range used), consistent with the idea that DPN interacts with equal affinity with high and low affinity agonist binding states of hMOR (data not shown).
Table II. Affinity values (Kd or Ki, nM) of agonists (DAMGO and Morphine) and antagonists (DPN and CTAP) in untreated (U) or modified (D or C+) membranes in absence or presence of Gpp(NH)p (100 µM).
The binding profiles and characteristics of hMOR with D membranes are reported in A and . The affinity and the number of binding sites (Bmax) for [3H]DPN and [3H]DAMGO were weakly affected by cholesterol depletion.
Figure 1. DAMGO affinity parameters. (A) Saturation binding experiments for one campaign of measurements with DAMGO, in the absence (full symbols, black curves) or in the presence (empty symbols, grey curves) of Gpp(NH)p on U (•,○), D (▪,□) or C+ (♦,⋄) plasma membranes. (B) Relative differences of [3H]-DAMGO Bmax (%) between the absence and the presence of 100 µM Gpp(NH)p. All results are means for three independent experiments performed in duplicate. *Significantly difference from U sample at p<0.005.
![Figure 1. DAMGO affinity parameters. (A) Saturation binding experiments for one campaign of measurements with DAMGO, in the absence (full symbols, black curves) or in the presence (empty symbols, grey curves) of Gpp(NH)p on U (•,○), D (▪,□) or C+ (♦,⋄) plasma membranes. (B) Relative differences of [3H]-DAMGO Bmax (%) between the absence and the presence of 100 µM Gpp(NH)p. All results are means for three independent experiments performed in duplicate. *Significantly difference from U sample at p<0.005.](/cms/asset/2c9f8159-8a2e-43d5-8f33-c181682acfa5/imbc_a_320505_f0001_b.gif)
We next compared the DAMGO binding characteristics for U and D membranes in the presence and in the absence of Gpp(NH)p (a G-protein decoupling agent). As expected, treatment with Gpp(NH)p decreased DAMGO binding to U membranes to some extent (, ), thus emphasizing the role G-protein coupling plays in stabilizing the receptor in a high affinity agonist binding state. Interestingly, the effect of GppNHp was magnified following cholesterol depletion, with at least 30% in the DAMGO Bmax values. This suggests that cholesterol depletion together with G-protein uncoupling drive the receptor into a low-affinity agonist-binding conformation (B, ). Finally to strengthen this conclusion, C+ membranes recovered the characteristics of high-affinity DAMGO binding. Our results thus indicate that cholesterol maintains the high affinity binding state for DAMGO even in absence of G-protein coupling.
Loading D membranes with ergosterol (E+) did not fully restore DAMGO binding (B) although microviscosity of E+ membranes was similar to that of C+ membranes (). These results are consistent with previous findings for S. cerevisiae membranes showing distinct effects of cholesterol and ergosterol with respect to the µ opioid receptor functionality. The differences observed in the binding experiments could be explained by the capacity of both sterols to generate different constraints in the depth of the membrane leading to distinct hMOR conformations Citation[34].
We evaluated the affinity of various ligands for U, D, and C+ membranes to determine whether the effect of cholesterol on binding is influenced by the nature of the ligand (agonist/antagonist) or by its molecular structure (alkaloid/peptide). The alkaloid agonist morphine and the agonist peptide DAMGO had similar binding patterns (). For both, the Ki and Kd affinity values in the presence of Gpp(NH)p was 2.5-fold higher for D membranes than for U membranes. As in the DAMGO experiments, a recovery of morphine binding was observed with C+ membranes. Cholesterol depletion weakly affected the antagonist CTAP binding. These findings suggest that whatever the nature of the agonist (peptide or alkaloid; ), changes in the cholesterol content modulate binding in the same manner, despite the fact that these ligands interact with different parts of the T7hMOR Citation[32], Citation[35]. These findings thus further demonstrate that cholesterol is needed for high-affinity agonist, but not antagonist, binding.
Effect of cholesterol on G protein coupling of T7hMOR
We used [35S]-GTPγS binding measurements to determine whether the cholesterol content modulates the ability of agonists to trigger G-protein activation Citation[36]. We did not detect any basal GTPγS binding suggesting that T7hMOR was not constitutively active in our cell system (data not shown). DAMGO stimulated [35S]-GTPγS binding to U membranes with a half-maximal effective concentration (EC50) of 28n± 5 M ().
