Abstract
The Amino acid-Polyamine-Organocation (APC) superfamily is the main family of amino acid transporters found in all domains of life and one of the largest families of secondary transporters. Here, using a sensitive homology threading approach and modelling we show that the predicted structure of APC members is extremely similar to the crystal structures of several prokaryotic transporters belonging to evolutionary distinct protein families with different substrate specificities. All of these proteins, despite having no primary amino acid sequence similarity, share a similar structural core, consisting of two V-shaped domains of five transmembrane domains each, intertwined in an antiparallel topology. Based on this model, we reviewed available data on functional mutations in bacterial, fungal and mammalian APCs and obtained novel mutational data, which provide compelling evidence that the amino acid binding pocket is located in the vicinity of the unwound part of two broken helices, in a nearly identical position to the structures of similar transporters. Our analysis is fully supported by the evolutionary conservation and specific amino acid substitutions in the proposed substrate binding domains. Furthermore, it allows predictions concerning residues that might be crucial in determining the specificity profile of APC members. Finally, we show that two cytoplasmic loops constitute important functional elements in APCs. Our work along with different kinetic and specificity profiles of APC members in easily manipulated bacterial and fungal model systems could form a unique framework for combining genetic, in-silico and structural studies, for understanding the function of one of the most important transporter families.
Introduction
Recent studies have described the crystal structures of four bacterial transporters, which despite the fact that they belong to evolutionary distinct protein families with no primary amino acid sequence similarity and different substrate specificities, share a similar structure. These carriers are the amino acid transporter LeuT Citation[1], a member of the of Neurotransmitter Sodium Symporter (NSS) family; the benzylhydantoin Mhp1 transporter Citation[2], a member of the Nucleobase Cation Symporter (NCS1) family 1; the galactose transporter vSGLT Citation[3], a member of the Solute Sodium Symporters (SSS) family; and the glycine-betaine permease BetP Citation[4], a member of the Betaine Choline Carnitine transporter (BCCT) family. All four newly described transporters have a very similar structural core consisting of two V-shaped domains (with 5 transmembrane domains [TMD] each) intertwined in an antiparallel topology. In all cases, a substrate binding site is located in a nearly identical position formed by amino acid residues at and near the unwound part of two broken helices, which are the first helices from each 5-TMD domain. In LeuT and Mhp1 the substrate binding broken helices correspond to TMD1 and TMD6, in vSGLT to TMD2 and TMD7, and in BetP in TMD3 and TMD8. The ion binding sites necessary for energy coupling map close to substrate binding site, but their position in the different transporters is more variable. In these proteins, two extra TMDs, which are not part of the inverted 5-TMD repeat, are N- or/and C-terminally located and seem to be involved in functions other than transport, possibly protein trafficking, stability or regulation (for a comment see Reference Citation[5]). The structure of a second member of the NSS family, the Pyrococcus horikoshii glutamate transporter Gltph, has common features with the above carriers, as far as it concerns the functional role of the unwound portion of its TMD7 and TMD8 segments Citation[6].
It is relevant that the structures determined for each protein represent four different conformations: ‘Outward-facing open’ (LeuT, Mhp1); ‘outward-facing occluded’ (LeuT, Mhp1); ‘intermediate, occluded from both sides’ (BetP); and ‘inward-facing occluded’ (vSGLT). As the deduced structures of the four transporters have identical topologies, the different conformations can be interpreted as specific, static junctures of one and the same dynamic structure Citation[2], Citation[4], Citation[7], Citation[8]. A fifth conformation corresponding to an ‘inward-facing open’ structure could be readily hypothesized. This creates a sequence of snapshots that previews the protein movements associated with transport catalysis. This almost complete sequence Citation[5] supports an alternating-access model similar, but not identical, to that proposed and experimentally supported for members of the Major Facilitator Superfamily (MFS), such as the lactose permease LacY or the glycerol permease GlpT Citation[9–11]. More specifically, the different conformations of the 5-TMD repeat transporters indicate that the switching motion between the two low-energy open states might be slow involving an additional low-energy intermediate state resulting from the concerted movement of TMDs around the substrate and ion binding sites, rather than an abrupt rocker-switch mechanism Citation[2], Citation[4], Citation[7], Citation[9–11]. Furthermore, the occluded and open version of these structures also support the existence of gating domains or dynamic filters in both the outward-facing and inward-facing conformations as a mechanism to add additional specificity or to prevent the leakage of substrates outwards Citation[5].
Using an in silico method (MemGen), which uses hydropathy profile alignments to predict structural similarity of membrane proteins not related in sequence, Lolkena and Slotboom Citation[12] very recently proposed that the SSS and NSS families are indeed similar in structure, supporting the crystallographic studies. The same method also predicts the same structural core for transporters of the Amino acid-Polyamine-Organocation (APC) Superfamily and one of the largest families of secondary transporters for which no crystal structure is available. The APC superfamily is the main family of amino acid transporters found in all domains of life including members that function as solute/cation symporters and solute/solute antiporters Citation[13]. They are found in bacteria, archaea, fungi, eukaryotic protists, plants and animals. Most APCs are predicted to comprise 12 transmembrane α-helical domains (TMD). Some homologues of APC transporters in Saccharomyces cerevisiae proved to function as sensors of amino acids rather than transporters Citation[14]. Mammalian APCs are implicated in several amino acid transport-related processes including neuroadaptation Citation[15], redox imbalance Citation[16], nitrous oxide synthesis Citation[17], macrophage activation and proliferation Citation[18], growth and survival of cancer cells Citation[19], primary inherited cystinuria and lysinuric protein intolerance Citation[20–22], or act as virus receptors, such as the ecotropic retroviral leukemia receptor (ERR), the human retroviral receptor (HRR), the T-cell early activator (Tea), and the Kaposi sarcoma-associated herpesvirus receptor Citation[23], Citation[24]. Low-resolution (6.5 Å) transmission electron microscopy (TEM) data on SteT (serine/threonine) and AdiC (arginine/agmatine), two detergent-solubilized prokaryotic antiporters, gave moderate structural information on APC transporter superfamily Citation[25], Citation[26].
In this article, by using sensitive homology threading, comparative modelling and mutational evidence concerning members of the APC family, we tentatively identify the amino acid binding site and other functional elements in this family of carriers. Our observations also lead to predictions concerning residues that might finely determine the specificity of these permeases towards different amino acids. When this manuscript was under revision the crystal structure of the AdiC antiporter became available, strongly supporting conclusion appearing herein (see also Note added in proof).
Materials and methods
In silico analysis, homology threading and modelling
Sequences were collected from NCBI (http://www.ncbi.nlm.nih.gov) and aligned using http://bioinfo.genotoul.fr/multalin/multalin.html program. Homology threading was performed using the HHPred toolkit program Citation[27]. The following APC protein sequences were used as inputs: PrnB, Can1p, Gap1p, GabP, PheP, AroP, CadB, AdiC, xCT. In all cases, best scores were obtained with the Mhp1 and LeuT structures, and recently published AdiC structure (see Note in proof). Structural models of APCs were constructed based on the Mhp1 (2JLN), the LeuT (2A65/3F3A) and the AdiC (3H6B) structures (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html), using the MOE (2004.03) molecular modelling suite (http://www.chemcomp.com). Multiple Alignments were performed with a hierarchical clustering program (http://bioinfo.genotoul.fr/multalin/multalin.html).
