Abstract
Two-component signal transduction systems are the main mechanism by which bacteria sense and respond to their environment, and their membrane-located histidine protein kinases generally constitute the sensory components of these systems. Relatively little is known about their fundamental mechanisms and precise nature of the molecular signals sensed, because of the technical challenges of producing sufficient quantities of these hydrophobic membrane proteins. This study evaluated the heterologous production, purification and activities of the 16 intact membrane sensor kinases of Enterococcus faecalis. Following the cloning of the genes into expression plasmid pTTQ18His, all but one kinase was expressed successfully in Escherichia coli inner membranes. Purification of the hexa-histidine ‘tagged’ recombinant proteins was achieved for 13, and all but one were verified as intact. Thirteen intact kinases possessed autophosphorylation activity with no added signal when assayed in membrane vesicles or as purified proteins. Signal testing of two functionally-characterized kinases, FsrC and VicK, was successful examplifying the potential use of in vitro activity assays of intact proteins for systematic signal identification. Intact FsrC exhibited an approximately 10-fold increase in activity in response to a two-fold molar excess of synthetic GBAP pheromone, whilst glutathione, and possibly redox potential, were identified for the first time as direct modulators of VicK activity in vitro. The impact of DTT on VicK phosphorylation resulted in increased levels of phosphorylated VicR, the downstream response regulator, thereby confirming the potential of this in vitro approach for investigations of modulator effects on the entire signal transduction process of two-component systems.
| Abbreviations | ||
| DDM | = | dodecyl-β-D-maltoside |
| DTT | = | dithiothreitol |
| EDTA | = | ethylenediamine tetraacetic acid |
| ESI-MS | = | electrospray ionization mass spectrometry |
| GBAP | = | gelatinase biosynthesis-activating pheromone |
| GSH | = | reduced glutathione |
| GSSG | = | oxidized glutathione |
| HK | = | histidine kinase |
| IPTG | = | isopropyl -D-1-thiogalactopyranoside |
| LB | = | Luria-Bertani |
| LDAO | = | lauryldimethylamine-oxide |
| Ni2+-NTA | = | nickel-nitrilotriacetic acid |
| PAS | = | Per-Arnt-Sim |
| RR | = | response regulator |
| SDS-PAGE | = | sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| TCS | = | two-component systems |
| TM | = | transmembrane |
Introduction
Enteroccocus faecalis is a Gram-positive, commensal bacterium commonly found in the gastrointestinal tract of many mammals, including humans Citation[1]. Since the late 1970s enterococci have been increasingly reported as significant causative agents of serious and life-threatening nosocomial infections in both the USA and Europe, including endocarditis, nosocomial bacteraemia, surgical wound infections, and urinary tract infections Citation[2]. This rise in the occurrence of infection accredited to E. faecalis has also been accompanied by intrinsic and increased resistance to most currently approved antibacterial agents, including the ‘last-line’ glycopeptide vancomycin Citation[3], Citation[4].
E. faecalis uses two-component signal transduction systems (TCSs) to monitor and respond to changes in its environment. These systems are found in almost all bacteria and are used to regulate a variety of cellular processes ranging from sporulation, biofilm formation, chemotaxis, and virulence to antibiotic production and resistance Citation[5–8]. TCSs consist of two proteins, a signal-sensing histidine protein kinase (HK) usually in the cell membrane that senses an environmental change, and a cytoplasmic cognate response regulator (RR) that effects an appropriate response, usually regulation of gene expression. The HK responds to the environmental stimulus by autophosphorylating on a conserved histidine residue in the cytoplasmic autokinase domain. The phosphate is then transferred to an aspartate residue on the partner RR, which is then able to effect a response, such as modulation of transcription through increased affinity for its target promoter DNA, thereby eliciting a cellular response to a changing environment (e.g., Citation[9]).
Most bacterial species possess multiple TCS pairs and therefore these species are able to sense and respond to a variety of different environmental cues. In the completed genome sequence of E. faecalis strain V583, 17 TCS pairs have been identified, along with one orphan RR Citation[10]. Using the HK classification scheme described previously Citation[11], Hancock and Perego sub-grouped the 17 HK proteins. All but one of the HKs were predicted to be localized in the membrane Citation[10]. Similarity searches Citation[10], systematic inactivation of RR components Citation[12–15] and investigations using infection models Citation[14], Citation[15] revealed important insights into the potential functions of these two-component signalling systems, some of which are associated with virulence and/or responses to antibacterial agents and other stresses. These are summarized below, and described in more detail in the Supplementary Information which can be found online at http://www.informaworld.com/smpp/title∼content=t713693962.
Some systems of this bacterium have been implicated in virulence using infection models, including EF1704-1703 (HK-RR04), EF3290-3289 (HK-RR05) (also required for intrinsic -lactam resistance), EF1261-1260 (HK-RR06) and EF1051-1050 (HK-RR10) Citation[14], Citation[15]. The systems that have been functionally characterized are: EF1820-1822 (HK-RR15), which corresponds to the Fsr quorum-sensing system that responds to the autoinduced peptide GBAP Citation[16–19], and has roles in both virulence Citation[20–22] and biofilm formation Citation[12], Citation[23], Citation[24]; EF2298-2299 (HK-RR11), VanSBRB, that regulates VanB-type vancomycin resistance Citation[10], Citation[25], though in this case the precise molecular signal is unknown; and EF1194-1193 (HK-RR07) VicKR. Similarity and knock-out studies of the response regulator component of this latter system revealed that it is likely to be a member of the YycFG/Vic subfamily of systems that is essential for cell viability Citation[10], Citation[12–14], Citation[26], Citation[27]. The precise signal to which the VicK sensor component responds has not yet been identified, though it may be a redox signal generated within electron transport Citation[27], and the protein to known to be modulated by other membrane proteins in the cell with which it interacts Citation[28–30]. Likewise, the target genes for the VicKR system in E. faecalis remain to be elucidated. Recently we described the crystal structure of the DNA-binding domain of the response regulator component VicR Citation[31].
Amongst the remaining systems of E. faecalis is EF1863-1864 (HK-RR08) which is homologous to VncSR associated with vancomycin tolerance in Streptococcus pneumoniae Citation[32] and is one of the six E. faecalis systems suggested to be involved in responses to oxidative and other cellular stresses Citation[12], and EF3197-3196 (HK-RR02), which is highly homologous to LytSR of Staphylococcus aureus involved in cell wall integrity Citation[33].
Little is known about the roles of the remaining systems, and even in the cases of many of the better-described systems above, the precise roles and in particular the molecular environmental signals sensed by the sensor kinase components and subsequent signal transduction mechanism across the membrane remain incompletely understood. One reason for this is the technical challenges associated with studies of the intact forms of these membrane sensory proteins, which are hydrophobic in nature. Therefore, previous studies of many kinases employed the cytoplasmic HK domains only, in soluble forms often lacking their sensing domains, including a full study of the genome complement of all the TCSs of E. coli Citation[34]. We were the first to demonstrate the successful heterologous overexpression in E. coli and purification of any intact membrane sensor kinase Citation[35]; the RegB (PrrB) redox-responsive sensor kinase of photosynthetic Rhodobacter sphaeroides was active in membranes and when purified retained autophosphorylation, phosphotransfer and phosphatase activities Citation[35]. We demonstrated that intact purified RegB responded to the reducing agent dithiothreitol (DTT), increasing autophosphorylation, using an in vitro approach Citation[36]. To determine how widely applicable such an approach might be, and to learn more about the roles of such kinases in processes such as virulence, antibiotic resistance and redox state, we evaluated the successes of expressing and purifying the full genome complement of membrane sensor kinases of E. faecalis.
Here we evaluate the systematic cloning, expression, purification and activity assays for each of the 16 membrane kinases. Using two of these systems, FsrC and VicK, we also report the successful identification or confirmation of molecular signals for each purified protein, indicating that this in vitro approach for signal identification is feasible and reliable in future systematic screening programs for the intact, heterologously-expressed membrane sensor kinases.
Materials and methods
Overexpression plasmid constructs for 16 HKs
Each HK gene was amplified using Vent polymerase or Pfu Turbo and E. faecalis strain V583 chromosomal DNA as template and the primer pairs listed in . Amplified DNA was confirmed using 1% (w/v) agarose gels and the products were excised and purified before digestion with PstI and EcoRI or NdeI. Plasmid pTTQ18His was digested with the same enzymes. Digested genes and plasmid were visualized in agarose gels, followed by band excision and purification before subsequent ligation with T4 DNA ligase. Competent E. coli DH5α (Invitrogen), Top10 (Invitrogen) or XL10 Gold (Stratagene) cells were transformed and selection of plasmid-containing clones was done using Luria-Bertani (LB) medium Citation[37] containing 50 µg/ml carbenicillin. The EF0373 (HK13) has an internal EcoRI site, and was cloned as a NdeI-, PstI-ended fragment. As the EF1863 (HK08) gene contains an internal EcoRI site and the EF0570 (HK12) gene contains an internal PstI site, a two step cloning process was employed. The correct orientation and sequence of all constructs was confirmed by DNA sequencing. All clones and strains used are described in .
Table I. Primers used in the study.
Table II. Bacterial strains and plasmids.
The vicR gene was amplified by polymerase chain reaction using upstream primer VICR-F 5'-GATCACATATGAAGAAAATTTTAGTAGTTGA-3' and downstream primer VICR-R 5'-AGCTGGATCCTAAAATTAACAGATTGAAAAAAG-3' and genomic template as described above. The 0.75 kb product was digested with NdeI and BamHI (sites underlined in the primer sequences) and cloned directly into pET14b resulting in pETvicR. The correct sequence of the cloned gene was verified by DNA sequencing, and the expressed protein was verified by ESI-MS, N-terminal sequencing and Western blotting.
Expression trials
E. coli BL21 (DE3) cells were transformed with plasmids derived from single colony-purified clones, and cultured on LB agar plates containing 100 µg/ml carbenicillin overnight at 37°C. Single colonies were used to inoculate 5–10 ml cultures of LB broth, M9 minimal medium or 2TY medium Citation[37] containing 100µg/ml carbenicillin, which were incubated aerobically at 37°C overnight. 1 ml volumes were used to inoculate 50 ml of the same media, again containing 100 µg/ml carbenicillin. These were cultured aerobically at 37°C until they reached an optical density of 0.4–0.6 measured at 600 nm. Expression was induced by the addition of varying amounts, 0–1 mM, of isopropyl -D-1-thiogalactopyranoside (IPTG). Optimal conditions were also investigated using different temperatures and periods of culturing post-induction. Cells were harvested at 4°C by centrifugation (5000 g for 20 min) and the pellets frozen at -80°C.