Figure 2. [35S]-GTPγS binding of DAMGO on untreated (U), cholesterol depleted (D), cholesterol complemented (C+) or cholesterol over-complemented membranes (C+ + ) prepared as described in Materials and methods. [35S]-GTPγS binding was determined with 0.1 nM [35S]-GTPγS in the presence of increasing concentrations of ligands (0.1 nM to 1 µM). All values are means for three independent experiments performed in triplicate.
![Figure 2. [35S]-GTPγS binding of DAMGO on untreated (U), cholesterol depleted (D), cholesterol complemented (C+) or cholesterol over-complemented membranes (C+ + ) prepared as described in Materials and methods. [35S]-GTPγS binding was determined with 0.1 nM [35S]-GTPγS in the presence of increasing concentrations of ligands (0.1 nM to 1 µM). All values are means for three independent experiments performed in triplicate.](/cms/asset/0f28ce2a-16cd-4aef-87f5-83cd600680ad/imbc_a_320505_f0002_b.gif)
Cholesterol depletion of U membranes diminished the potency of DAMGO to promote nucleotide binding (EC50=69±9 nM) and reduced by 50±5% the efficacy (Emax) of DAMGO-induced G protein coupling (). When the cholesterol content of D membranes was re-established, potency and efficacy of DAMGO to stimulate [35S]-GTPγS binding were restored. This indicates that the changes we describe in the ability of DAMGO to trigger G protein activation following cholesterol depletion are consequences of the alteration of the cholesterol content rather than non-specific effects of MβCD. A slight decrease of the EC50 values for DAMGO was observed for C+ or C+ + membranes, supporting the view that cholesterol improves the capacity of the receptor to activate G-proteins. Morphine was slightly less potent (EC50=43nM) than DAMGO as previously described Citation[37], but the morphine-induced [35S]-GTPγS binding to D, C+ and C+ + membranes paralleled those obtained for DAMGO (data not shown).
These experiments show that cholesterol is needed for a certain pool of T7hMOR to activate G- proteins in response to the agonist.
Effect of cholesterol content on total protein, PL composition and T7hMOR membrane localization
To investigate whether T7hMOR pharmacology is related to its localization in cholesterol-enriched microdomains, we isolated DRMs from U and D CHO/T7hMOR membranes by accurate detergent treatment (TX100 Citation[38]) and ultracentrifugation on a sucrose gradient (). With 0.25% of cold TX100, an opaque annulus formed in the 20% sucrose region (fraction 4) of the gradient, corresponding to DRMs-U of CHO total membrane preparations. DRMs-D were also extracted from D membranes (A, 3B, 3C). These experimental conditions revealed that, almost all the hMOR (>95%) was extracted from the pellets of U and D membranes. These DRM fractions (A fractions 3, 4, 5) co-localized with normalized alkaline phosphatase activity (AP), consistent with previously published results Citation[3]. However, the AP quantities in DRM fractions, relative to that in N-DRM fractions, were higher in U membranes (2.8) than in D membranes (1.5), indicating that cholesterol stabilizes the DRMs. The amounts of proteins (B) and lipids (data not shown) localized in DRMs represented less than 10% in U membranes and less than 5% in D membranes, as compared to the total amount of these molecules before detergent extraction. The Chol/PL ratios in these DRMs were higher than in the N-DRM fractions 6, 7, 8, (B), as described in previous reports Citation[2]. Comparison of the Chol/PL ratios between U and D membranes (C) revealed a decrease of 66% (DRM), 52% (N-DRM), 59% (pellet), and 57% (TM). The small loss of PL and total proteins between U and D membranes (data not shown) explains why the Chol/Prot and Chol/PL ratios vary similarly and why PL/Prot ratios are unchanged (C).