Construction of PrnB mutant strains
Mutations Gly56Ala, Gly56Ser, Thr57Ser and Thr57Ala were constructed by oligonucleotide directed mutagenesis on plasmid pA4 Citation[35], which is a version of plasmid pBC9 containing the prnB open reading frame fused in-frame with the gfp open reading frame via a specific four amino acid linker Citation[35].
The forward oligonucleotide primers used for mutagenesis were:
G56A: 5′CATCGGAGGATGTATTGCCACTGGTCTCTTCGTCG3′,
G56S 5′CCATCGGAGGATGTATTAGCACTGGTCTCTTCGTC3′,
T57A 5′CGGAGGATGTATTGGCGCTGGTCTCTTCGTCGG3′, and
T57S 5′CGGAGGATGTATTGGCAGTGGTCTCTTCGTCGG3′.
The reverse ologonucleotides were complementary to the forward primers. Mutations were confirmed by sequencing (ABI 310 Genetic Analyser at the Institute of Biology, NCSRD, Athens, Greece). A direct method for integrating prnB alleles into the prnB genomic locus has been described in Citation[35]. Briefly, upon transformation Citation[53] of the mutant strain prn397, which carries a large deletion extending from within the prnB open reading frame to within the neighbouring prnC open reading frame, with linearized pA4 plasmids containing the prnB alleles, transformants are isolated on proline as the sole nitrogen source. Strain prn397 lacks both PrnB and PrnC (L-D1-pyrroline-5-carboxylate dehydrogenase) activities and does not grow at all on media containing proline as a sole nitrogen source, as the absence of PrnC results in strong proline toxicity. Integration of the mutated prnB alleles in the prnB locus was confirmed by Southern blot analysis and PCR Citation[35], Citation[54]. Confocal laser microscopy (Bio-Rad MRC 1024, Laser Sharp Version 3.2 Bio-Rad Software) was used to verify cellular expression of PrnB alleles. Samples were prepared as described previously Citation[55]. All four mutants were normally expressed in the plasma membrane of germlings and young hyphae.
Functional analysis of PrnB mutants
Minimal (MM) and Complete (CM) media and growth conditions for Aspergillus nidulans have previously been described Citation[56]. The novel mutant strains were tested on their ability to grow on L-proline (10 mM) as sole nitrogen source under various pH and temperature conditions. In all conditions tested, mutants, G74S and T75A, scored as prnB− strains, whereas mutants G74A and T75S scored as prnB+ strains. Uptake studies, performed as previously described Citation[33], confirmed that mutants G74S and T75A have no PrnB-mediated proline transport, whereas mutants G74A and T75S have nearly wild-type transport kinetics (see text).
Results and discussion
APC carriers are structurally similar to the ‘5-TMD antiparallel repeat’ transporters
We carried out a homology detection and structure prediction search using the HHpred toolkit Citation[27] using selected members of the APC family. The selected proteins were chosen to represent prokaryotic and eukaryotic transporters of different specificities for which mutational analyses or low-resolution models are available (; Citation[28–34]. The scores obtained are shown in Supplementary Table S1 (online version only). All APC members analyzed showed high structural similarity (>92.4% probability, E-value:>0.47, p-value:>2.4×10−5) with Mhp1, LeuT and BetP (results not shown). All of them gave highest scores with the Mhp1 benzyl-hydantoin transporter (>99.6% probability, E-value:>3.3×10−17, p-value:>1.7×10−15), but the local structural similarity in the domains involved in substrate binding (see below) was highest with the LeuT amino acid transporter. As a control for the sensitivity of HHpred toolkit we also ran the Mhp1 and LeuT sequences, which showed, as expected, total structural identity with themselves (100% probability) and very high similarity with each other (98.1% probability, E-value: 0.00026, p-value: 1.3×10−8). This showed that APCs are equally similar in structure to Mhp1 and LeuT, to the similarity shared in LeuT and Mhp1.
Table I. APC transporters with mutational analyses.
shows as an APC example, the predicted structure of PrnB, the major proline transporter of A. nidulans, modelled on LeuT. A very similar structure was obtained when PrnB was modelled on the Mhp1 structure (not shown). Similar structures were also obtained with Can1p, PheP, CadB, GabP and xCT (See ; results not shown).
Figure 1. Predicted 3D structure of PrnB modelled on the inward-facing occluded crystal structure of leuT (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html;2A65/3F3A). Left: Ribbon representation of the LeuT Leucine transporter from Aquifex aeolicus (RCSB entry: 2A65, solved at 1,65 A). Middle: The predicted 3D structure of PrnB. Domains involved in substrate binding and transport are highlighted in yellow (TMD1), blue (TMD3), orange (TMD6) and turquoise (TMD8). Functional loops L2 and L8 are in magenta and black respectively. Right: The PrnB model superimposed with its LeuT template. The LeuT template is in red, whereas the PrnB model is in green colour. This Figure is reproduced in colour in Molecular Membrane Biology online.
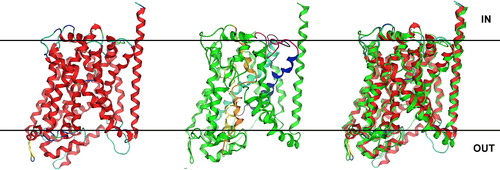
The overall architecture of APC proteins reveals 12 membrane-spanning helices ( and ), as was previously predicted on the basis of hydropathy profiles and determined employing fusions with topological reporters. In fact, the previously published two-dimensional predictions are very similar to the result obtained herein using the homology threading approach Citation[31], Citation[35], Citation[36]. Only the boundaries of TMDs need to be moderately revised, particularly for TMD6 and especially TMD8, which are significantly longer (approximately 10–18 amino acid residues towards the cytoplasmic side of the permeases) than predicted. More importantly, 3D modelling showed that the 12 TMDs are arranged in two intertwined V-shaped repeating units (TMDs 1–5 and TMDs 6–10) connected by a relatively long loop (21–29 residues long), followed by two additional helices (TMD11 and 12). This structural core is almost identical to that of LeuT () and Mhp1 (not shown).