Mixed E. coli membranes were prepared by water lysis Citation[38] and samples (30 µg) were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) using 15% and 4% (w/v) acrylamide in the resolving and stacking gels respectively. Proteins from similar gels were also blotted onto Fluorotrans Transfer membranes (Pall Corp.) for detection of the hexa-Histidine tag of expressed proteins in Western blot analysis using the INDIATM HisProbe-HRP (Perbio Science UK Ltd.) and Supersignal West Pico Chemiluminescent substrate (Pierce) detection system.
Preparation of inner membrane vesicles
A total of 4–6 l cultures of E. coli cells harbouring each plasmid were grown aerobically in 500–750 ml batches in 2-l flasks and induced with IPTG. Cells were harvested by centrifugation and stored as described above. Cells were defrosted, resuspended in 20 mM Tris-HCl (pH 7.5) containing 0.5 mM EDTA, and lysed by explosive decompression using a cell disruptor to produce predominantly inside-out membrane vesicles that were purified by separation on sucrose gradients Citation[38]. Freshly-prepared inner membrane vesicles in 20 mM Tris-HCl (pH 7.5), were used in activity assays, or stored at -80°C for subsequent purification of His-tagged protein. Total protein concentration in the inner membrane vesicles was determined by either Schaffner and Weissmann's assay Citation[39], or by BCA assay (Pierce) performed in 96-well plate format using a Fluostar Optima plate reader at a wavelength of 550 nm.
Detergent solubilization trials
100 µg samples of inner membrane proteins were incubated with 100 µl solubilization buffer comprising 20 mM Tris HCl (pH 7.9), 10 mM imidazole, 20% (v/v) glycerol, 300 mM NaCl and 1% (w/v) detergent. The detergents trialed included: n-hexyl-β-D-maltoside, n-octyl-β-D-maltoside, n-nonyl-β-D-maltoside, n-decyl-β-D-maltoside, n-undecyl-β-D-maltoside, n-dodecyl-β-D-maltoside, n-hexyl-β-D-glucopyranoside, n-heptyl-β-D-glucopyranoside, n-octyl-β-D-glucopyranoside, n-nonyl-β-D-glucopyranoside, n-decyl-β-D-glucopyranoside, n-dodecyl-β-D-glucopyranoside, cyclohexyl-n-hexyl-β-D-maltoside, Triton X-100, ethyleneglycol monooctylether (C8E1), octaethyleneglycol monododecylether (C12E8), nonaethyleneglycol monododecylether (C12E9), N,N-dimethyl dodecylamine oxide (LDAO) and N,N-dimethyl decylamine oxide (DDAO), N,N-dimethyl octylamine oxide (ODAO), N-octyltetraoxyethylene, N-octyl pentaoxyethylene and methyl-6-O-(N-heptylcarbamoyl)-a-D-glucopyranoside (HECAMEG). Following incubation for 1 h at 4°C, with gentle rotation, 2×50 µl samples were tested for successful protein solubilization by retaining one sample on ice whilst the remaining 50 µl were centrifuged at 100,000 g for 30 min to pellet any protein which remained insoluble after treatment. In general, 15 µl of total and soluble fractions were analysed by SDS-PAGE using 12% gels, whilst in the case of lower-expressing proteins Western blotting was also used to visualize expressed proteins. Successful solubilization was judged by the presence of HK protein bands in the lanes containing centrifuged solubilized fractions.
Protein solubilization and purification
Membrane vesicle preparations were incubated for 1–2 h at 4°C in solubilization buffer (20 mM Tris HCl (pH 7.9), 20 mM imidazole (pH 7.9), 2 mM β-mercaptoethanol, 20% v/v glycerol, 250–300 mM NaCl and 1% detergent), to a final concentration of between 2.5–5 mg/ml total inner membrane protein. Protein solutions were then centrifuged at 100,000 g for 30 min to remove all remaining insoluble material, and the soluble proteins incubated between 0.5–15 h with 1 ml nickel-NTA resin (Qiagen) at 4°C with gentle rotation. The resin was centrifuged at 1,000 g for 1 min, resuspended in wash buffer (typically 10 mM Tris HCl (pH 7.9), 20 mM imidazole (pH 7.9), 2 mM β-mercaptoethanol, 10–20% v/v glycerol and 0–0.05% DDM, and in most cases 250 mM NaCl), and transferred to a 5 ml column. Increasing concentrations of imidazole, 0.1 M, 0.2 M, 0.3 M and 0.5 M in wash buffer were trialed to elute some proteins and samples were analysed for the presence of protein by SDS-PAGE. Elution fractions containing protein were concentrated using Vivaspin Centrifugal Concentrators (Sartorius) and exchanged into required buffers for further analyses.
All purified proteins were subjected routinely to verification as intact and undegraded using Western blotting (to verify the presence of the hexa-his tag engineered at the C-terminus of each protein) together with N-terminal sequencing, and/or using ESI-MS.
To obtain soluble recombinant VicR protein, plasmid pPETvicR was transformed into the expression host E. coli BL21 (DE3) and expression and purification carried out as described previously Citation[35]. The protein accounted for 15% of total soluble proteins (data not shown). The integrity of purified VicR was verified by: (i) N-terminal sequence analysis of purified VicR, which gave the predicted sequence G-S-S-H-H-H; and (ii) electrospray mass spectrometry analysis, which identified a mass of 28989 Da that closely matches the predicted mass of 28990 Da.
Autophosphorylation assays for inner membrane vesicles
These were performed as described previously Citation[35], Citation[36]. Briefly, inner membrane proteins (200 µg) were incubated at room temperature (24°C) in assay buffer containing a final concentration of 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 50 mM KCl, 0.005% DDM and 1 mM dithiothreitol (DTT). Total reaction volumes of 200 µl were prepared and the reactions initiated by the addition of 50 µM ATP containing 50 µCi [γ − 32P] or [γ − 33P] ATP. Samples (20 µl) were removed at timed intervals between 0 and 60 min, and reactions stopped by the addition of 6 µl 4×SDS-PAGE sample loading buffer (60 mM Tris (pH 6.8), 10% (v/v) glycerol, 2% (w/v) SDS, 0.005% bromophenol blue and 3% β-mercaptoethanol). Samples were prepared for SDS-PAGE analysis by incubation for 30 min at 37°C to ensure full solubilization of proteins prior to loading gels containing 4% and 12% stacking and resolving gels respectively Citation[37]; alternatively samples were stored at -80°C for subsequent analysis. Following gel electrophoresis as described previously Citation[35], gels were briefly washed in deionized water and dried under vacuum at 70–80°C. Labelled protein bands were visualized by autoradiography using BioMax film (Kodak) exposed for 1–7 days. Labelled protein quantitation was undertaken using a Fuji BAS 1000 phospho-imaging analyzer system (Fujifilm Co., Japan) and AIDA 2D analytical software (Raytest).
Assays that included glutathione and accompanying controls employed an additional 500 mM Tris HCl pH 7.6 to maintain assay pH.
Autophosphorylation assays using purified proteins
Assays were carried out as described previously in buffers without added DDM Citation[35]. Briefly, 800 pmoles purified protein (1.5–5 mg/ml stocks) in storage buffer (10 mM Tris-HCl (pH 7.9), 25% glycerol and 2mM β-mercaptoethanol) were incubated at room temperature (24°C) in reaction buffer containing 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2 and 50 mM KCl. 150 µl total reaction volumes were used for each protein and the reactions were initiated by the addition of 37.5 µCi [γ − 33P] ATP at a final concentration of 50 µM. 15 µl samples were removed at intervals between 0.3 min and 1 h and terminated by the addition of 5 µl 4×SDS-PAGE sample loading buffer (stop buffer) as described above. For the VicK and FsrC proteins, the effects of individual candidate signal molecules were tested using 60–80 pmoles protein per 15 µl reaction mix. Signal and protein were incubated at 24°C for 20 min, prior to the addition of radiolabelled ATP to initiate the autophosphorylation reaction, which proceeded for 40 or 60 min before reactions were terminated with stop buffer. All samples were analysed by SDS-PAGE as described above for assays of membrane-bound proteins. In the case of VicK, purified protein was stored in buffer without added -mercaptoethanol. Assays using purified proteins with glutathione employed an additional 500 mM Tris HCl pH 7.6 to maintain assay pH. Stock solutions of synthetic GBAP, L-Gln—L-Asn—L-Ser—L-Pro—L-Asn—L-Ile—L-Phe—L-Gly—L-Gln—L-Trp—L-Met possessing a lactone ring formed by C-terminal L-Met and L-Ser and which is identical in structure to natural GBAP, were prepared in 20% (w/v) acetonitrile and stored at -20°C as described previously Citation[40].
Western blotting
Proteins separated by SDS-polyacrylamide gel electrophoresis were transferred to FluorotransTM membrane (Pall BioSupport UK) by semi-dry electroblotting for 2 h at 30 V. Membranes were washed once in TBSTT (20 mM Tris-HCl pH 7.5, containing 500 mM NaCl, 0.05% Tween 20 and 0.2% Triton X100) for 10 min, followed by overnight incubation in TBSTT containing 3% bovine serum albumin. Membranes were then washed twice in TBSTT for 10 min each and incubated for 1 h with 10 ml 1:5000-diluted INDIATM HisProbe-HRP (Perbio Science UK Ltd), a nickel-activated derivative of horse radish peroxidase for detection of histidine-tagged proteins. Following four washes with 10 ml TBSTT for 10 min each, membranes were incubated for 5 min in 10 ml Supersignal West Pico Chemiluminescent substrate and exposed to film for 5–60 sec.
Electrospray ionization mass spectrometry (ESI-MS)
50 µl purified HK protein samples (∼3 mg/ml) were prepared for electrospray ionization mass spectrometry by mixing with 150 µl of analytical grade methanol. 50 µl of chloroform were added and mixed to form a single phase, then 150 µl of water was added to initiate phase separation. Following centrifugation at 10,000 g for 2 min the aqueous phase was removed and 100 µl methanol was added. Following a second identical centrifugation step and removal of the solution the pellet of precipitated protein was dried under a flow of nitrogen. Size-exclusion chromatography using SephadexTM LH-20 (GE Healthcare) was used to complete the preparation of HK proteins as described previously Citation[41].