Figure 3. Characterization of sucrose gradient fractions from U or D T7hMOR/CHO membranes. (A) According to their normalized alkaline phosphatase (U: •), (D: ○) titration and sucrose concentration (U: □, D: X); (B) According to their cholesterol/phospholipid (Chol/PL) ratio contents (U: •), (D: ○), (▴) corresponds to the distribution of total proteins after a sucrose gradient. U and D total membranes gave identical results; (C) Lipid characteristics of various membrane fractions correspond to: a = DRMs (Fractions 3 + 4+5), b = N-DRMs (Fractions 6 + 7+8), c = pellet (Fraction 9) and d = total membranes, before (filled bars) and after cholesterol depletion (black bars) for cholesterol to phospholipid ratios (mol/mol), µ-moles of cholesterol per mg of proteins, and µ-moles of phospholipids per mg of proteins. Bars correspond to standard deviations of seven experiments before cholesterol depletion and six after. *The absence of PL in fractions 1 and 2 did not allow Chol/PL ratios to be calculated.
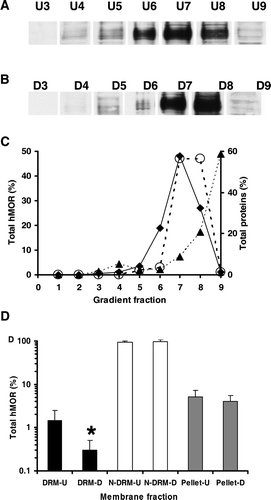
The presence of T7hMOR in the gradient fractions 3–9 from T7hMOR/CHO membranes before and after cholesterol depletion was studied by electrophoresis on SDS polyacrylamide gel and western blot analysis (A and 4B). We quantified the T7hMOR bands (C), and the T7hMOR abundance was normalized to the total amount of proteins recovered in the DRM and N-DRM fractions from U and D membranes (D). This showed that more than 93% and 95% of T7hMOR was found in N-DRM from U and D membranes, respectively. Small quantities of T7hMOR (less than 1.5% and 0.3%, respectively) were detected in DRM, and about 5% and 4% were detected in the pellets (D).
Figure 4. Western blot analysis of the distributions of T7hMOR in CHO membranes before (U) and after cholesterol depletion (D). (A) Western blot of hMOR in membrane fractions of solubilized membranes: lanes U3, U4, U5 (DRMs-U), lanes U6, U7, U8 (N-DRMs-U) and lane U9 (pellet of the gradient). After electrophoresis on SDS polyacrylamide gels, western blot analysis identified T7hMOR at approximately 70 kDa. We had previously verified the absence of T7hMOR from non-transfected CHO/K1 membranes. (B) Quantification of western blot of hMOR in gradient fractions from cholesterol-depleted membranes: lanes D3, D4, D5 (DRM), lanes D6, D7, D8 (N-DRM) and lane D9 (pellet) C) T7hMOR distribution, as percentages, in sucrose gradient fractions 1–9. (♦) and (○) indicate the distribution of hMOR in 15 µg of total proteins of U and D membranes respectively analyzed by Western blot (see Material and methods for the quantification); (▴) corresponds to the distribution of total proteins after a sucrose gradient. U or D total membranes gave identical results. (D) The corrected hMOR distribution in a sucrose gradient of combined U and D membrane fractions according to the criteria: DRMs (fractions 3–5), N-DRMs (fraction 6 to 8), and pellet (fraction 9). Western blot are performed with HRP conjugate anti-T7 antibody used as the probe (1/5000). All results are presented as means for three independent experiments performed in duplicate. *Significantly difference from DRU-U at p<0.01. See text for a quantification of hMOR distribution.
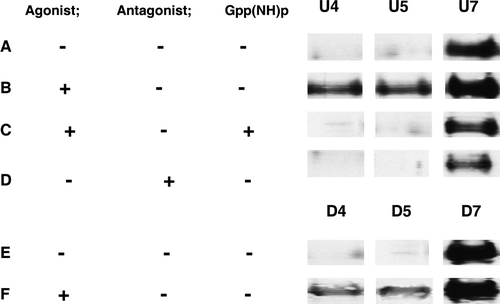
Western blot analysis showed that T7hMOR () redistributed towards the DRMs after the addition of DAMGO (12±3%), although 83±10% was still present in the N-DRMs (B). However, this DRM redistribution was inhibited when membranes were treated with DAMGO plus Gpp(NH)p (<1% in DRM) (C) and did not occur after treatment with the antagonist CTAP (D). DAMGO-stimulation of T7hMOR redistribution to DRMs-D was weaker than in DRMs-U (3% of T7hMOR, E and 5F), thus indicating that some hMOR in a high affinity agonist binding state are localized in DRMs and that depletion abolishes strongly this phenomenon.