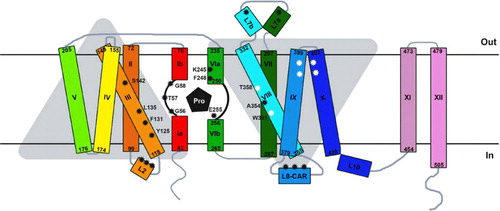
Figure 2. Schematic two-dimensional topology of PrnB in the plane of the membrane. The Figure is based on the corresponding 3D model shown in . Amino acid residues important for substrate binding and transport are (TMD1, TMD3, TMD6 and TMD8) are shown (black asterisks). Among those, G56, T57, G58 in TMD1 and F245 in TMD6 are nearly absolutely conserved in APCs (see ). K245 is uniquely conserved in proline transporters. Non-annotated black asterisks denote approximate positions of other functionally important residues in L2 and L8 in several APCs. White asterisks in TMD8, TMD9, TM10 denote residues important for function, mostly in PheP. This Figure is reproduced in colour in Molecular Membrane Biology online.
Mutational evidence supports the involvements of TMD1, TMD3, TMD6 and possibly TMD8, in substrate binding in APCs
Several bacterial and fungal APC transporters have been identified and studied in detail at the level of transport kinetics, specificity and regulation of expression. These transporters may be specific for one or more L amino acids and their kinetics of transport and regulation of expression may also vary significantly Citation[28], Citation[29]. In addition, mutational analyses are extant for several of them, such as PheP, AroP, GabP, PrnB and Can1p. Mutational analyses have also been carried out for the human xCT and the bacterial CadB antiporters (for references see ). Although randomly selected or designed mutations affecting the function of APC transporters map in various segments in different proteins, five regions seem of primary importance. These, based on the homology threading approach presented herein, are predicted to be TMD1, the cytoplasmic loop between TMD2-TMD3 (L2), TMD3, TMD6 and the cytoplasmic loop between TMD8-TMD9 (L8). A limited number of interesting functional mutation maps also in TMD8, TMD9 and TMD10, and in loops L1, L6, L7.
In the models created here (see and ), TMD1 and TMD6 of APCs correspond to the two partially ‘broken’ helices which form an essential part of substrate and ion binding site, as directly established in the crystal structures of all five 5-TMD antiparallel repeat transporters obtained in the presence of their substrates. The short, unwound, and thus flexible, segment of each of these two TMDs not only hosts the main residues interacting directly with substrates and the symported ion, but in addition allows key residues from other domains (TMD3 and TMD8 in Mhp1, LeuT and possibly APCs; see later) to interact with substrates Citation[7]. Importantly, the proposed direct involvement of TMD6 and TMD8 in substrate/cation binding and translocation in the Mhp1 carrier is nicely supported by mutational data in the homologous Fcy2p permease of S. cerevisiaeCitation[37].
Mutational evidence for a direct role of TMD1 in the formation of the substrate and ion biding site in APCs existed only for PheP. In this permease, a series of Glycine residue (Gly35, Gly38, Gly40, Gly44) within TMD1 (see ) are critical for function, but not for insertion of the protein in the plasma membrane Citation[38], Citation[39].
Among those, Gly38 is absolutely conserved in APCs, Gly40 is nearly absolutely conserved, whereas the other two are less conserved. Gly38 lies in the short segment of two to three amino acids (residues 37–39) that ‘breaks’ the TMD1 helix. Second-site suppressors of primary mutations in Gly38 or Gly 44 map, reciprocally, to the same residues, and led to transporter versions with partially restored activity for phenylalanine, but not for tyrosine. Suppressors of Gly35 or Gly40 mutations mapped in TMD3 or TMD2 and very close to the cytoplasmic border of TMD8 with L8, respectively. Among those, only suppressors in TMD2 (substitutions of Tyr60) and TMD3 (substitutions of Tyr107) restore activity for both substrates of PheP (phenylalanine and tyrosine), whereas suppressors in TMD8/L8 (e.g., Ser323Phe) only lead to phenylalanine transport activity. These results suggest that at least Gly38 at the broken segment of TMD1 and the following Gly40 are major residues for PheP function, probably by interacting directly with substrates. This is further supported herein, as we show that Gly40 in PheP corresponds to a Gly residue shown in the LeuT structure to make most of the contacts with amino acid substrates (see later and ). The genetic interaction of Gly35 with Tyr107 supports the proposed molecular synergy of TMD1 and TMD3 in substrate binding and transport. Furthermore, the Gly40-Tyr60 interaction supports the close TMD1-TMD2 topological interaction shown in the crystal structures of similar transporters. This latter interaction however, seems to be crucial for structure rather function, as primary mutation in TMD2 affect insertion in the plasma membrane and several combinations of different amino acids in these positions fully restore PheP transport activity. The significance of suppressors of TMD1 mutations mapping in L8 will become apparent later.
Table II. Functional residues in PrnB (APCs) corresponding to residues involved in substrate binding in Mhp1 and LeuT.
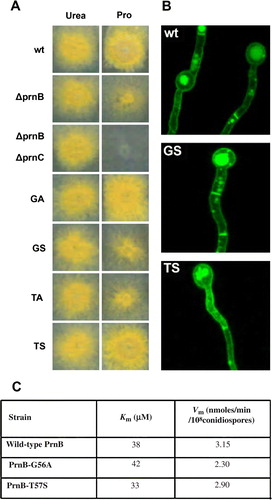
To further investigate the general role of TMD1 in APC function, we mutated by standard reverse genetic approaches residues Gly56 and Thr57 of PrnB Citation[35], and analyzed the kinetic behaviour of the corresponding PrnB versions. These residues are both absolutely conserved in APC members and Gly56 defines the short break in TMD1 (equivalent to Gly38 in PheP) (). All mutated versions of PrnB constructed (Gly56Ala, Gly56Ser, Thr57Ala, Thr57Ser) were normally expressed in the plasma membrane of A. nidulans, as this was evidenced using a functional PrnB-GFP fusion (). Growth tests on media containing L-proline as the sole nitrogen source showed that the conservative changes Gly56Ala and Thr57Ser do not affect significantly the function of PrnB. Uptake studies using radiolabelled proline confirmed that these mutants conserve nearly wild-type affinity and transport capacity for proline. In contrast substitutions Gly56Ser and Thr57Ala led to total loss of PrnB activity as this was shown with growth tests and uptake studies. These results, shown in A and C, clearly establish a crucial role of Gly56 and Thr57 in PrnB function and, together with results on PheP, constitute strong genetic evidence for the functional role of the segment around the short unwound segment of TMD1 in APCs. How these absolutely conserved residues are involved in the recognition or/and transport of several amino acids by different APC members is discussed later.
Figure 3. Functional characterization of novel PrnB mutants in TMD1. (A) Growth tests on proline, (B) Cellular expression, and (B) Kinetic profile. In (A) notice the leaky growth of a ΔprnB control strain due to the existence of a secondary proline transporter, compared to the total absence of growth in a strain also lacking the ability to catabolize proline (ΔprnB ΔprnC). Mutants Gly56Ser and Thr57Ala (GS, TA) score similar to ΔprnB, whereas mutants Gly56Ala and Thr57Ser (GA, TS) score similar to a prnB+ (wt) strain. In (B), apparent loss-of-function PrnB alleles Gly56Ser and Thr57Ala show normal expression in the plasma membrane, septa and vacuoles. In (C), functional PrnB alleles Gly56Ala and Thr57Ser show nearly wild-type kinetics of proline transport. This Figure is reproduced in colour in Molecular Membrane Biology online.