N-terminal sequencing
Purified HK proteins were submitted for N-terminal sequencing on FluorotransTM membrane (Pall BioSupport, UK) following electroblot transfer of proteins from SDS-polyacrylamide gels, and visualization using Coomassie Blue G-250 staining. Proteins were excised from the membrane for sequencing using Edman degradation.
Protein determination
Protein determinations were performed as already described above for inner membrane vesicles. Otherwise, spectrophotometry at a wavelength of 280 nm was undertaken and results compared to the absorbance for each HK predicted by the ProtParam tool on the ExPASy website (http://ca.expasy.org/tools/protparam.html).
Results
To clone each full-length HK gene, the polymerase chain reactions we carried out as described in Methods using the specific primer pairs shown in with E. faecalis V583 chromosomal DNA as template, and fragments cloned into the membrane protein expression vector pTTQ18His. Descriptions of the resulting plasmids are shown in . Each purified plasmid was transformed into E. coli BL21 (DE3) for expression, purification and activity trials as described below.
Fifteen out of 16 intact sensor kinases are expressed in E. coli membranes
In general, the expression of most membrane proteins is expected to be significantly lower than that of soluble proteins. Therefore the conditions reported previously Citation[35], Citation[38], Citation[42] for successful culture growth and gene expression of other bacterial membrane proteins were tested initially, and optimized for some proteins, to determine if these were also suitable to obtain significant or detectable amounts of the intact membrane sensor kinases. In general, such conditions (use of LB culture medium, induction of expression using 0.5–1 mM IPTG, post-induction temperature of 30°C) proved successful for the overexpression of the majority of sensors. For proteins exhibiting low expression, optimization of culture and expression conditions were investigated in growth trials using different media (LB, M9 minimal, and 2TY medium), different concentrations of the IPTG inducer for expression of the inserted kinase gene in pTTQ18His, and different time periods and temperatures post-induction prior to cell harvesting. Expression of each sensor kinase was assessed as described in Methods using total E. coli membranes prepared by water lysis and analysed by SDS-PAGE and Western blot analysis to detect the engineered C-terminal hexa-histidine tag possessed by each recombinant protein. The results are summarized in .
Table III. Expression conditions, solubilization and activities of the 16 intact membrane sensor kinases of E. faecalis.
Overall, most sensors were optimally expressed in LB media, with only one protein (EF1261 [HK06]) expressed at higher levels in the richer 2TY medium and none of the low-expressing proteins exhibiting better expression in minimal medium compared with the other media (). Suitable inducer concentrations and post-induction temperatures amongst the proteins ranged from 0.2–1 mM IPTG (the highest used in this study) and 15, 30 or 37°C post-induction temperatures (). In general the expression time employed post-induction was 3 h, with only HK08 and HK16 benefiting from overnight incubation at the lower temperature of 15°C.
The observed molecular masses of the majority of recombinant proteins in SDS-PAGE and Western blot analysis appeared less than those predicted ( and ). We therefore verified that the expressed proteins represented intact, undegraded proteins. Using the 13 proteins that could be purified (see below), ESI-MS and/or N-terminal sequencing, together with the positive signals observed in Western analysis that detects an intact C-terminus of the protein through the hexa-histidine tag motif, was undertaken. In all but three cases (EF3197 (HK02) for which no N-terminal sequence or MS data could be obtained, EF0570 (HK12), which could not be solubilized from membranes, and EF1335 (HK16), which was not eluted from Ni2 + -NTA columns), the presence of intact proteins was successfully confirmed. Therefore the observed mass differences of these proteins are attributable to their anomalous behaviour in SDS-PAGE analysis, as is typically observed for hydrophobic membrane proteins Citation[38]. It is noteworthy that, in general, the greatest mass differences were observed for proteins that possess higher numbers of predicted transmembrane regions (TMs), such as EF1820 (HK15) and EF1335 (HK16) (). These proteins are predicted to possess six and seven putative TMs, respectively, and their apparent masses in SDS-PAGE analysis of total membrane preparations were indeed significantly smaller than expected (35 and 32 kDa compared with 53 and 52 kDa, respectively). Likewise, the apparent mass of EF3197 (HK02) (also six predicted TMs) was 53 kDa compared with a predicted mass of 67 kDa ().
Figure 1. Expression of the 16 intact membrane sensor kinases of E. faecalis in E. coli BL21 (DE3). Cells harbouring pTTQ18His containing the recombinant gene for each of the intact membrane kinases were cultured as described in Methods, and mixed membrane proteins prepared by water lysis. Samples (15 µg) were separated and analysed by: (A) SDS-PAGE and visualized using Coomassie blue staining (upper panel); and (B) and (C), Western blotting to detect the engineered hexa-histidine tag at the C-terminus of each protein. Panel C shows a longer exposure required for detection of the successful expression of EF2298 (HK11). M, molecular mass standards. U, mixed membranes of an uninduced culture of E. coli BL21 (DE3)/pTTQHK01.This Figure is reproduced in colour in Molecular Membrane Biology online.
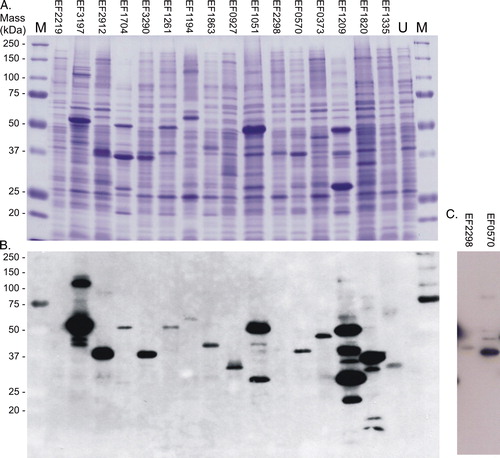
shows that of the 16 intact membrane sensor kinases cloned in this study, 15 were successfully overexpressed. Only EF2219 (HK01) was not detectable in E. coli BL21 (DE3) membranes under any of the test conditions. Partial degradation was observed for EF1209 (HK14), and there was no improvement upon addition of protease inhibitors into the water-lysis preparation procedures ().
Eleven out of 15 proteins retain autophosphorylation activity in E. coli membranes in the absence of signal
Inner membrane vesicles expressing each sensor kinase were prepared as described in Methods, and 200 µg of inner membrane protein were used in autophosphorylation assays with radiolabelled ATP. At intervals following ATP addition, 20 µg samples were removed to stop buffer, and the level of phosphorylated protein at each time point evaluated as described in Methods. shows the results for the 11 kinases that produced detectable autophosphorylation activities in these assays. EF0927 (HK09), EF2298 (HK11), EF0570 (HK12) and EF1335 (HK16) showed no detectable activity on at least three attempts, suggesting either inactivity or absence of an activating signal. The membrane-localized kinases exhibit a range of different kinetics in these assays (B). Nine of the proteins (EF3197 (HK02), EF1704 (HK04), EF1261 (HK06), EF1194 (HK07), EF1863 (HK08), EF1051 (HK10), EF0373 (HK13), EF1209 (HK14) and EF1820 (HK15) attained maximum autophosphorylation within the initial 20 minutes, and for EF3197 (HK02), EF1704 (HK04), EF1194 (HK07), EF1863 (HK08), EF1051 (HK10), EF0373 (HK13) and EF1209 (HK14), this was followed by rapid dephosphorylation (B); EF1051 (HK10), EF0373 (HK13) and EF1209 (HK14) all autophosphorylate particularly rapidly, within the first minute or so following assay initiation.
Figure 2. Autophosphorylation activities of intact sensor kinases in inner membrane vesicles. Inner membrane proteins (200 µg) were incubated at 24°C as described in Methods, and reactions initiated by the addition of 50 µM ATP containing 50 Ci [γ − 32P] or [γ − 33P] ATP. Samples (20 µl) were removed at the time points indicated, reactions terminated by the addition of 6 µl 4×SDS-PAGE sample loading buffer, and proteins separated by SDS-PAGE. Following electrophoresis, gels were washed, dried, and exposed to an image plate for quantitative analysis using a Fuji BAS1000 imaging analyzer system (Fujifilm Co., Japan). (A) Image and (B) quantitative representation of the data for each radiolabelled phosphorylated protein during autophosphorylation. The identities of the phosphorylating proteins (indicated by the arrows) were verified by comparisons with Western data. Graphs show the pixel counts relative to the maximum value attained during the assays. The numbers above each panel indicate the time sampled (minutes) after reaction initiation.
![Figure 2. Autophosphorylation activities of intact sensor kinases in inner membrane vesicles. Inner membrane proteins (200 µg) were incubated at 24°C as described in Methods, and reactions initiated by the addition of 50 µM ATP containing 50 Ci [γ − 32P] or [γ − 33P] ATP. Samples (20 µl) were removed at the time points indicated, reactions terminated by the addition of 6 µl 4×SDS-PAGE sample loading buffer, and proteins separated by SDS-PAGE. Following electrophoresis, gels were washed, dried, and exposed to an image plate for quantitative analysis using a Fuji BAS1000 imaging analyzer system (Fujifilm Co., Japan). (A) Image and (B) quantitative representation of the data for each radiolabelled phosphorylated protein during autophosphorylation. The identities of the phosphorylating proteins (indicated by the arrows) were verified by comparisons with Western data. Graphs show the pixel counts relative to the maximum value attained during the assays. The numbers above each panel indicate the time sampled (minutes) after reaction initiation.](/cms/asset/febc5b01-64b4-4400-8f6c-b94f366cd8fd/imbc_a_336155_f0002_b.gif)

EF3290 (HK05) phosphorylated maximally after approximately 30 min, followed by slow dephosphorylation, still retaining approximately 60% of its maximum phosphorylation level at the end of the 60-min assay period. Uniquely, levels of phosphorylated EF2912 (HK03) increased steadily throughout the assay time and did not reach a maximum within 60 min.