Figure 5. Western blot of hMOR in membrane fractions 4 and 7 of sucrose gradient from Triton X100 solubilized membranes. Equal amounts of proteins (15 µg) were loaded in all lanes. (A): U membranes in a basal state. (B): after DAMGO addition. (C): U membranes after DAMGO + Gpp(NH)p addition. (D): U membranes after CTAP addition. (E): D membranes in a basal state. (F): D membranes after DAMGO addition. See text for quantification of hMOR distribution.
Effect of cholesterol depletion on the lipid and fatty acid compositions of DRM membrane fractions
The lipid composition (percentages of lipid classes and fatty acid composition) of total membranes (TM), DRMs, N-DRMs and pellets from U and D membranes was determined (). TLC was used to study these various fractions and the findings for U and D membranes were compared (A). Sphingolipids (SM1, SM2) were found in TM and DRMs of U and D membranes. Glycolipids (GL1, GL2) were partially lost in D membranes after cold TX100 treatment (A). Quantitative analysis of the TLC results (B) revealed that: (i) the lipid compositions of total U and D membranes are comparable (data not shown) and correspond to the filled bars in B; (ii) the percentages of PC and PE from DRMs in U and D membranes were not significantly different; (iii) the percentages of PS and GL were around 10% and 70% respectively lower in DRM-D than in DRM-U (B); (iv) in contrast, SM was 30% more abundant in D than in U membranes.
Figure 6. Lipid characteristics of TM, DRM, N-DRM and pellet (P) of U and D membranes. (A) TLC plates: TM-U and TM-D were membrane extracts from U and D membranes; DRM-U, N-DRM-U and P-U are from of U membranes, respectively; DRM-D, N-DRM-D and P-D are from of D membranes. (B) The percentage repartitions of lipid classes normalized for each deposited samples are shown: TM-U or TM-D (filled bars), DRM-U (open bars), DRM-D (grey bars). (C) Percentages of saturated or unsaturated fatty acid of TM-U (filled bars), DRM-U (open bars), DRM-D (grey bars), pellet (bicolor bars). Insert: Zoom of the zone C20:1-C24:0. CX:Y indicated: X: the number of carbon atoms in the fatty acid chain; Y: the number of double bonds. The ratios of unsaturated to saturated fatty acids for TM-U, DRM-U, DRM-D were: 1.7±0.1, 1.0±0.15, and 1.1±0.2, respectively. GL1, GL2 can correspond to glyco-lipids or spingoglycolipids that we were not able to differentiate.
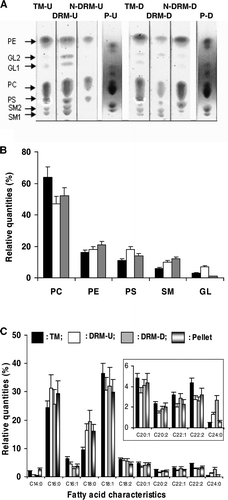
GC analysis of fatty acid indicated that total U and D membranes had similar compositions (data not shown) and revealed that three major fatty acids (9-cis-octadecenoic acid [C18:1], hexadecanoic acid [C16:0] and octadecanoic acid [C18:0]) made up more than 70% of total fatty acids. In DRMs, only tetradocanoic acid (C24:0), which is mainly found in sphingolipid species Citation[39], was substantially enhanced after depletion (C).
Discussion
The aim of this study was to evaluate the influence of cholesterol on T7hMOR function and the lateral distribution of this molecule in mammalian cell membranes. We carried out binding assays on U and D membranes with various ligands. We also explored the relationship between the pharmacology of T7hMOR and its localization in different membrane microdomains. We first checked that the binding profile of T7hMOR was similar to that previously described for the wild-type receptor Citation[32]. Indeed, the two receptors have similar affinities for the antagonists DPN and CTAP and the agonists DAMGO and morphine. There were twice more DPN binding sites than DAMGO sites, suggesting that only 50% of the receptors were in a high-affinity agonist-binding conformation in U membranes.