Re-evaluation of PrnB, Bap2p, Hip1p and PheP (see ) mutations strongly supported the prediction that residues in TMD6 of APCs are also part of the substrate and ion binding site. PrnB mutations concerning residues Lys245 and Phe248, which map one or two helix turns upstream from the unwound segment in TMD6 (see ) have been shown to affect specifically the affinity of PrnB for proline Citation[35]. Replacement of the equivalent Phe residue by a Ser in Bap2p and Hip1p led to transporters with reduced amino acid uptake capacity, resistant to low pH and with the novel ability to take up K+ ions Citation[40], Citation[41]. Furthermore, changes of the equivalent Phe220 in PheP are able to suppress primary mutations concerning the critical residue Arg317 in TMD8 Citation[38] (see below). This Phe residue is nearly absolutely conserved in the APC family suggesting that it might be critical for substrate, as well as, ion binding. In contrast, Lys245 of PrnB is only conserved in Put4p, the only other APC transporters specific for proline, suggesting that this amino acid is directly involved in determining the specificity of these carriers (see also later). More mutational evidence for a functional role of the broken part of TMD6 comes from PheP. Several substitutions of Glu226, located just at the end of the unwound segment in TMD6, lead to total loss of transport activity Citation[42]. A second Glu residue (Glu234) at the border of TMD6 with the following cytoplasmic loop L6 has also been shown to be important for PheP function. Interestingly, it has been postulated Citation[38] and supported by the current model that Glu234 might interact with Arg317 in TMD8. Of significant interest is also the reported finding that effective second-site suppressors of mutations in either of these residues involves changes to Met116, closely linked to Glu118 in TMD3, implicated in the substrate binding pocket. A few other severe loss-of-function mutations in PheP map, besides TMD1, TMD3, TMD6 and TMD8, in TMD5 (Lys168) and TMD9 (Pro341) Citation[42], Citation[46]. Further evidence for the importance of TMD6 is also reflected in the observation that one of the most severe mutations, reducing 22-fold the affinity of CadB for its substrates, also concerns the same Glu residue at the end of the unwound region of TMD6 (Glu204) Citation[33]. Most of the other CadB loss-of-function mutations map in L2 and L8 (see later).
Figure 4. Alignments of functional TMDs and loops. In bold residues that proved, by mutational analysis, important for function at least in more than two APCs. Also in bold, the consensus sequences of TMDs and loops. Consensus sequences and unwound regions in TMD1 and TMD6 are highlighted in grey. Also in grey highlight are the specific residues differentiating the two proline permeases (PrnB and Put4p). Numbering of amino acids is shown on the right and on the left in each protein segment. Sequence accession numbers are shown in . Other accession number are: Put4p:NP_014993; Lyp1p: NP_014131; Tat1P: NP_009625; Agp1p: NP_009905; Dip5p:NP_015058; LysP: AAA17053; AgtA: ACC77607.

The functional role of TMD3 (see ) in APCs is supported not only through suppressor selection of TMD1 mutations Citation[38], Citation[39], but also with AroP-PheP chimeric proteins and primary functional mutations in PheP Citation[42], Citation[43] and PrnB Citation[35] While mutations in TMD3 of PheP (Glu118) or AroP-PheP chimeras (Ala107, Val114) can affect transport activities, what is mostly interesting is that mutations in TMD3 could also alter the specificity of these proteins. A key residue in determining specificity was found to be Tyr103 in AroP or Phe111 in PheP. For example, substitution Phe111Tyr enables PheP to transport tryptophan. The same studies also showed that other residues in TMD3 may also contribute to specificity. This is in excellent agreement with the structural proximity of TMD3 to the unwound region of TMD1, as shown in . What is also important is that none of the TMD3 mutations, similarly to TMD1 mutations, and unlike TMD2 mutations, affects folding and insertion of PheP or AroP in the plasma membrane. In PrnB, Ile119Asn and Gly12Trp at the border of TMD3 with the following cytoplasmic loop (L2) were genetically selected as severe loss of function mutations. Finally, further evidence for the role of TMD3 in substrate binding comes from the genetic identification of residues enlarging the substrate specificity of both Can1p and Gnp1 Citation[44]. In particular, substitutions of Tyr173 (Can1) and Trp239 (Gnp1) rescue the inability of a gap1Δ strain to take up citrulline. In the same suppressor screen, all other mutations restoring citrulline uptake were exclusively obtained in Can1p and concern residues in L1, L2, L6, L7 and TMD10. Among those suppressors two, concerning the conserved Pro148 and Pro313 residues, which map in L2 and L6, have the broader effect on Can1p specificity (discussed later).
Evidence for a critical TMD8 () in APC function comes from the observation that both Tyr313 and Arg317 have been shown to be critically important for PheP function. As already discussed above, extensive second-site suppression studies of these residues have led to frequent changes in Met116, but also in other TMD3 residues located towards the periplasmic side of this domain, supporting a synergistic function of TMD3 and TMD8 Citation[38]. Second-site suppressors of TMD8 mutations in PheP also led to the isolation of mutations clustered in the periplasmic side of TMD9 and TMD10 Citation[38]. Interestingly, TMD10 in Mhp1 and vSGLT was shown to be part of the main outward-facing cavity formed principally by TMD1, TMD3, TMD6 and TMD8, and more importantly, its periplasmic part seems to be part of a gating mechanism through bending into the outward-facing cavity and thus occluding substrate escape outwards Citation[2]. Cys-scanning mutagenesis further supported a functional role of TMD8 in substrate or ion binding in APCs Citation[45]. This powerful approach has shown that Cys327 in xCT is accessible to modification, but inactivation is protected by substrates. Furthermore, Cys327 substitutions (C327S and C327A) increase glutamate Km two-fold, suggesting. Besides Cys327, Ala337 is another residue in TMD8 of xCT that seems to affect function (50% reduction in transport capacity).
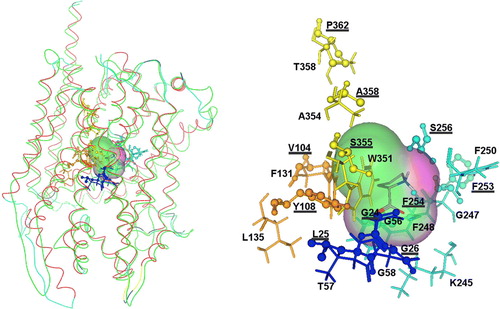
Based on the above conclusions, highlights the predicted substrate (proline) binding site of an APC transporter (PrnB) folded and superimposed on LeuT.
Figure 5. Left: Interactions of APCs with amino acid substrates and the PrnB paradigm. Superimposition of the LeuT (red line) and PrnB (green line) structures and predicted interactions with proline. Right: Substrate interacting residues are shown in balls and sticks representation for LeuT and as sticks representation for PrnB. Residues taken from the X-ray structure of LeuT (rcsb code: 2A65) are underlined. Moreover all amino acids involved in substrate binding () are colour-coded according to the conventions of (TMD1 blue; TMD3 orange; TMD6 turquoise; TMD8 yellow). This Figure is reproduced in colour in Molecular Membrane Biology online.