Fourteen out of 15 intact kinases are solubilized from E. coli membranes
DDM has been widely used previously for solubilizing bacterial membrane proteins Citation[38], Citation[42], and was also successful here for solubilizing almost all the proteins in this study, except for EF1863 (HK08) and EF0570 (HK12); LDAO was successfully used for solubilization of EF1863 (HK08), whilst none of the detergents tested for this protein were successful for the solubilization of EF0570 (HK12) from E. coli inner membranes ().
Thirteen of the 15 expressed proteins are successfully purified
Following the solubilization trials described above, the appropriate detergents summarized in were used at 1.0 or 1.2% final concentration to solubilise each intact kinase from E. coli membranes as described in Methods. Purification of the hexa-histidine tagged proteins was performed by nickel affinity purification as described in Methods; only EF1335 (HK16) could not be eluted successfully from the nickel affinity column.
The results of one-step purification are shown in . The purities of EF1704 (HK04), EF1261 (HK06) and EF1051 (HK10) were highest reaching 87, 78 and 90%, respectively, as judged by densitometry measurements. The remaining proteins were not as pure, ranging from 19% for EF0373 (HK13) to 74% for EF0927 (HK09) (). As described above, all purified proteins were confirmed by Western blotting using the His6 probe to verify that these were the expected recombinant Histidine-tagged proteins. Their observed masses were in agreement with those obtained in mixed membranes ( and ; ).
Figure 3. One-step purification of the expressed intact membrane sensor kinases of E. faecalis. Following solubilization, proteins were purified by nickel affinity as described in Methods. Samples (10 µg) were analysed by: (A) separation on SDS-polyacrylamide gels and staining with Coomassie Blue, and% purity determined by densitometry; and (B) Western blot analysis using an INDIATM His probe. M, molecular mass markers. This Figure is reproduced in colour in Molecular Membrane Biology online.
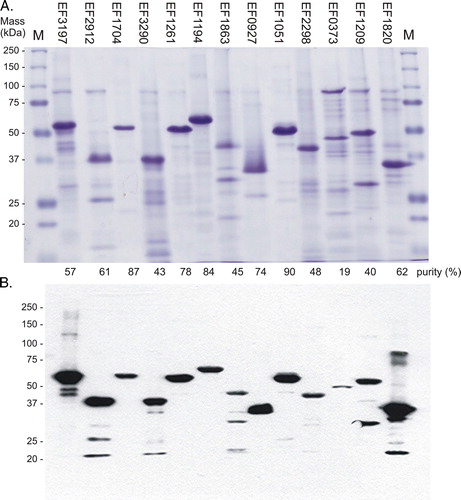
There was evidence of possible degradation amongst some of the proteins, since the Western blot revealed the presence of minor proteins of lower mass in preparations of EF3197 (HK02), EF2912 (HK03), EF3290 (HK05), EF1863 (HK08), EF1209 (HK14) and EF1820 (HK15) FsrC, and to a lesser extent for EF1051 (HK10) and EF2298 (HK11). The one-step purification of EF1194 (HK07) VicK also resulted in protein of high purity, at 84%. However, with minor changes to the purification protocol such as reduced time for solubilization using higher detergent concentrations of 1.2%, the protein was readily purified to > 95% purity and was therefore used at this purity for subsequent signal identification (see below).
Twelve out of 13 purified proteins retain autophosphorylation activity in the absence of signal
summarizes the autophosphorylation activities obtained for 10 of the purified intact kinases that were active following successful expression and purification from E. coli inner membranes. Two further proteins (EF2298 [HK11] and EF1209 [HK14]) were also active as purified proteins, though only weakly and are therefore not shown. Of the 11 intact kinases that were active when membrane-bound, all but one (EF1863 [HK08]) remained active following purification. In the case of EF1863 (HK08), the loss of detectable activity may be attributable to the presence of 0.1% LDAO in the assays, which was required to prevent aggregation of this protein. We have observed that detergent can be inhibitory for the activities of some purified kinases, and therefore assays are routinely undertaken in the absence of added detergent post-solubilization if possible.
Figure 4. Autophosphorylation activities of purified intact sensor kinases. Purified proteins (600–1000 pmoles) were added to reaction buffer, and autophosphorylation initiated using radiolabelled ATP in a total reaction volume of 150 µl, as described in Methods. Sample volumes (15 µl) were removed at the time points indicated. (A) Image and (B) quantitative representation of the data for each radiolabelled phosphorylated protein during autophosphorylation. Phosphorylating proteins (shown by the arrows) were verified by mass determinations and comparisons with Western data. Graphs show the pixel counts relative to the maximum value attained during the assays.
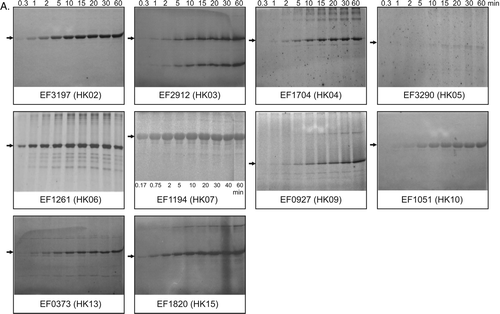

EF0927 (HK09), which previously produced no detectable autophosphorylation when assayed in membranes, produced detectable levels of autophosphorylation activities when purified protein was used (). In the case of EF2298 (HK11) VanSB, in which activity levels were low, there was no detectable increase upon addition into the assays of a 10-fold molar excess of vancomycin, a candidate signal for this kinase (data not shown). We therefore conclude that vancomycin is not an activating signal for this kinase under these conditions.
Comparisons of autophosphorylation kinetics exhibited by each kinase throughout the sixty minute assay when membrane-bound compared with purified proteins, revealed that only EF2912 (HK03) and EF1261 (HK06) possessed similar kinetics using both purified and membrane-bound versions. In the case of most other kinases, specifically EF3197 (HK02), EF1704 (HK04), EF3290 (HK05), EF1194 (HK07), EF1051 (HK10), EF0373 (HK13) and EF1820 (HK15), autophosphorylation activities of purified forms reached maximum levels later in the assays, with little subsequent dephosphorylation observed in the time period compared to that observed for the proteins when membrane-bound ( and ). Although the different concentrations present will presumably affect kinetics, these data may also suggest that autophosphorylation rates are faster when the proteins are bound into a membrane environment or have not undergone the extraction and purification procedures. Alternatively, it is also possible that the membrane environment or even some interfering membrane-bound E. coli phosphatases present in the vesicles assayed, may dephosphorylate or promote the intrinsic phosphatase activities that are typically associated with many sensor proteins.
EF1820 (HK15) FsrC and EF1194 (HK07) VicK are the kinase components of two of the three systems with established roles in this bacterium (the third being EF2298 (HK11) which exhibits low activity as described above) Citation[12], Citation[16–25]. Therefore these two intact active kinases (hence forward referred to as FsrC and VicK, respectively) were selected for further studies to determine if signal identification and signal confirmation may be achieved using heterologously-expressed intact membrane sensor kinases.
Signal identification: The peptide pheromone GBAP stimulates autophosphorylation of purified intact FsrC
FsrCA (EF1820-EF1822, HK-RR15) is a quorum-sensing TCS that positively regulates the expression of infection-related extracellular proteases, gelatinase and a serine protease Citation[16–19] and regulates biofilm formation in E. faecalis Citation[23], Citation[24]. An activating signal for the sensor is the peptide gelatinase biosynthesis-activating pheromone, GBAP Citation[16]. GBAP has been synthesized chemically in an active form, as demonstrated by its ability to activate gelatinase biosynthesis in E. faecalis cells Citation[40]. To demonstrate that synthetic GBAP induces activation through direct interactions with the FsrC sensor kinase, and indeed to demonstrate that intact FsrC is able to respond to its expected GBAP signal in vitro, purified FsrC was pre-incubated with different concentrations of synthetic GBAP for 20 min prior to initiation of autophosphorylation assays (Materials and methods). GBAP stimulated FsrC autophosphorylation by approximately 10-fold (A) in response to increasing concentrations of GBAP up to approximately 10 µM, a two-fold molar excess of GBAP over FsrC. Higher GBAP concentrations up to 85.3 µM elicited little further increase in levels of phosphorylated FsrC (A).
Figure 5. Effect of the GBAP peptide pheromone on autophosphorylation activity of FsrC and other purified sensor kinases. Reactions contained 80 pmoles purified protein preincubated in reaction buffer for 20 min in the presence or absence of synthetic GBAP, prior to the initiation of autophosphorylation reactions in a total reaction volume of 15 µl. After 60 min, samples were removed to 5 µl stop buffer. The quantity of 32P associated with each protein was determined by SDS-PAGE followed by autoradiography and phosphoimager analysis. All reactions (including controls) contained a final concentration of 3.4% acetonitrile (used to dissolve GBAP). (A) FsrC-P levels in response to increasing GBAP. Means of duplicate data are presented (except for 0.7, 21.3 and 170.7 µM GBAP datapoints, for which single measurements were made), and averaged datapoints varied by±0.07–5.43%. (B) Comparison of responses to 10.7 µM GBAP by six purified sensors. Single data measurements are shown for all proteins except FsrC (EF1820) which shows the mean and standard deviation derived from triplicate data. Autoradiographs of individual data are also shown.
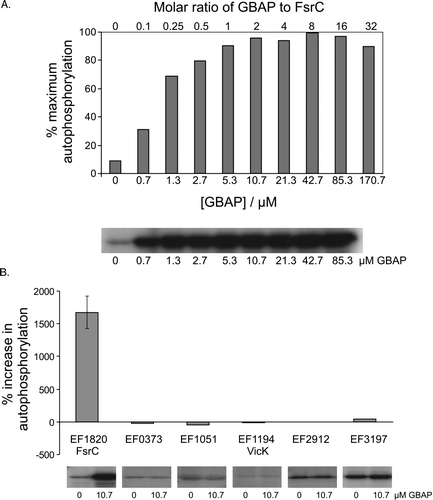
To determine if GBAP exhibits specificity for the FsrC protein or whether it stimulates the activities of other intact E. faecalis sensor kinases, we also tested the effects of GBAP on VicK and five other purified intact kinases which have no known or predicted roles in quorum sensing. Using 10.7 µM GBAP, there was very little change in the activities of any of the other proteins (B), confirming that GBAP exerts a strong, significant effect specifically on FsrC. These data therefore provide the first evidence for direct and specific interactions between GBAP and FsrC, and indeed for any peptide pheromone with its cognate quorum sensor kinase.