Effect of cholesterol contents on the T7hMOR pharmacology
Cholesterol depletion markedly decreased high affinity binding of the agonists DAMGO and morphine in the presence, but not in the absence, of Gpp(NH)p. This indicates that sensitivity to cholesterol depletion is mainly observed in the absence of G-protein coupling. Further evidence of this phenomenon was that this effect was abolished by the restoration of the cholesterol content (C+ membranes). In contrast, cholesterol depletion did not affect binding of the antagonists DPN and CTAP. These results are consistent with our previous findings with S. cerevisiae membranes Citation[19] and suggest that cholesterol alone, independently of G-proteins, can maintain hMOR in a high affinity agonist binding state. An increased affinity for agonists, even in the absence of G-proteins, has also been reported for constitutively active mutant receptors Citation[40], but [35S]-GTPγS binding showed that there was no constitutively active T7hMOR in our cell system. In the absence of Gpp(NH)p, cholesterol depletion changed the binding of agonists only slightly, but dramatically altered the receptor-mediated activation of G-proteins in response to agonists. This strongly suggests that different conformational states exist for agonist binding and G-protein activation, and that these states are differentially affected by cholesterol depletion. This is consistent with previous work showing that GPCRs exhibit different conformational states for their functioning Citation[41].
Using fluorescence polarization experiments, we found that pTMA-DPH was dependent on sterol content, indicating that MβCD-mediated depletion of cholesterol was effective for membrane fluidification and that the effects were reversible after complementation. However, the pTMA-DPH values cannot account for the subtle local changes in microviscosity which modify T7hMOR function locally Citation[17]. The different pharmacological profiles of T7hMOR in E+ and C+ membranes, which have similar pTMA-DPH values, are consistent with this.
Characterization of lipids in DRM and N-DRM from U and D membranes
Particular lipid compositions can generate membrane microdomains, in which certain proteins are confined. In the case of GPCRs, their activation and receptor-mediated signalling are favoured. We therefore developed an experimental approach to isolate lipid microdomains based on their resistance to dissolution in cold non ionic detergents.
As previously reported Citation[4], the composition of DRMs, N-DRMs and the pellet varied with the nature and the concentration of detergents used. Furthermore, the cholesterol content modulates the efficiency of detergent extraction Citation[42]. Our results are consistent with these findings and the alkaline phosphatase content of DRMs decreasing following cholesterol depletion, suggested that cholesterol depletion enhanced the efficiency of detergent extraction (A). We also found that the extent of cholesterol depletion was in the same range for DRMs and N-DRMs (69% and 50%, respectively), revealing some DRMs were still present in D membranes. These findings may be accounted for by cholesterol not being completely removed and by the enrichment of SM in DRMs-D and DRMs-U. Indeed, the residual cholesterol (∼40% of the total initial cholesterol) may be sufficient to stabilize rafts, owing to the strong intermolecular hydrogen bond cholesterol establishes with SM Citation[43], impeding membrane solubilization by detergents. The enrichment of both DRMs-D and DRMs-U in long saturated C18:0 and C24:0 acetyl chains, which are considered to be major constituents of SM fatty acids Citation[39], is consistent with this hypothesis. However, like the cholesterol content, the amounts of the glycolipids GL1 and GL2 markedly differ in DRMs-D and in DRMs-U (A and 6B); it is therefore likely that despite the lack of difference in lipid head group composition, the two DRMs differ substantially in nature ().
Localization and quantification of T7hMOR in various membrane fractions
With U membranes in the basal state, T7hMOR was mostly (93.3%) localized in N-DRMs, with very little (1.5%) in DRMs. The proportion of these receptors in DRMs-U increased (12.5%) after DAMGO activation (C). Furthermore, addition of Gpp(NH)p showed that the redistribution of T7hMOR to DRMs-U was dependent upon coupling to G-proteins (C), and antagonist treatments (D) did not relocalize T7hMOR to DRMs-U. These various results indicate that the fraction of T7hMOR (12.5%) redistributing toward DRMs-U represents receptor conformations that are in a high-affinity agonist binding state. Given that the number of agonist binding sites is half that of antagonist binding sites, the 12.5% of T7hMOR localizing in DRMs in response to DAMGO, corresponds to 25% of the receptors in a high-affinity agonist binding state. Our results appear to differ from those of Zhao et al. Citation[44] who, using a detergent-free preparation of rafts from total cell membranes, found MOR to be present principally in the rafts both in the basal state and after agonist activation. So these differences may be explained as it is suggested in many reports, by rafts have various compositions which depend on cell type, on the nature of starting membrane materials Citation[25] and by the protocols used for their isolation. If detergents are used, the lipid and protein composition of rafts depends on the detergent nature Citation[42] and on its concentration Citation[45]. Our results also suggest that the remaining receptors with a high affinity for agonists (75%) are located in N-DRMs-U (where the Chol/PL ratio is ∼0.37 [C]). However, consistent with our findings for S. cerevisiae membranes Citation[19], the receptors in this membrane fraction remain cholesterol-dependent, because no high affinity for agonists was detected when cholesterol was absent Citation[19]. Additionally, it is more than likely that some of these receptors also require cholesterol for activating G-proteins in response to agonists, as revealed by the extent of [35S]-GTPγS binding that decreases after cholesterol depletion (50% of receptors with high affinity agonist binding). This result is not consistent with the 75% of hMOR detected in the N-DRM fraction by western blot analysis. This implies that certain membrane microdomains, containing T7hMOR in an activated form leading to a high agonist binding affinity, have been extracted from N-DRMs-U.