Loops as key functional elements in APC transporters: The prominent role of L2 and L8
Specific loops connecting the membrane-embedded segments of a protein should, in principle, be responsible for the dynamic structural alterations of the transporter body associated with drastic movements of several TMDs in order to create the outward-facing, the intermediate, or the inward-facing conformations. In addition, re-entrant or flexible loops might directly act as components of gating mechanisms both at the outward- and inward-facing conformers. The solved structures of LeuT, Mhp1 and other similar transporters have indeed shown that substantial helical and loop segments on the solvent-exposed surfaces of these carriers interact with TMDS and might have functional role in substrate recognition and transport Citation[1], Citation[2]. Between TMD2 and TMD3, a re-entrant loop (L2) including a helical part occludes the intracellular face of LeuT. Between TMD7 and TMD8, L7 forms a broken helix which makes numerous contacts with TMD1. Loops that make contacts with TMDs are also formed in all similar transporters.
Two cytoplasmic loops, L2 and L8, proved particularly important for APC function (see and ). Most loss-of-function L2 and L8 mutations do not effect insertion to the plasma membrane which in turn means that they do not affect folding. Interestingly, L2 and L8 in different APC proteins, unlike several TMDs, do not show any significant primary amino acid sequence similarity. The importance of L2 becomes apparent from the observation that in several mutational studies on APCs (e.g., Can1p, Gnp1p, CadB, xCT) functional or specificity mutations have been repeatedly identified within this region. Furthermore, studies of biotinylation and accessibility to sulfhydryl reagents in the human xCT carrier revealed that L2 is a putative re-entrant loop and His110, which is located at the apex of this loop, should lie close to the substrate binding or translocation pathway Citation[45]. This strongly suggests that L2 could be part of a cytoplasmic-facing gating mechanism, similar to the one proposed for the transporters with solved structures. If so, mutations in L2 might affect transport either positively (loss or relaxed of gating in the inward-facing conformer) or negatively (permanent or increased occlusion in the inward-facing conformer), but at the same time, L2 mutants might conserve wild-type substrate or ligand binding constants. This is exactly the case in xCT (e.g., His110Cys, increased transport; Citation[45]) and in PrnB (e.g., Gly120Trp, reduced transport; Citation[35]) carriers.
The importance of L8 has also been underlined by missense mutations or insertions or duplications of amino acids in PrnB, PheP and mostly GabP Citation[32], Citation[35], Citation[38], Citation[39], Citation[46–49], which lead to loss of transport function or specificity shifts, but also by the isolation of an L8 mutation suppressing TMD1 mutations in PheP Citation[39]. In the latter case, the mutation in L8 could specifically restore the uptake for one substrate (phenylalanine) but not of another (tyrosine). This region has been also shown to be functionally significant in the mammalian mouse cationic amino acid transporters MCAT Citation[50]. Interestingly, L8 corresponds to a consensus amphipathic region (CAR) shared by many APC transporters, as well as, the non-homologous mammalian GABA transporters in the nervous system Citation[32], Citation[35], Citation[38], Citation[47–49]. The nature of the CAR has suggested that it might be part of the substrate translocation pathway and that substitutions, insertions or duplications of amino acid residues might affect its function or alter its topology relatively to other domains involved in amino acid binding and transport. Similar to L2, loss-of-function mutations in L8 of PrnB Citation[35] or GabP Citation[49] seem to affect substrate transport, without affecting binding constants, suggesting a role in transport per se rather than substrate binding.
On how specificity is determined in APCs
Most APC carriers are specific for one or more amino acids. At one extreme is the case of Gap1p which recognizes all amino acids and several amino acid analogues, and at the other end is PrnB which strictly recognizes L-proline. In addition, some APC members show substrate affinities approaching nM levels, while others recognize amino acids at concentrations close to the mM range. How such differences are determined and how can we approach this question now that modelling provides putative 3D structures of APCs? Basically, specificity and affinity profiles should depend on the sum of interactions a given amino acid can perform with specific amino acids of the APC binding site. This does not exclude more complex situations where specificity also depends on molecular filters of gating domains distinct from the substrate binding site Citation[5], Citation[51]. Anyway, knowing the kinetic and specificity details of several APC permeases plus the putative domains that form the substrate binding pocket, allows us to make important predictions. shows sequence alignments of TMD1, TMD3, TMD6 and TMD8 (the helices that are expected to host the amino acid and ion binding sites) in APC amino acid permeases. TMD1 is the most conserved helix and residues located in its unwound segment are nearly absolutely conserved in all APCs. A consensus motif of the core of TMD1 would be GG/TXIGTGLF, where X is a small, mostly hydrophobic amino acid, and in bold are the nearly absolutely conserved residues of the unwound segment. This exceptional conservation and the presence of Gly residues led to a previous belief that TMD1 might be involved in protein folding through its involvement in key inter-helical interactions, rather than functioning as a substrate binding and specificity determinant. This idea was in line with the observation that substitutions of the Gly residues were critical for APC activity and with genetic evidence (second-site suppressors) showing that TMD1 indeed interacts with other TMDs in PheP (TMD2 and TMD3) Citation[39]. While the interaction of TMD1 with TMD2 was shown to be important for stabilizing PheP, modelling and the analysis of several TMD1 mutations presented herein also support an important role of TMD1 in substrate binding.
The fact that the residues which seem to be involved in substrate binding, such as the Gly and Thr residues in the unwound segment of TMD1, are nearly absolutely conserved in APCs, which however recognize different amino acids as substrates, is only an apparent paradox. An explanation comes from the crystal structure of the LeuT carrier solved in complex with six different amino acid substrates each time. As described above, LeuT is not an APC member but it has an almost identical structural core with APCs Citation[7]. In all LeuT-amino acid crystal complexes, a Gly residue (Gly26) defining the end of the unwound region of TMD1 (equivalent to Gly58 in PrnB or Gly40 in PheP; and ) makes most of the contacts with the α-carboxy groups of the bound amino acid.
The same α-carboxy groups are also bound by Leu25 in TMD1 (equivalent to the essential Thr57 in PrnB, conserved in all APCs; and ), Tyr108 in TMD3 (equivalent to an aliphatic residue important for function in PheP, L135 in PrnB; and ), and Phe254 in TMD6 (equivalent to a crucial Phe in all APCs, F248 in PrnB; and ). One more crucial residue in TMD3 of APCs corresponds to F131 in PrnB (F111 in PheP or Y103 in AroP). The α-amino group of amino acid substrates is also bound by Phe253 and Ser256, two residues close to the unwound segment of TMD6 (residues 258-261). Interestingly, the unwound segment of TMD6 is one amino acid longer uniquely in the specific proline transporters, PrnB and Put4p (). These two permeases are also characterized by the presence of a crucial Lys residue (Lys245 in PrnB) just upstream from the unwound segment of their TMD6, in a position very similar to Phe253 or Phe254 of LeuT. Proline is an imino acid having a secondary instead of primary amino group, and thus, fine differences in the unwound region of TMD6 might have evolved to accommodate this structural modification. The local unwinding of TMD1 and TMD6 is critical as it permits direct hydrogen bonding of substrates with the backbone and side chains of residues lining the binding pocket. The aliphatic side chains of the substrates of LeuT (Leu, Ala, Gly, Met, Tyr and Trp), by contrast to the α substituents, were found to be cradled within a hydrophobic pocket created by side chains of residues in TMD3, TMD6 and TMD8.