Signal identification: Reductants dithiothreitol (DTT) and glutathione significantly increase autophosphorylation of VicK
Ng & Winkler Citation[27] reported a link between the class I YycFG/VicKR TCSs and the capacity for electron transport, and suggested that YycG may directly (or indirectly through YycH Citation[28]) sense the redox state of electron transport. To identify definitively the signals for intact VicK (a Class I member), we firstly investigated whether redox potential acts as a direct modulator of intact VicK activity using DTT as chemical reductant as well as reduced glutathione as a physiologically-relevant reducing agent. DTT was used as in vitro reducing agent since it has been used extensively in previous studies of redox-sensing proteins (e.g., Citation[35], Citation[36], Citation[43]). Using both purified and membrane-bound VicK proteins in which the predicted intracellular PAS, ATP-binding and kinase domains are all exposed in the ATP-requiring assays, levels of VicK-P increased significantly in response to increasing concentrations of DTT (up to 50 mM) in the assays (A, 6B), suggesting that VicK responds to redox signals in vitro and may therefore be a redox sensor in vivo. Low but detectable levels of VicK phosphorylation occurred under air-oxidizing conditions in the absence of DTT, but purified VicK-P levels were increased by 432% and 347% in response to 5 and 20 mM DTT respectively, whilst VicK-P levels in membranes increased by 170% and 296% with 5 and 25 mM DTT (A, 6B). Hence, the purified version of the intact protein was more responsive to the membrane-permeant signalling agent than the membrane-bound form, possibly indicating either an increase in signal sensitivity upon protein purification, or a barrier role played by the membrane in DTT access to the protein. It is also possible that it is the extracellular sensing domain that senses DTT and that this difference reflects an incomplete equilibration of DTT across the membrane and into the interior of the vesicles.
Figure 6. Effects of dithiothreitol (DTT) and reduced (GSH) and oxidized (GSSG) glutathione on autophosphorylation activity of intact VicK. Each reaction contained either membrane-bound VicK in E. coli membranes (20 µg) (A), (C) & (E), or 60–80 pmoles purified VicK (B) and (D), which were incubated in reaction buffer for 20 min in the presence or absence of DTT (A) and (B), or 5 and 25 mM concentrations of GSH or GSSG (C), (D) & (E), prior to the initiation of autophosphorylation reactions in a reaction volume of 15 µl. Reactions were terminated with stop buffer after 2 min (membrane-bound VicK [A] and [C]), or 40 min (purified protein [B] and [D]) when VicK was autophosphorylating, or after 40 min for membrane-bound protein, when VicK is dephosphorylating (E). The quantity of 33P associated with VicK was then determined by SDS-PAGE followed by autoradiography and phosphoimager analysis. Reactions were performed in triplicate. Autoradiographs show one individual blot; numbers beneath are the phosphorimager data presented as a mean of the triplicate data.
![Figure 6. Effects of dithiothreitol (DTT) and reduced (GSH) and oxidized (GSSG) glutathione on autophosphorylation activity of intact VicK. Each reaction contained either membrane-bound VicK in E. coli membranes (20 µg) (A), (C) & (E), or 60–80 pmoles purified VicK (B) and (D), which were incubated in reaction buffer for 20 min in the presence or absence of DTT (A) and (B), or 5 and 25 mM concentrations of GSH or GSSG (C), (D) & (E), prior to the initiation of autophosphorylation reactions in a reaction volume of 15 µl. Reactions were terminated with stop buffer after 2 min (membrane-bound VicK [A] and [C]), or 40 min (purified protein [B] and [D]) when VicK was autophosphorylating, or after 40 min for membrane-bound protein, when VicK is dephosphorylating (E). The quantity of 33P associated with VicK was then determined by SDS-PAGE followed by autoradiography and phosphoimager analysis. Reactions were performed in triplicate. Autoradiographs show one individual blot; numbers beneath are the phosphorimager data presented as a mean of the triplicate data.](/cms/asset/64b3f53f-4391-433b-9905-cbbde5f085fb/imbc_a_336155_f0006_b.gif)
To determine whether the effects of redox potential towards VicK autophosphorylation could function in vivo, DTT was replaced with the alternative and more physiologically-relevant reductant, glutathione (GSH). Whether using membrane-bound VicK in vesicles or as purified VicK, the presence of reduced GSH at the physiologically-relevant concentration of 5 mM, resulted in modest but significant increases in VicK-P levels in the autophosphorylation assays (C, 6D). The largest effect was observed with purified protein, with a 60% increase in VicK-P levels using 5 mM GSH compared with the 37% increase obtained using membrane-bound protein, suggesting that the protein may be slightly less responsive to GSH when membrane-bound. At the higher concentration of 25 mM, the effects of GSH were less pronounced (C, 6D). Unexpectedly, oxidized glutathione (GSSG) also exerted significant increases in purified VicK autophosphorylation rates, and these were either similar to, or slightly greater than those obtained using GSH. This suggests that glutathione itself, rather than its reduced/oxidized state, exerts a positive effect on VicK activity. Indeed, the effects of both GSH and GSSG on membrane-bound VicK were significantly more pronounced when the assays were terminated after 40 min (instead of 2 min). At this point, VicK is significantly dephosphorylated (), contrasting with the autophosphorylating protein assayed in A–D. Under such conditions, the physiologically-relevant concentration of 5 mM GSH elicited a 360% increase in VicK-P, whilst GSSG produced a 226% increase (E), again confirming an effect of glutathione per se.
The effects of 10 other agents that could potentially serve as signals to modulate activity of VicK in vivo were also tested, particularly as VicK possesses two putative PAS domains that could be involved in sensing more than one signal. These included respiratory quinones Citation[43], Citation[44] and reduced and oxidized versions of menaquinone (vitamin K) (in 50-fold molar excess) (included because E. faecalis possesses dimethylmenaquinones Citation[45–47]), 10 mM NAD+ or NADH, 10 mM sodium phosphate as a source of a 1875-fold molar excess of phosphate ions Citation[48], 10 mM ammonium chloride as a source of ammonium ions Citation[49] and 10 mM lactate, acetate, fumarate or pyruvate Citation[50], Citation[51]. In all cases these agents brought about no significant change in VicK-P levels in autophosphorylation assays (data not shown).
Sensor responses exert an effect on downstream signal transduction in vitro: Elevated VicK-P levels resulting from DTT result in higher levels of VicR-P during phosphotransfer
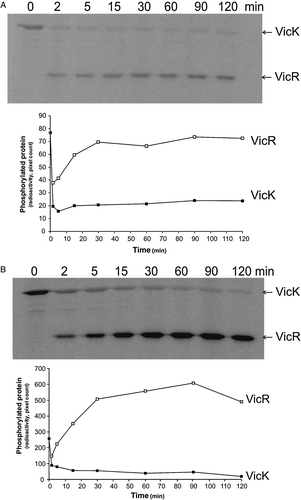
When VicK was permitted to autophosphorylate for 15 min in the presence of 0.5 mM or 100 mM DTT and VicR was then added at a final concentration of 1.5:1 VicK:VicR, in both cases phosphotransfer resulted in a steady loss of VicK-P and increasing levels of VicR-P throughout the 2 h of the assay (). The presence of higher DTT resulted in higher levels of VicR-P levels throughout the phosphotransfer assays, thereby demonstrating that the increased levels of phosphorylated VicK induced by DTT has a concomitant impact on downstream levels of VicR-P. also shows that the kinetics of phosphotransfer or maintenance of the VicR-P signalling state in the presence of low DTT differ with those observed previously for PrrB; as described above, levels of VicR-P increase steadily with time during phosphotransfer (), unlike PrrA-P levels which actually decrease with time Citation[35].
Figure 7. Effect of redox potential on phosphotransfer from VicK to VicR in vitro using DTT as reducing agent. 420 pmoles VicK were permitted to autophosphorylate for 15 min in the presence of either 0.5 mM or 100 mM DTT in 130 µl reaction volumes. 13 µl samples were removed to stop buffer prior to the addition of 252 pmoles VicR to the remaining 378 pmoles VicK (1.5:1 VicK:VicR). Samples (50 pmoles) were removed to stop buffer at regular intervals up to 2 h. Autoradiograph and graphical representation of quantitative data obtained using (A) 0.5 mM DTT, and (B) 100 mM DTT.
Discussion
To understand how changes in a myriad of environmental conditions are perceived by bacterial cells and how these signals are networked and coordinated into overall cellular responses requires a fundamental knowledge of all the sensory proteins present in individual cells. In the main, these sensory proteins are histidine protein kinases (or sensor kinases) and most bacteria possess many different HKs to match the varied environmental cues encountered. Laboratory production of these membrane sensory proteins has previously been one of the bottlenecks in determining their structures and mechanisms. Here we have evaluated the heterologous production and activities of the 16 intact membrane sensor kinases of E. faecalis. Following the systematic cloning of the genes for each protein into the membrane protein expression vector pTTQ18His, we demonstrated that of the 16 proteins, all but one (EF2219 [HK01]) were successfully expressed in E. coli membranes (). The success of pTTQ18 as an expression plasmid for membrane proteins has been well documented previously Citation[38], Citation[42] and in a recent comparison of eight vectors for expression of 37 membrane protein targets, pTTQ18 showed the greatest number of targets expressed Citation[52].
The levels of E. faecalis HK expression varied, depending on the protein, from 3.5–16.5% of mixed membrane proteins, significantly higher than the estimated 100 or so copies per cell of each sensory kinases in vivo, and therefore to be regarded as an amplified level of expression. In this study the choice of host strain, expression medium, induction temperature, concentration of inducer and post-induction time were based on our previous experiences with expression of intact RegB Citation[35], other bacterial membrane proteins Citation[42], and on limited trials to optimise conditions for this set of sensor kinases. In general, LB broth at induction temperatures of 30 or 37°C with 0.5 or 1 mM IPTG and 3 h post-induction time were successful for the overexpression of the majority of E. faecalis sensors (). High success rates were also obtained for the proportion of intact sensor kinases that could be subsequently solubilized from E. coli membranes and purified by nickel affinity purification. Fourteen out of 15 proteins were successfully solubilized and 13 purified.