After cholesterol depletion, hMOR was detected mainly in N-DRMs-D, in which the Chol/PL ratio was 0.19; a very small quantity (<3%) of T7hMOR was found in DRMs-D even after DAMGO activation (F). This suggests that the agonist-induced redistribution of hMOR to DRMs depends on the cholesterol level, which is consistent with our main conclusion that cholesterol stabilizes T7hMOR in a high-affinity agonist binding state.
Comparisons of membrane localization and pharmacological properties of T7hMOR
Due to their high concentrations of cholesterol and sphingolipids with long saturated hydrocarbon chains, DRMs-U have a thicker lipid bilayer than N-DRMs-U Citation[6]. Conformational changes of δ-opioid receptors following agonist activation have been reported to trigger a specific increase in the mean membrane thickness (∼10% of the thickness of DOPC membrane for DOR, i.e., ∼0.5 nm) Citation[46]. We recently confirmed specific modulations of δ-opioid receptor – containing membrane thickness after agonist stimulation, using the fluorescence properties of a pyrene-labeled cholesterol Citation[47]. Similarly, the changes in hMOR conformation associated with agonist activation may result in lipid/protein hydrophobic mismatches able to generate new rafts or to drive activated hMOR towards existing raft zones. We therefore suggest that the T7hMOR found in DRMs-U after agonist activation are associated with greater membrane thickness than that of associated with the remaining receptors in N-DRMs-U. These differences in T7hMOR thickness may be related to different patterns of pharmacological behaviour. The thicker conformation may correspond to an active hMOR conformation able to couple G-proteins that is abolished when cholesterol levels fall.
Finally, our findings show that cholesterol is required for the stabilization of a receptor conformation with a high affinity for agonists and for triggering G-protein activation in response to agonists. They also suggest that different surrounding lipid environments, with different cholesterol levels and membrane thicknesses, may lead hMOR to adopt at least two active molecular conformations required for agonist binding and G protein activation. As suggested in a recent work on hβ2-AR Citation[23], cholesterol may regulate interactions between hβ2-AR and G proteins either by cholesterol-mediated association (dimerization) or modulating subcellular signalling. These different pathways could lead to sequential coupling to different G proteins (in association with different conformation of receptor) Citation[23].
The findings of our study based solely on stationary approaches are confirmed by FRAP experiments revealing that hMOR diffusion is restricted to different submicrometre domains after agonist binding Citation[48]. We are therefore now carrying out SPT experiments with tagged hMOR, to improve our understanding of these mechanisms and receptor membrane distribution during signalling processes.