A basic idea on how APCs recognize their substrates arises from all observations made herein and is exemplified in using the PrnB proline transporter. Substrates are basically recognized through hydrogen bond interactions between fully or partially conserved residues in the proximity of the unwound regions of TMD1 (Gly56, Thr57, Gly58) and TMD6 (Gly247, F248, F250) and the α-carboxy and α-amino groups of amino acids (see also ). In such interactions, the role of TMD1 seems more rigid as the residues involved are absolutely conserved, whereas those of TMD6 (besides F248) are more variable. Interactions of TMD3 residues (Phe131, Leu135) with α-carboxy groups stabilize binding. Specificity, and probably the affinity, towards different amino acids seems to be determined by variations in interactions with TMD6 and mostly by interactions (Van der Waals or H bonds) of the substrate side chains with variable residues lining the substrate pocket (TMD3: Tyr125, Phe131, Leu135, Ser142; TMD6: Lys245, Gly247, Phe250; TMD8: Trp351, Ala354).
Comparing TMD3, TMD6 and TMD8 of fungal amino acid transporters allows us to predict which particular amino acids may determine specificity. The most obvious differences related to specificity are in or close to the unwound region of TMD6 (see ). Except from those discussed above in respect to the two proline transporters (the presence of a Lys residue and a longer unwound region), we also noticed that very promiscuous members or those transporting aromatic amino acids, such as Gap1p, Agp1p, Bap3p, Gnp1p, Val1p, Tat1p, AroP, PheP have a small flexible residue such as Ala or Gly in the position corresponding to Ser256 in LeuT, which was shown to be involved in binding and to be close to the side chain of substrates (). At the same position, proline transporters have a larger aliphatic residue (Phe or Leu). All transporters specific strictly for basic amino acids (Can1p, Lyp1p, Alp1p, LysP) have a Gln residue, which is always combined with an Asn residue six residues upstream. Hip1p (histidine transporter) and GabP (GABA transporter) have a Ser or Met residue, respectively. The methionine transporter has a Val residue. The acidic amino acid transporters Dip5p and AgtA have a Thr or a Leu residue. In other words, there is an impressive correlation between specificity and the presence of different residues in the unwound part of the TMD6 of APCs. Finally, we also noticed a difference between the two proline permeases. PrnB lacks a Gly residue (replaced by Ser254) at the end of the unwound segment that all other transporters, including Put4p, conserve. This might account for its extreme specificity for L-proline, unlike the situation in Put4p which, besides L-proline, also recognizes proline analogues and GABA Citation[36].
Conclusions
The striking structural resemblance of APC members with several evolutionary distinct transporters with different specificities, despite low similarity in primary amino acid sequence (15–16% identity), provides not only a unique framework for the re-evaluation of previous mutational studies, but also allows the design and analysis of novel mutations to test the static crystal structures. It is important that at present there is no other family, besides APCs, conforming to the 5-TMD repeat structural motif, for which there is a similar wealth of mutational data. The modelling shown herein was obtained using the outward-facing conformations of LeuT or Mhp1, the two structures giving the highest similarity with all APCs tested. The fact that LeuT is an amino acid carrier proved extremely valuable in extrapolating information on how APCs might also recognize amino acids. Our analysis strongly suggests that APCs and LeuT, and possibly other neurotransmitters, utilize similar molecular interactions for recognizing and transporting amino acids.
The present analysis leads to a number of significant observations. First, the tentative substrate binding of APCs, as predicted by modelling, is nicely supported by mutations. What is impressive is that mutations in APCs corresponding to residues shown to interact directly with substrates in the solved structures of structurally similar transporters (), affect function as would be expected for residues involved in substrate binding. More specifically, mutations within or in the vicinity of the unwound segments of TMD1 and TMD6 lead to dramatically reduced APC function compatible with loss of substrate binding or alter substrate specificity. In addition, mutations in TMD6 affect ion selectivity. Mutations in TMD3 and TMD8 also affect substrate binding, specificity and transport. The importance of residues found principally in TMD1, but also in TMD6 and TMD3, in interactions with the α-carboxy and α-amino groups, common in all amino acid substrates, is fully supported by the evolutionary conservation and specific substitutions of amino acid residues in these domains. Specificity and affinity determinants contributing to the unique kinetic profile of each APC homologue are predicted to map in TMD3, TMD6 and TMD8, helices that line the permeation pathway and come into contact with the side chains of different substrates. The particularities of the extremely specific proline transporters and the promiscuity of general amino acid permeases are also predicted to lie on subtle differences in TMD6, and possibly in TMD3 and TMD8.
Very recently, a similar to approach of re-evaluation of mutations, in the light of the structure of LeuT, in two cation-activated amino acid transporters/channels was reported Citation[52]. These carriers are classified in the same family with LeuT (NSS), distinct from the APC family. This study provides further evidence that TMD1 and TMD6 are the domains hosting principal residues involved in substrate and ion binding and transport, whereas residues in TMD3, TMD8 and L7 play auxiliary but significant functional roles. Interestingly, mutations affecting specificity concern partially conserved residues mapping in TMD6, TMD8 or one helix turn upstream from the unwound segment of TMD1 (see below).
This work also provides strong evidence for a functional and specificity role of two cytoplasmic loops. The importance of L2 is also suggested by the recently solved structures. This partially helical segment has been shown to be a re-entrant loop acting as an important gating element from the cytoplasmic side of LeuT, Mhp1 and similar transporters. Mutational evidence from several APC members supported the notion that this domain acts in a similar manner in all 5-TMD repeat carriers. The functional importance of L8, a segment overlapping with an extended amphipathic region (CAR) in APCs is supported genetically. The same loop in other similar carriers is much smaller and it importance is not obvious from the solved structures. We propose that this region might be unique functional element specific for APC carriers. The genetic work on the APCs has also showed that some mutations in other TMDs (TMD2, TMD9 and TMD10) and loops (L1, L6, L7) can also affect function and specificity [44,57 and references therein]. In most cases this is justified from the structural analysis of similar transporters (e.g., L7, TMD10), whereas in some cases it concerns an indirect structural effect (e.g., TMD2). Also in agreement with the structural work, no functional mutation has been assigned to TMD11 and TMD12 of APCs, two domains that are not part of the carrier core catalyzing transport.