Producing active proteins is crucial for in vitro studies of signal identification and interactions. Unlike truncated soluble versions, some full-length sensor kinases that possess intact signal sensing domains might be expected to phosphorylate only in the presence of their environmental signal. For other intact sensor kinases the converse may hold true, and the presence of the signal may be required to repress the autophosphorylation activities of the kinase or enhance any phosphatase activity of the sensor towards its cognate response regulator, as is the case for RegB (PrrB) Citation[53]. Indeed, it is also possible that the activities of some kinases such as VicK that belong to essential two-component systems, are never switched off entirely in vivo, and rather they are up- or down-regulated from a constitutive basal level, so activities are present even in the absence of activating signals.
In the majority of cases, the nature of the signal(s) is unknown. Therefore, in this study we determined the autophosphorylation activities displayed by the membrane-bound and purified sensor kinases in the absence of known signals, as a starting point for assessing activities of these overexpressed intact proteins. Eleven of the 15 membrane-bound proteins and 12 of the 13 purified proteins were active in autophosphorylation assays. Remarkably, 13 of the 15 expressed proteins were active in E. coli membranes and/or as purified proteins. Their activities may be attributable to absence in these assays of any repressive ‘signals’ required to regulate these sensors in vivo, as discussed above. However, this is unlikely to hold true for all these sensors, since some are predicted/known to respond to signals that activate kinase activity, rather than repress it, as in the case of FsrC, which senses the GBAP pheromone ( and ) Citation[16], Citation[19], Citation[40]. Indeed, in our present studies with FsrC and VicK, we demonstrated that levels of autophosphorylation were significantly increased upon addition into activity assays of activating signals ( and ). These findings not only confirm direct interactions between each sensor and its signal in this in vitro approach, but also demonstrate the potential of such an approach in future studies of signal identification using other sensor kinases.
In the case of FsrC, the strong stimulatory effect on autophosphorylation activity produced by a synthetic version of the peptide inducer GBAP was also a specific effect, since five other kinases included in the assays exhibited little or no response to the pheromone (). Signal specificity is assumed to be a fundamental feature of two-component systems. Interestingly, the Fsr system, unlike its Agr counterparts in Staphylococcus aureus, is particularly highly conserved amongst E. faecalis strains Citation[18].
Of the 12 agents tested as potential signals for intact VicK, the findings that only increasing concentrations of DTT and of physiological concentrations of glutathione significantly increase activities of the intact kinase (), provide the first direct in vitro signal identification for this intact kinase. The responses of VicK to the reducing agent DTT were similar to those observed using other redox-responsive proteins including PrrB Citation[35], Citation[36] and OxyR Citation[54], and therefore our data indicate that VicK may also sense redox potential. It is possible that the chelation properties of DTT might also exert an effect on VicK activity, though the only predicted role for metals is in the Mg2 + dependent ATPase functions which would be expected to reduce VicK activity, not increase it as observed here. Finally, we also demonstrated that the increased levels of VicK-P caused by DTT resulted in concomitant increases in phosphotransfer and VicR-P levels (), demonstrating that these in vitro assays can also be used to examine signal effects on subsequent signal transduction through TCS pathways.
Reduced glutathione (GSH) was included in VicK activity assays as an alternative reducing agent. The results with GSH supported the DTT data; the presence of the physiologically-relevant concentration of 5 mM GSH also significantly increased VicK autophosphorylation activity, confirming a possible role for redox potential in modulating VicK, a protein that possesses two putative PAS domains (). Unusually for many Gram-positive bacteria, E. faecalis possesses significant levels of glutathione Citation[55], suggesting that redox control of VicK might be exerted at least in part by GSH itself in vivo. However, the findings here () that oxidized glutathione (GSSG), in addition to GSH, also significantly increased VicK∼P levels suggest a different or additional role for glutathione itself in VicK modulation. Glutathione is linked with protection against oxidative and other stresses in the cell in vivo Citation[56], Citation[57]. Glutathione may therefore play a protective role towards this essential kinase, possibly under stress conditions. The protective role for glutathione in bacteria has been attributed, at least in part, to the direct glutathionylation of proteins by GSSG Citation[57–60]. Since glutathione is located intracellularly in vivo, the region of VicK involved in glutathione-sensing must presumably be located in the cytoplasmic portion. This is supported by the membrane vesicle data in , in which the inside-out orientation supported increased VicK activity in response to membrane-impermeant glutathione, though we cannot exclude the possibility that in the case of the reduced form, GSH may enter the interior of these inside-out vesicles via the E. coli transporter CydDC, giving access to the putative extracellular sensing domain of the protein Citation[61].
In this study, all the intact kinases of virulence-associated systems (FsrCA, EF1704-1703 [HK-RR04] [PhoPR], EF3290-3289 [HK-RR05] [CroSR], EF1261-1260 [HK-RR06] and EF1051-1050 [HK-RR10] [EtaSR]) were successfully overexpressed and active in membranes or as purified proteins. The putative VncS homologue (EF1863 [HK08]) was also successfully expressed and active, and EF2298 (HK11) VanSB and EF1209 (HK14) were also observed to exhibit low autophosphorylation as purified proteins. The nature of the signals that these kinases sense can therefore be fully investigated in future in vitro experiments using the full-length versions of these membrane proteins. For example, EF2298-2299 (HK-RR11) VanSBRB and EF1863-1864 (HK-RR08) may sense changes in cell wall intermediates and components; successful expression of their intact sensory components here paves the way for future studies of mechanisms of vancomycin resistance and tolerance in this bacterium, and expands on previous studies with soluble versions of the kinase (e.g., Citation[62]).
Given the successful demonstration of GBAP as a direct and specific activating signal for FsrC, and the first successful identification of modulators of VicK autophosphorylation activities with subsequent downstream effects on levels of phospho-signalling to its partner response regulator VicR, further work is now underway using both membrane-bound and purified proteins to identify the signals for each of the kinases of E. faecalis to determine their full roles in virulence and antibiotic resistance in this pathogenic bacterium.
Supplementary information
In the completed genome sequence of E. faecalis strain V583, 17 two-component system (TCS) pairs have previously been identified, along with one orphan RR Citation[1]. Using the histidine kinase (HK) classification scheme described previously Citation[2], Hancock and Perego sub-grouped the 17 HK proteins, identifying two HKs as belonging to the group I sub-family, one group II member, 10 HKs belonging to group IIIa (which appears to be the most abundant class of kinases in the Gram-positive pathogens whose genomes have currently been sequenced), one in group IV and three that do not belong to any of the previously described classes of HKs Citation[1]. All but one of the HKs were predicted to be membrane localized Citation[1].
Similarity searches, comparisons with known TCSs in other bacteria Citation[1], systematic inactivation of the response regulator (RR) components Citation[65–68], and investigations using mouse peritonitis and peritonitis macrophage models Citation[3], Citation[6] revealed important insights into the potential functions of many systems, some of which are associated with virulence or responses to antibacterial agents and other stresses. However, in most cases the precise roles and in particular the molecular environmental signals to which these systems respond remain incompletely understood. The following is a summary of the main points of what is currently known or suggested about each system.
EF2219-2218 (HK-RR01) is homologous to a B. subtilis system located next to putative sugar and galactan utilisation genes, and whilst its role remains unknown, inactivation of the response regulator component RR01 was shown to result in increased sensitivity to cell wall active agent bacitracin Citation[5]. The TCS EF3197-3196 (HK-RR02) is highly homologous to LytSR of Staphylococcus aureus; the HK proteins possess 52% identity Citation[1], Citation[7], suggesting a possible role for the enterococcal system in cell wall integrity mediated through effects on the intrinsic murein hydrolase activity of the cells. However, Hancock and Perego found no change in autolysis rates in an RR02 mutant, and therefore such a role remains unconfirmed, though alterations in uptake of antibiotics such as erythromycin were observed in the mutant, suggesting some involvement in cell wall-related permeability functions Citation[5]. Mutations in the RR component of the TCS EF2912-2911 (HK-RR03) resemble those of EF2218 (RR01), exhibiting increased sensitivity to bacitracin; this TCS most closely resembles B. subtilis YcqE and Staphylococcus aureus VraS Citation[5], but again, the precise role remains unknown. The TCS EF1704-1703 (HK-RR04) is homologous to B. subtilis PhoPR Citation[1], Citation[3], Citation[8]; disruption of the err04 gene of this system resulted in increased heat sensitivity in strain JH2-2 (a phenotype also shared by two other RR mutants, err08 and err18) Citation[4] and a growth defect in strain V583 Citation[5]. This system was recently shown to be induced in phosphate deprivation conditions, and to be required for full survival in macrophages hence confirming a role in virulence Citation[6]. The EF3290-3289 (HK-RR05) system, CroSR, is required for intrinsic β-lactam resistance Citation[5], Citation[9]; a croR mutation caused significant growth defects and cell morphology alterations in strain JH2-2 Citation[4], and is implicated in virulence since the mutation also impairs survival in macrophages Citation[6]. EF1261-1260 (HK-RR06) is also linked with virulence, since mutations in the RR component RR06 result in decreased survival in macrophages and susceptibility to oxidative stress Citation[6]; this may be linked with membrane functions and architecture, since a RR06 mutation was also found to be more susceptible than wild type to heat and SDS Citation[5]. EF1194-1193 (HK-RR07) (VicKR) has been reported to be an essential system for E. faecalis, with high similarity to the essential YycFG systems found in B. subtilis, Staphylococcus aureus, Streptococcus pneumoniae and other low%GC Gram-positive species Citation[3–5], Citation[72–74]. Target genes have been identified for many species and include those involved in cell wall biosynthesis or cell membrane composition Citation[8], Citation[75–79], cell division (ftsAZ) Citation[18], virulence and genetic competence Citation[17], Citation[19], Citation[20] and biofilms Citation[20]. EF1863-1864 (HK-RR08) is most homologous to the VncSR system of S. pneumoniae implicated in vancomycin tolerance Citation[21], is linked to mobile genetic elements Citation[5] and in common with EF1704-1703 and EF3329 mutations, its disruption leads to heat sensitivity Citation[4]. EF0927-0926 (HK-RR09) is less well understood and shows no particular homology with any known systems in the database. Using both a mouse peritonitis model and peritoneal macrophages, the EtaSR (EF1051-1050 or HK-RR10) system has been shown to contribute to virulence Citation[3], Citation[6] and is most homologous to the LisKR and CsrSR systems of other Gram-positive bacteria. Mutants display increased acid sensitivity Citation[3] and heat resistance correlated with increased DnaK and GroEL levels in these strains Citation[4]. EF2298-2299 (HK-RR11), VanSBRB, regulates VanB-type vancomycin resistance and its inactivation renders E. faecalis significantly more sensitive to vancomycin Citation[5], Citation[22]. EF0570-0571 (HK-RR12) is similar to KdpDE found in several Gram-positive and Gram-negative organisms; in E. faecalis it is often located on a pathogenicity island though it is not present in all strains of E. faecalis suggesting that it is not essential for a commensal life style Citation[3], Citation[5], Citation[23]. EF0373-0372 (HK-RR13) expression was only detectable at a higher temperature of 50°C, suggesting a possible involvement in the E. faecalis stress response Citation[4]. The RR of the EF1209-1210 (HK-RR14) system is most similar to the CitBT family of response regulators involved in regulating citrate and/or C4 dicarboxylate levels; EF1210 inactivation resulted in a growth defect Citation[5] and increased sensitivity at 50°C, implicating it is the stress response Citation[4]. EF1820-1822 (HK-RR15) corresponds to the Fsr quorum-sensing system of E. faecalis Citation[86–89] and has a role in virulence Citation[90–92] and biofilm formation Citation[5], Citation[93–95]. Little is known of EF1335-1336 (HK-RR16), though in common with several other RR knockout strains of E. faecalis, the RR16 mutant displays faster doubling time during growth in the presence of subinhibitory concentrations of erythromycin, suggesting an effect in uptake of this class of drug Citation[5]. EF1632-1633 (HK-RR17) is the only system with a cytoplasmic histidine kinase and exhibits no significant similarity to known TCSs Citation[1]. Inactivation of orphan response regulator EF3329 (RR18) results in increased heat sensitivity Citation[4], and a phenotype similar to that of RR16 Citation[5].