Acknowledgements
This work was supported by grants from ARC (Association pour la Recherche contre le Cancer), G. Gaibelet was supported by a CNRS fellowship. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J 2004; 378: 281–292
- Ge M, Gidwani A, Brown HA, Holowka D, Baird B, Freed JH. Ordered and disordered phases coexist in plasma membrane vesicles of RBL-2H3 mast cells. An ESR study. Biophys J 2003; 85: 1278–1288
- London E. Insights into lipid raft structure and formation from experiments in model membranes. Curr Opin Struct Biol 2002; 12: 480–486
- Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. Resistance of cell membranes to different detergents. Proc Natl Acad Sci USA 2003; 100: 5795–5800
- Ipsen JH, Karlstrom G, Mouritsen OG, Wennerstrom H, Zuckermann MJ. Phase equilibria in the phosphatidylcholine-cholesterol system. Biochim Biophys Acta 1987; 905: 162–172
- Gandhavadi M, Allende D, Vidal A, Simon SA, McIntosh TJ. Structure, composition, and peptide binding properties of detergent soluble bilayers and detergent resistant rafts. Biophys J 2002; 82: 1469–1482
- Gimpl G, Fahrenholz F. Cholesterol as stabilizer of the oxytocin receptor. Biochim Biophys Acta 2002; 1564: 384–392
- Scanlon SM, Williams DC, Schloss P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry 2001; 40: 10507–10513
- Nunez MT, Glass J. Reconstitution of the transferrin receptor in lipid vesicles. Effect of cholesterol on the binding of transferrin. Biochemistry 1982; 21: 4139–4143
- Pike LJ, Casey L. Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking. Biochemistry 2002; 41: 10315–10322
- Westover EJ, Covey DF, Brockman HL, Brown RE, Pike LJ. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J Biol Chem 2003; 278: 51125–51133
- Polozova A, Litman BJ. Cholesterol dependent recruitment of di22:6-PC by a G protein-coupled receptor into lateral domains. Biophys J 2000; 79: 2632–2643
- Niu SL, Mitchell DC, Litman BJ. Manipulation of cholesterol levels in rod disk membranes by methyl-beta-cyclodextrin: effects on receptor activation. J Biol Chem 2002; 277: 20139–20145
- Niu SL, Mitchell DC, Lim SY, Wen ZM, Kim HY, Salem N, Jr, Litman BJ. Reduced G protein-coupled signaling efficiency in retinal rod outer segments in response to N-3 fatty acid deficiency. J Biol Chem 2004; 279: 31098–31104
- Pang L, Graziano M, Wang S. Membrane cholesterol modulates galanin-GalR2 interaction. Biochemistry 1999; 38: 12003–12011
- Harikumar KG, Puri V, Singh RD, Hanada K, Pagano RE, Miller LJ. Differential Effects of modification of membrane cholesterol and sphingolipids on the conformation, function, and trafficking of the G protein-coupled cholecystokinin receptor. J Biol Chem 2005; 280: 2176–2185
- Emmerson PJ, Clark MJ, Medzihradsky F, Remmers AE. Membrane microviscosity modulates mu-opioid receptor conformational transitions and agonist efficacy. J Neurochem 1999; 73: 289–300
- Farahbakhsh ZT, Deamer DW, Lee NM, Loh HH. Enzymatic reconstitution of brain membrane and membrane opiate receptors. J Neurochem 1986; 46: 953–962
- Lagane B, Gaibelet G, Meilhoc E, Masson JM, Cezanne L, Lopez A. Role of sterols in modulating the human mu-opioid receptor function in Saccharomyces cerevisiae. J Biol Chem 2000; 275: 33197–33200
- Pucadyil TJ, Chattopadhyay A. Cholesterol modulates ligand binding and G-protein coupling to serotonin (1A) receptors from bovine hippocampus. Biochim Biophys Acta 2004; 1663: 188–200
- Gimpl G, Klein U, Reilander H, Fahrenholz F. Expression of the human oxytocin receptor in baculovirus-infected insect cells: high-affinity binding is induced by a cholesterol-cyclodextrin complex. Biochemistry 1995; 34: 13794–13801
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 1995; 34: 13784–13793
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007; 318: 1258–1265
- Gimpl G, Fahrenholz F. Human oxytocin receptors in cholesterol-rich vs. cholesterol-poor microdomains of the plasma membrane. Eur J Biochem 2000; 267: 2483–2497
- Cézanne L, Navarro L, Tocanne JF. Isolation of the plasma membrane and organelles from Chinese hamster ovary cells. Biochim Biophys Acta 1992; 1112: 205–214