Last but not least, what is very important is that all the above conclusions and predictions can be easily tested in model systems such as S. cerevisiae, A. nidulans or E. coli. Rational design of mutations, genetic screens, Cys-scanning mutagenesis, cross-linking and other biochemical approaches, as well as docking studies, can be applied to fully understand how structure defines function and specificity in a very important family of amino acid transporters.
Note added in proof
When this manuscript was under revision the crystal structure (3.6 Å) of the AdiC (PDB 3H6B), a member of the LAT sub-family of the APC antiporter family became available. The overall fold is very similar to that of LeuT, Mhp1, vSGLT or BetP confirming our modelling approach. Homology threading of PrnB and PheP showed extremely similar structures with AdiC (not shown). The reported AdiC structure is in its outward-facing conformation, exposing a conserved acidic pocket to the periplasm, similarly to the LeuT and Mhp1 structures published. The authors also presented functional analysis of mutants corresponding to amino acid substitutions that are proposed to define the essential substrate binding residues and a possible conserved mechanism for antiporter activity. On the whole, structural data on AdiC are in excellent agreement with our observations. The authors emphasize the role of TMD6 in substrate binding and specificity and the role of TMD10 in gating. AdiC residues proposed to interact with substrates, lying deep in the binding pocket, are Asn22 and Ser26 close to the break of TMD1 (equivalent to the essential Gly53/Thr57 in PrnB or Gly35/Thr39 in PheP), Tyr93 in TMD3 (Tyr127 in PrnB or Tyr107 in PheP), the conserved Glu208 in the break of TMD6 (equivalent to the essential Glu226 in PheP or Glu204 in CadB) and Tyr365 in TMD10. The absolutely conserved Glu208 was proposed to act as a pH sensor for all APcs. Evidence for the importance of these residues in substrate binding and transport was obtained with mutations leading to loss of AdiC function. Other potentially important residues lining the surface of the proposed substrate binding pocket are the conserved Gly residues in TMD1, Tyr87 and Tyr101 in TMD3, Gly206, Val207, Ser20, Ala210, Val217 and Asn219 in TMD6, Cys286, Ser289, Leu290, Trp293 in TMD8 and Val358 in TMD10, many of them also predicted as important residues with our approach. A second structure of AdiC at 3.2 A resolution was published during the finalization of acceptance of our manuscript Citation[58]. The protein is captured in an outward-open, substrate-free conformation with a transmembrane architecture essentially similar to that described by Gao et al (2009) Citation[57] and the APC structures modelled in our manuscript. The structural data now available provide new opportunities for speculation and experimental design in easily manipulated model systems which we hope to pursuit for validating and extending ideas on how APC proteins function
Acknowledgements
We thank Dr S. Amillis for help in making Figures and A. Bobori for help in characterizing some of the novel PrnB mutants. We also thank Prof. C. Scazzocchio for critically reading the manuscript. Finally, we thank Dr E. Mikros and members of his lab for their help in modelling. Work at Athens was supported by NCSR ‘Demokritos’ grants to I.V. and D.Vl. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−dependent neurotransmitter transporters. Nature 2005; 437: 215–223
- Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, Carpenter EP, Rutherford NG, Hadden JM, O'Reilly J, Ma P, Saidijam M, Patching SG, Hope RJ, Norbertczak HT, Roach PC, Iwata S, Henderson PJ, Cameron AD. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 2008; 322: 709–713
- Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na + /sugar symport. Science 2008; 321: 810–814
- Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature 2009; 458: 47–52
- Diallinas G. Biochemistry. An almost-complete movie. Science 2008; 322: 1644–1645
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004; 431: 811–818
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008; 322: 1655–1661
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci USA 2008; 105: 10338–10343
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003; 301: 610–615
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003; 301: 616–620
- Zhou Y, Guan L, Freites JA, Kaback HR. Opening and closing of the periplasmic gate in lactose permease. Proc Natl Acad Sci USA 2008; 105: 3774–3778
- Lolkema JS, Slotboom DJ. The major amino acid transporter superfamily has a similar core structure as Na + -galactose and Na + -leucine transporters. Mol Membr Biol 2008; 25: 567–570
- Saier MH, Jr. Families of transmembrane transporters selective for amino acids and their derivatives. Microbiology 2000; 146: 1775–1795
- Forsberg H, Ljungdahl PO. Sensors of extracellular nutrients in Saccharomyces cerevisiae. Curr Genet 2001; 40: 91–109
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 2003; 6: 743–749
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T, Takahashi S, Bannai S. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 2005; 280: 37423–37429
- Nicholson, B, Manner, CK, Kleeman, J, MacLeod, CL. 2001. J Biol Chem, 276: 15881–15885.
- Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, McLeod C, Palacín M, Modolell M, Lloberas J, Celada A. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J Immunol 2006; 176: 5918–5924
- Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime?. Semin Cancer Biol 2005; 15: 254–266
- Calonge MJ, Gasparini P, Chillaron J, Chillon M, Gallucci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Di Silverio F, Barcelo P, Estivill X, Zorzano A, Nunes V, Palacin M. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cysteine. Nat Genet 1994; 6: 420–425
- Torrents D, Mykkänen J, Pineda M, Feliubadaló L, Estévez R, de Cid R, Sanjurjo P, Zorzano A, Nunes V, Huoponen K, Reinikainen A, Simell O, Savontaus ML, Aula P, Palacín M. Identification of SLC7A7, encoding y + LAT-1, as the lysinuric protein intolerance gene. Nat Genet 1999; 21: 293–296
- Borsani G, Bassi MT, Sperandeo MP, De Grandi A, Buoninconti A, Riboni M, Manzoni M, Incerti B, Pepe A, Andria G, Ballabio A, Sebastio G. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet 1999; 21: 297–301
- Reizer J, Finley K, Kakuda D, MacLeod CL, Reizer A, Saier MH, Jr. Mammalian integral membrane receptors are homologous to facilitators and antiporters of yeast, fungi, and eubacteria. Protein Sci 1993; 2: 20–30
- Kaleeba JA, Berger EA. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: Cystine transporter xCT. Science 2006; 311: 1921–1924