Acknowledgements
We are grateful to Professor Michael S. Gilmore, Department of Microbiology & Immunology, University of Oklahoma Health Sciences Center, USA, for provision of E. faecalis V583, and to Halina Norbertczak and Peter Roach for preparations of EF1051 (HK10)-harbouring E. coli membranes in a fermenter facility. We thank Lianne Davis, Claire Harris, Tacita Nye and Rida Saeed for optimization and activity data for some of the kinases, and Denise Ashworth, Jeff Keen, Alison Ashcroft and Lynsey Jones (University of Leeds) for DNA sequencing, N-terminal sequencing and ESI-mass spectrometry services respectively, provided by the Wellcome Trust. MKP-J and PJFH are grateful to the BBSRC (BB/D001641/1), and PJFH to the European Union (EMeP grant LSHG-CT-2004-504601) for financial support. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Hancock LE, Gilmore MS. Pathogenicity of enterococci. Gram-positive pathogens, V Fischetti, R Novick, J Ferretti, D Portnoy, J Rood. ASM Press, Washington DC 2000
- Mundy LM, Sahm DF, Gilmore M. Relationships between enterococcal virulence and antimicrobial resistance. Clin Microbiol Rev 2000; 13: 513–522
- Huycke MM, Sahm DF, Gilmore MS. Multiple-drug resistant enterococci: The nature of the problem and an agenda for the future. Emerg Infect Dis 1998; 4: 239–249
- Shepard BD, Gilmore MS. Antibiotic-resistant enterococci: The mechanisms and dynamics of drug introduction and resistance. Microb Infect 2002; 4: 215–224
- Hoch JA, Silhavy TJ. Two-component signal transduction. ASM Press, Washington DC 1995
- West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 2001; 26: 369–376
- Hakenbeck R, Stock JB. Analysis of two-component signal transduction systems involved in transcriptional regulation. Meth Enzymol 1996; 273: 281–300
- Szurmant H, White RA, Hoch JA. Sensor complexes regulating two-component signal transduction. Curr Op Struct Biol 2007; 17: 706–715
- Laguri C, Phillips-Jones MK, Williamson MP. Solution structure and DNA binding of the effector domain from the global regulator PrrA (RegA) from Rhodobacter sphaeroides: insights into DNA binding specificity. Nucl Acids Res 2003; 31: 6778–6787
- Hancock LE, Perego M. Two-component signal transduction in Enterococcus faecalis. J Bacteriol 2002; 184: 5819–5825
- Fabret C, Feher VA, Hoch JA. Two-component signal transduction in Bacillus subtilis: How one organism sees its world. J Bacteriol 1999; 181: 1975–1983
- Hancock LE, Perego M. Systematic inactivation and phenotypic characterisation of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol 2004; 186: 7951–7958
- Le Breton Y, Boël G, Benachour A, Prévost H, Auffray Y, Rincé A. Molecular characterization of Enterococcus faecalis two-component signal transduction pathways related to environmental stresses. Environ Microbiol 2003; 5: 329–337
- Teng F, Wang L, Singh KV, Murray BE, Weinstock GM. Involvement of PhoP-PhoS homologs in Enterococcus faecalis virulence. Infect Immun 2002; 70: 1991–1996
- Muller C, Sanguinetti M, Riboulet E, Hebert L, Posteraro B, Fadda G, Auffray Y, Rince A. Characterization of two signal transduction systems involved in intracellular macrophage survival and environmental stress response in Enterococcus faecalis. J Mol Microbiol Biotech 2008; 14: 59–66
- Nakyama J, Cao Y, Horii T, Sakuda S, Akkermans ADL, de Vos W, Nagasawa H. Gelatinase biosynthesis-activating pheromone: A peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 2001; 41: 145–154
- Qin X, Singh KV, Weinstock GM, Murray BE. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect Immun 2000; 68: 2579–2586
- Qin X, Singh KV, Weinstock GM, Murray BE. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J Bacteriol 2001; 183: 3372–3382
- Nakayama J, Chem SM, Oyama N, Nishiguchi K, Azab EA, Tanaka E, Kariyama R, Sonomoto K. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheremone propeptide corresponding to staphylococcal AgrD. J Bacteriol 2006; 188: 8321–8326
- Mylonakis E, Engelbert M, Qin X, Sifri CD, Murray BE, Ausubel FM, Gilmore MS, Calderwood SB. The Enterococcus faecalis fsrB gene, a key component of the fsr quorum-sensing system, is associated with virulence in the rabbit endophthalmitis model. Infect Immun 2002; 70: 4678–4681
- Engelbert M, Mylonakis E, Ausubel FM, Calderwood SB, Gilmore MS. Contribution of gelatinase, serine protease, and fsr to the pathogenesis of Enterococcus faecalis endophthalmitis. Infect Immun 2004; 72: 3628–3633
- Zeng J, Teng F, Murray BE. Gelatinase is important for translocation of Enterococcus faecalis across polarized human enterocyte-like T84 cells. Infect Immun 2005; 73: 1606–1612
- Hancock LE, Perego M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol 2004; 186: 5629–5639
- Pillai SK, Sakoulas GM, Eliopoulos GM, Moellering RC, Murray BE, Inouye RT. Effects of glucose on fsr-mediated biofilm formation in Enterococcus faecalis. J Infect Dis 2004; 190: 967–970
- Evers S, Courvalin P. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR(B) two-component regulatory system in Enterococcus faecalis V583. J Bacteriol 1996; 178: 1302–1309
- Fabret C, Hoch JA. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J Bacteriol 1998; 180: 6375–6383
- Ng W-L, Winkler ME. Singular structures and operon organizations of essential two-component systems in species of Streptococcus. Microbiol UK 2004; 150: 3096–3098
- Szurmant H, Nelson K, Kim E-J, Perego M, Hoch JA. YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilis. J Bacteriol 2005; 187: 5419–5426
- Szurmant H, Fukushima T, Hoch JA. The essential YycFG two-component system of Bacillus subtilis. Meth Enzymol 2007; 422: 396–417
- Szurmant H, Mohan MA, Imus PM, Hoch JA. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J Bacteriol 2007; 189: 3280–3289
- Trinh CH, Liu Y, Phillips SEV, Phillips-Jones MK. Structure of the response regulator VicR DNA-binding domain. Acta Crystallog Sect D 2007; 63: 266–269
- Novak R, Henriques B, Charpentier E, Normark S, Tuomanen E. Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature 1999; 399: 590–593
- Brunskill EW, Bayles KW. Identification and molecular characterization of a putative regulatory locus that affects autolysis in Staphylococcus aureus. J Bacteriol 1996; 178: 611–618
- Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, Ishihama A. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J Biol Chem 2005; 280: 1448–1456
- Potter CA, Ward A, Laguri C, Williamson MP, Henderson PJF, Phillips-Jones MK. Expression, purification and characterisation of full-length heterologously expressed histidine protein kinase RegB from Rhodobacter sphaeroides. J Mol Biol 2002; 320: 201–213
- Potter CA, Jeong E-L, Williamson MP, Henderson PJF, Phillips-Jones MK. Redox-responsive in vitro modulation of the signalling state of the isolated PrrB sensor kinase of Rhodobacter sphaeroides NCIB 8253. FEBS Lett 2006; 580: 3206–3210
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual, C Nolan. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY 1989
- Ward A, Sanderson NM, O'Reilly J, Rutherford NG, Poolman B, Henderson PJF. The amplified expression, identification, purification, assay and properties of hexahistidine-tagged bacterial membrane transport proteins. Membrane transport – a practical approach, SA Baldwin. Oxford University Press, Oxford 1999
- Schaffner W, Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 1973; 56: 502–514
- Nakayama J, Cao Y, Horii T, Sakuda S, Nagasawa H. Chemical synthesis and biological activity of the gelatinase biosynthesis-activating pheromone of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem 2001; 65: 2322–2325
- Venter H, Ashcroft AE, Keen JN, Henderson PJF, Herbert RB. Molecular dissection of membrane-transport proteins: Mass spectrometry and sequence determination of the galactose-H+ symport protein, GalP, of Escherichia coli and quantitative assay of the incorporation of [ring-2-C-13]histidine and (NH3)-N-15. Biochem J 2002; 363: 243–252
- Saidijam M, Psakis G, Clough JL, Meuller J, Suzuki S, Hoyle CJ, Palmer SL, Morrison SM, Pos MK, Essenberg RC, Maiden MCJ, Abu-bakr A, Baumberg SG, Neyfakh AA, Griffith JK, Stark MJ, Ward A, O'Reilly J, Rutherford NG, Phillips-Jones MK, Henderson PJF. Collection and characterisation of bacterial membrane proteins. FEBS Lett 2003; 555: 170–175
- Swem LR, Gong X, Yu CA, Bauer CE. Identification of a ubiquinone-binding site that affects autophosphorylation of the sensor kinase RegB. J Biol Chem 2006; 281: 6768–6775
- Georgellis D, Kwon O, Lin ECC. Quinones as the redox signal for the Arc two-component system of bacteria. Science 2001; 292: 2314–2315
- Baum RH, Dolin MI. Isolation of 2-solanesyl-1,4-naphthoquinone from Streptococcus faecalis. J Biol Chem 1965; 240: 3425–3433