- Zak B, Dickenman RC, White EG, Burnett H, Cherney PJ. Rapid estimation of free and total cholesterol. Am J Clin Pathol 1954; 24: 1307–1315
- Leborgne N, Cézanne L, Teulières C, Canut H, Tocanne JF, Boudet AM. Lateral and rotational mobilities of lipids in specific cellular membranes of Eucalyptus gunnii. Plant Physiol 1992; 100: 246–254
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res 1997; 38: 2264–2272
- Huang P, Xu W, Yoon SI, Chen C, Chong PL, Liu-Chen LY. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol 2007; 73: 534–549
- Prendergast FG, Haugland RP, Callahan PJ. 1-[4-(trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene: synthesis, fluorescence properties and use as fluorescence probe of lipid bilayers. Biochemistry 1981; 20: 7333–7338
- Van Blitterswijk WJ, Van Hoeven RP, Van der Meer BW. Lipid structural order parameters (reciprocal of fluidity) in biomembranes derived from steady-state fluorescence polarization measurements. Biochim Biophys Acta 1981; 644: 323–332
- Gaibelet G, Capeyrou R, Dietrich G, Emorine LJ. Identification in the mu-opioid receptor of cysteine residues responsible for inactivation of ligand binding by thiol alkylating and reducing agents. FEBS Lett 1997; 408: 135–140
- Gaibelet G, Meilhoc E, Riond J, Saves I, Exner T, Liaubet L, Nurnberg B, Masson JM, Emorine LJ. Nonselective coupling of the human mu-opioid receptor to multiple inhibitory G-protein isoforms. Eur J Biochem 1999; 261: 517–523
- Cantor RS. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem Phys Lipids 1999; 101: 45–56
- Fukuda K, Kato S, Mori K. Location of regions of the opioid receptor involved in selective agonist binding. J Biol Chem 1995; 270: 6702–6709
- Wieland C, Jakobs KH, Wieland T. Altered guanine nucleoside triphosphate binding to transducin by cholera toxin-catalysed ADP-ribosylation. Cell Signal 1994; 6: 487–492
- Gray RE, Munks MW, Haynes RR, Olsen GD. Mu opioid receptor efficacy and potency of morphine-6-glucuronide in neonatal guinea pig brainstem membranes: comparison with transfected CHO cells. Brain Res Bull 2001; 54: 499–505
- Garner AE ,Smith A ,Hooper NM. 2007. Visualization of detergent solubilisation of membranes: Implications for the isolation of rafts Biophys J. doi:10:1529/biophysj.1107.114108.
- Bagnat M, Keranen S, Shevchenko A, Simons K. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc Natl Acad Sci USA 2000; 97: 3254–3259
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem 1993; 268: 4625–4636
- Berchiche YA, Chow KY, Lagane B, Leduc M, Percherancier Y, Fujii N, Tamamura H, Bachelerie F, Heveker N. Direct assessment of CXCR4 mutant conformations reveals complex link between receptor structure and G(alpha)(i) activation. J Biol Chem 2007; 282: 5111–5115
- Babiychuk B, Draeger A. Biochemical characterization of detergent-resistant membranes: a systematic approach. Biochem J 2006; 397: 407–416
- Li XM, Momsen MM, Smaby JM, Brockman HL, Brown RE. Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry 2001; 40: 5954–5963
- Zhao H, Loh HH, Law PY. Adenylyl cyclase superactivation induced by long-term treatment with opioid agonist is dependent on receptor localized within lipid rafts and is independent of receptor internalization. Mol Pharmacol 2006; 69: 1421–1432
- André A, Gaibelet G, Le Guyader L, Welby M, Lopez A, Lebrun C. 2008. Membrane partitioning of various delta-opioid receptor forms before and after agonist activations: the effect of cholesterol. Biochim Biophys Acta: DOI: 10.1016/j.bbamem.2008.1003.1017.
- Salamon Z, Cowell S, Varga E, Yamamura HI, Hruby VJ, Tollin G. Plasmon resonance studies of agonist/antagonist binding to the human delta-opioid receptor: new structural insights into receptor-ligand interactions. Biophys J 2000; 79: 2463–2474
- Le Guyader L, Le Roux C, Mazères S, Gaspard-Iloughmane H, Gornitzka H, Millot C, Mingotaud C, Lopez A. Changes of the membrane lipid organization characterized by means of a new cholesterol-pyrene probe. Biophys J 2007; 93: 4462–4473
- Saulière A, Gaibelet G, Millot C, Mazères S, Lopez A, Salomé L. Diffusion of the mu opioid receptor at the surface of human neuroblastoma SH-SY5Y cells is restricted to permeable domains. FEBS Lett 2006; 580: 5227–5231