- Reig N, del Rio C, Casagrande F, Ratera M, Gelpí JL, Torrents D, Henderson PJ, Xie H, Baldwin SA, Zorzano A, Fotiadis D, Palacín M. Functional and structural characterization of the first prokaryotic member of the L-amino acid transporter (LAT) family: A model for APC transporters. J Biol Chem 2007; 282: 13270–13281
- Casagrande F, Ratera M, Schenk AD, Chami M, Valencia E, Lopez JM, Torrents D, Engel A, Palacin M, Fotiadis D. Projection structure of a member of the amino acid/polyamine/organocation transporter superfamily. J Biol Chem 2008; 283: 33240–33248
- Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005; 21: 951–960
- Sophianopoulou V, Diallinas G. Amino acid transporters of lower eukaryotes: regulation, structure and topogenesis. FEMS Microbiol Rev 1995; 16: 53–75
- Regenberg B, Düring-Olsen L, Kielland-Brandt MC, Holmberg S. Substrate specificity and gene expression of the amino-acid permeases in Saccharomyces cerevisiae. Curr Genet 1999; 36: 317–328
- Pi J, Pittard AJ. Topology of the phenylalanine-specific permease of Escherichia coli. J Bacteriol 1996; 178: 2650–2655
- Cosgriff AJ, Pittard AJ. A topological model for the general aromatic amino acid permease, AroP, of Escherichia coli. J Bacteriol 1997; 179: 3317–3323
- Hu LA, King SC. Functional sensitivity of polar surfaces on transmembrane helix 8 and cytoplasmic loop 8–9 of the Escherichia coli GABA (4-aminobutyrate) transporter encoded by gabP: Mutagenic analysis of a consensus amphipathic region found in transporters from bacteria to mammals. Biochem J 1998; 330: 771–776
- Soksawatmaekhin W, Uemura T, Fukiwake N, Kashiwagi K, Igarashi K. Identification of the cadaverine recognition site on the cadaverine-lysine antiporter CadB. J Biol Chem 2006; 281: 29213–29220
- Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martín R, Zorzano A, Borsani G, Palacín M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc-. Pflugers Arch 2001; 442: 286–296
- Tavoularis SN, Tazebay UH, Diallinas G, Sideridou M, Rosa A, Scazzocchio C, Sophianopoulou V. Mutational analysis of the major proline transporter (PrnB) of Aspergillus nidulans. Mol Membr Biol 2003; 20: 285–297
- Gilstring CF, Ljungdahl PO. A method for determining the in vivo topology of yeast polytopic membrane proteins demonstrates that Gap1p fully integrates into the membrane independently of Shr3p. J Biol Chem 2000; 275: 31488–31495
- Ferreira T, Brèthes D, Pinson B, Napias C, Chevallier J. Functional analysis of mutated purine-cytosine permease from Saccharomyces cerevisiae. A possible role of the hydrophilic segment 371–377 in the active carrier conformation. J Biol Chem 1997; 272: 9697–9702
- Pi J, Chow H, Pittard AJ. Study of second-site suppression in the pheP gene for the phenylalanine transporter of Escherichia coli. J Bacteriol 2002; 184: 5842–5847
- Dogovski C, Pi J, Pittard AJ. Putative interhelical interactions within the PheP protein revealed by second-site suppressor analysis. J Bacteriol 2003; 185: 6225–6232
- Wright MB, Ramos J, Gomez MJ, Moulder K, Scherrer M, Munson G, Gaber RF. Potassium transport by amino acid permeases in Saccharomyces cerevisiae. J Biol Chem 1997; 272: 13647–13652
- Farcasanu IC, Mizunuma M, Hirata D, Miyakawa T. Involvement of histidine permease (Hip1p) in manganese transport in Saccharomyces cerevisiae. Mol Gen Genet 1998; 259: 541–548
- Pi J, Wookey PJ, Pittard AJ. Site-directed mutagenesis reveals the importance of conserved charged residues for the transport activity of the PheP permease of Escherichia coli. J Bacteriol 1993; 175: 7500–7504
- Cosgriff AJ, Brasier G, Pi J, Dogovski C, Sarsero JP, Pittard AJ. A study of AroP-PheP chimeric proteins and identification of a residue involved in tryptophan transport. J Bacteriol 2000; 182: 2207–2217
- Regenberg B, Kielland-Brandt MC. Amino acid residues important for substrate specificity of the amino acid permeases Can1p and Gnp1p in Saccharomyces cerevisiae. Yeast 2001; 18: 1429–1440
- Gasol E, Jiménez-Vidal M, Chillarón J, Zorzano A, Palacín M. Membrane topology of system xc- light subunit reveals a re-entrant loop with substrate-restricted accessibility. J Biol Chem. 2004; 279: 31228–31236
- Pi, J, Dogovski, C, Pittard, AJ. Functional consequences of changing proline residues in the phenylalanine-specific permease of Escherichia coli. 1998. J Bacteriol, 180:5515–5519.
- Hu LA, King SC. Identification of the amine-polyamine-choline transporter superfamily ‘consensus amphipathic region’ as the target for inactivation of the Escherichia coli GABA transporter GabP by thiol modification reagents. Role of Cys-300 in restoring thiol sensitivity to Gabp lacking Cys. Biochem J 1999; 339: 649–655
- King SC, Hu LA, Pugh A. Induction of substrate specificity shifts by placement of alanine insertions within the consensus amphipathic region of the Escherichia coli GABA (gamma-aminobutyric acid) transporter encoded by gabP. Biochem J 2003; 376: 645–653
- Hu LA, King SC. Functional significance of the ‘signature cysteine’ in helix 8 of the Escherichia coli 4-aminobutyrate transporter from the amine-polyamine-choline superfamily. Restoration of Cys-300 to the Cys-less Gabp. J Biol Chem 1998; 273: 20162–20167
- Closs EI, Lyons CR, Kelly C, Cunningham JM. Characterization of the third member of the MCAT family of cationic amino acid transporters. Identification of a domain that determines the transport properties of the MCAT proteins. J Biol Chem 1993; 268: 20796–20800
- Papageorgiou I, Gournas C, Vlanti A, Amillis S, Pantazopoulou A, Diallinas G. Specific interdomain synergy in the UapA transporter determines its unique specificity for uric acid among NAT carriers. J Mol Biol 2008; 382: 1121–1135
- Castagna Castagna, M, Bossi, E, Sacchi, VF. 2009. Molecular physiology of the insect K-activated amino acid transporter 1 (KAAT1) and cation-anion activated amino acid transporter/channel 1 (CAATCH1) in the light of the structure of the homologous protein LeuT. Insect Mol Biol PMID: 19389142.
- Tilburn J, Scazzocchio C, Taylor GG, Zabicky-Zissman JH, Lockington RA, Davies RW. Transformation by integration in Aspergillus nidulans. Gene 1983; 26: 205–221
- Sambrook, J, Fritisch, EF, Maniatis, T. 1989. Molecular cloning: A laboratory manual. USA: Cold Spring harbour Laboratory.
- Tavoularis S, Scazzocchio C, Sophianopoulou V. Functional expression and cellular localization of a green fluorescent protein-tagged proline transporter in Aspergillus nidulans. Fungal Genet Biol 2001; 33: 115–125
- Cove DJ. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim Biophys Acta 1966; 113: 51–56
- Gao X, Lu F, Zhou L, Dang S, Sun L, Li X, Wang J, Shi Y. Structure and mechanisms of an Amino Acid Antiporter. Science 2009; 19(324 (5934))1565–1568
- Fang, Y, Jayaram, H, Shane, T, Kolmakova-Partensky, L, Wu, F, Williams, C, Xiong, Y, Miller, C. 2009. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Jul 5. [Epub ahead of print], doi:10.1038/nature08201.
Table SI. Homology threading scores using HHpred toolkit (Söding 2005) Citation[27].