- Collins MD, Jones D. The distribution of isoprenoid quinones in streptococci of serological groups D and N. J Gen Microbiol 1979; 114: 27–33
- Huycke MM, Moore D, Joyce W, Wise P, Shepard L, Kotake Y, Gilmore MS. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol Microbiol 2001; 42: 729–740
- Howell A, Dubrac S, Krogh Anderson K, Noone D, Fert J, Msadek T, Devine K. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid approach. Mol Microbiol 2003; 49: 1639–1655
- Clausen VA, Bae W, Throup J, Burnham MKR, Rosenberg M, Wallis NG. Biochemical characterization of the first essential two-component signal transduction system from Staphylococcus aureus and Streptococcus pneumoniae. J Mol Microbiol Biotech 2003; 5: 252–260
- Iuchi S, Lin ECC. Two-component signal transduction, JA Hoch, TJ Silhavy. ASM Press, Washington DC 1995; 223–231
- Rodriguez C, Kwon O, Georgellis D. Effect of D-lactate on the physiological activity of the ArcB sensor kinase in Escherichia coli. J Bacteriol 2004; 186: 2085–2090
- Surade S, Klein M, Stolt-Bergner PC, Muenke C, Roy A, Michel H. Comparative analysis and “expression space” coverage of the production of prokaryotic membrane proteins for structural genomics. Prot Sci 2006; 15: 2178–2189
- Oh J-I, Ko I-J, Kaplan S. Reconstitution of the Rhodobacter sphaeroides cbb3-PrrBA signal transduction pathway in vitro. Biochem (USA) 2004; 43: 7915–7923
- Storz G, Tartaglia LA, Ames BN. Transcriptional regulator of oxidative stress-inducible genes – direct activation by oxidation. Science 1990; 248: 189–194
- Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. Distribution of thiols in microorganisms: Mycothiol is a major thiol in most actinomycetes. J Bacteriol 1996; 178: 1990–1995
- Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol – disulfide status. Proc Natl Acad Sci USA 1999; 96: 6161–6165
- Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antiox Redox Signall 2006; 8: 753–762
- Lillig CH, Potamitou A, Schwenn JD, Vlamis-Gardikas A, Holmgren A. Redox regulation of 3′-phosphoadenylylsulfate reductase from Escherichia coli by glutathione and glutaredoxins. J Biol Chem 2003; 278: 22325–22330
- Green J, Paget MS. Bacterial redox sensors. Nat Rev Microbiol 2004; 2: 954–966
- Hondorp ER, Matthews RG. Oxidative stress inactivates cobalamin-independent methionine synthase (MetE) in Escherichia coli. PLoS Biol 2004; 2: 1738–1753
- Pittman MS, Robinson HC, Poole RK. A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm. J Biol Chem 2005; 280: 32254–32261
- Wright GD, Holman TR, Walsh CT. Purification and characterization of VanR and the cytosolic domain of VanS – a two-component regulatory system required for vancomycin resistance in Enterococcus faecium BM4147. Biochemistry 1993; 32: 5057–5063
- Hancock LE, Perego M. Two-component signal transduction in Enterococcus faecalis. J Bacteriol 2002; 184: 5819–5825
- Fabret C, Feher VA, Hoch JA. Two-component signal transduction in Bacillus subtilis: How one organism sees its world. J Bacteriol 1999; 181: 1975–1983
- Teng F, Wang L, Singh KV, Murray BE, Weinstock GM. Involvement of PhoP-PhoS homologs in Enterococcus faecalis virulence. Infect Immun 2002; 70: 1991–1996
- Le Breton Y, Boël G, Benachour A, Prévost H, Auffray Y, Rincé A. Molecular characterization of Enterococcus faecalis two-component signal transduction pathways related to environmental stresses. Environ Microbiol 2003; 5: 329–337
- Hancock LE, Perego M. Systematic inactivation and phenotypic characterisation of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol 2004; 186: 7951–7958
- Muller C, Sanguinetti M, Riboulet E, Hebert L, Posteraro B, Fadda G, Auffray Y, Rince A. Characterization of two signal transduction systems involved in intracellular macrophage survival and environmental stress response in Enterococcus faecalis. J Mol Microbiol Biotech 2008; 14: 59–66
- Brunskill EW, Bayles KW. Identification and molecular characterization of a putative regulatory locus that affects autolysis in Staphylococcus aureus. J Bacteriol 1996; 178: 611–618
- Howell A, Dubrac S, Krogh Anderson K, Noone D, Fert J, Msadek T, Devine K. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid approach. Mol Microbiol 2003; 49: 1639–1655
- Comenge Y, Quintiliani R, Li L, Dubost L, Brouard JP, Hugonnet JE, Arthur M. The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J Bacteriol 2003; 185: 7184–7192
- Fabret C, Hoch JA. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J Bacteriol 1998; 180: 6375–6383
- Martin PK, Li T, Sun D, Biek DP, Schmid MB. Role of cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol 1999; 181: 3666–3673
- Throup JP, Koretke KK, Bryant AP, Ingraham KA, Chlaker AF, Ge Y, Marra A, Wallis NG, Brown JR, Holmes DJ, Rosenberg M, Burnham MKR. A genomic analysis of two-component signal transduction in Streptococcus pneumoniae. Mol Microbiol 2000; 35: 566–576
- Ng W-L, Robertson GT, Kazmierczak KM, Zhao J, Gilmour R, Winkler ME. Constitutive expression of PcsB suppresses the requirement for the essential VicR (YycF) response regulator in Streptococcus pneumoniae R6. Mol Microbiol 2003; 50: 1647–1663
- Ng W-L, Kazmierczak KM, Winkler ME. Defective cell wall synthesis in Streptococcus pneumoniae R6 depleted for the essential PcsB murein hydrolase or the VicR (YycF) response regulator. Mol Microbiol 2004; 53: 1161–1175
- Dubrac S, Msadek T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol 2004; 186: 1175–1181
- Mohedano ML, Overweg K, de la Fuente A, Reuter M, Altabe S, Mulholland F, de Mendoza D, Lopez P, Wells JM. Evidence that the essential response regulator YycF in Streptococcus pneumoniae modulates expression of fatty acid biosynthesis genes and alters membrane composition. J Bacteriol 2005; 187: 2357–2367
- Ng W-L, Tsui H-CT, Winkler ME. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphporylated VicR (YycF) response regulator in Streptococcus pneumoniae. J Bacteriol 2005; 187: 7444–7459
- Fukuchi K, Kasahara Y, Asai K, Kobayashi K, Moriya S, Ogasawara N. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiol SGM 2000; 146: 1573–1583
- Wagner C, de Saizieu A, Schonfeld H-J, Kamber M, Lange R, Thompson CJ, Page MG. Genetic analysis and functional characterisation of the Streptococcus pneumoniae vic operon. Infect Immun 2002; 70: 6121–6128
- Senadheera MD, Guggenheim B, Spatafora GA, Huang Y-CC, Choi J, Hung DCI, Treglown JS, Goodman SD, Ellen RP, Cvitkovitch DG. A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB and ftf expression, biofilm formation and genetic competence development. J Bacteriol 2005; 187: 4064–4076
- Novak R, Henriques B, Charpentier E, Normark S, Tuomanen E. Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature 1999; 399: 590–593
- Evers S, Courvalin P. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR(B) two-component regulatory system in Enterococcus faecalis V583. J Bacteriol 1996; 178: 1302–1309
- Shankar N, Baghdayan AS, Gilmore MS. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 2002; 417: 746–750
- Nakyama J, Cao Y, Horii T, Sakuda S, Akkermans ADL, de Vos W, Nagasawa H. Gelatinase biosynthesis-activating pheromone: A peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 2001; 41: 145–154
- Qin X, Singh KV, Weinstock GM, Murray BE. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect Immun 2000; 68: 2579–2586
- Qin X, Singh KV, Weinstock GM, Murray BE. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J Bacteriol 2001; 183: 3372–3382
- Nakayama J, Chem SM, Oyama N, Nishiguchi K, Azab EA, Tanaka E, Kariyama R, Sonomoto K. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheremone propeptide corresponding to staphylococcal AgrD. J Bacteriol 2006; 188: 8321–8326
- Mylonakis E, Engelbert M, Qin X, Sifri CD, Murray BE, Ausubel FM, Gilmore MS, Calderwood SB. The Enterococcus faecalis fsrB gene, a key component of the fsr quorum-sensing system, is associated with virulence in the rabbit endophthalmitis model. Infect Immun 2002; 70: 4678–4681
- Engelbert M, Mylonakis E, Ausubel FM, Calderwood SB, Gilmore MS. Contribution of gelatinase, serine protease, and fsr to the pathogenesis of Enterococcus faecalis endophthalmitis. Infect Immun 2004; 72: 3628–3633
- Zeng J, Teng F, Murray BE. Gelatinase is important for translocation of Enterococcus faecalis across polarized human enterocyte-like T84 cells. Infect Immun 2005; 73: 1606–1612
- Hancock LE, Perego M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol 2004; 186: 5629–5639
- Carniol K, Gilmore MS. Signal transduction, quorum-sensing, and extracellular protease activity in Enterococcus faecalis biofilm formation. J Bacteriol 2004; 186: 8161–8163
- Mohamed JA, Murray BE. Influence of the fsr locus on biofilm formation by Enterococcus faecalis lacking gelE. J Med Microbiol 2006; 55: 1747–1750