
Abstract
Geranylgeranyl diphosphate is a 20-carbon isoprenoid phospholipid whose lipid moiety can be post-translationally incorporated into proteins to promote membrane association. The process of geranylgeranylation has been implicated in anti-proliferative effects of clinical agents that inhibit enzymes of the mevalonate pathway (i.e. statins and nitrogenous bisphosphonates) as well as experimental agents that deplete geranylgeranyl diphosphate. Inhibitors of geranylgeranyl diphosphate synthase are an attractive way to block geranylgeranylation because they possess a calcium-chelating substructure to allow localization to bone and take advantage of a unique position of the enzyme within the biosynthetic pathway. Here, we describe recent advances in geranylgeranyl diphosphate synthase expression and inhibitor development with a particular focus on the molecular mechanisms that link geranylgeranyl diphosphate to cell proliferation via geranylgeranylated small GTPases.
Introduction
Post-translational attachment of a lipid group to a protein is commonly used by cells to promote association of the protein with a biological membrane (Palsuledesai and Distefano, Citation2015). The enzymes of protein prenylation specifically utilize intermediates of cellular isoprenoid biosynthesis as substrates to modify one or two cysteine residues near the C-terminus of the protein. Prenylation may refer to incorporation of the 15-carbon molecule farnesyl diphosphate (farnesylation) or the 20-carbon geranylgeranyl diphosphate (geranylgeranylation) onto a protein. The specific modification depends upon the amino acid sequence found at the C-terminus of that particular protein. Both types of prenylation are directed by the CaaX motif – composed of a cysteine followed by three additional amino acid residues (Reid et al., Citation2004), though in some Rab proteins geranylgeranylation can occur at two cysteine residues with other consensus sequences.
Because many small GTPases of the Ras superfamily are prenylated, inhibition of protein prenylation has been viewed as a potential therapeutic target for diseases in which GTPases contribute to the pathogenesis (Berndt et al., Citation2011, Hottman and Li, Citation2014, Cox et al., Citation2015). The majority of these studies have examined disrupting farnesylation of Ras family members H-, N- and K-Ras, which are known oncogenes. Other small GTPases, such as RhoA, also have been reported to play roles in survival and proliferation of malignant cells (Qiu et al., Citation1995, Ghosh et al., Citation1999, Allal et al., Citation2000, Yoshida et al., Citation2009) and altering the membrane localization of RhoA can reduce proliferation (Adnane et al., Citation1998, Li et al., Citation2002, Denoyelle et al., Citation2003, Fromigue et al., Citation2006, Tang et al., Citation2006, Zhu et al., Citation2013). While these and other studies have clearly shown that disruption of prenylation can reduce proliferation of malignant cells, the clinical potential of prenylation inhibitors for treatment of disease is often limited because: (1) Prenylated proteins can be critical to the function of healthy normal cells; (2) direct inhibition of prenylation enzymes in cells can be overcome by activation of alternative prenylation pathways; and (3) prenylation and the membrane association that it promotes is not always required for protein activity. Although much effort has been devoted to pre-clinical development of prenyl transferase inhibitors, these compounds have not yet gained clinical relevance.
In contrast, bisphosphonate-based inhibitors of cellular isoprenoid biosynthesis and protein prenylation have achieved clinical success (Clezardin et al., Citation2011, Ebetino et al., Citation2011). Although they can be associated with mechanism-based side-effects such as osteonecrosis in a small percentage of patients, these drugs greatly reduce the risk of fractures resulting from osteoporosis (Russell et al., Citation2008). Additionally, they are now frequently used for treatment of cancer-related skeletal complications (Ibrahim et al., Citation2003) that arise from both solid tumor metastases (Gnant, Citation2012) and hematological diseases such as myeloma (Terpos et al., Citation2014) where they prevent growth of the malignant cells in the bone environment. The nitrogen-containing bisphosphonates appear to function primarily by blocking isoprenoid biosynthesis (Amin et al., Citation1992, Shipman et al., Citation1998) thereby preventing protein prenylation (van Beek et al., Citation1999b, Bergstrom et al., Citation2000). In osteoclasts, this results in loss of bone resorption but in malignant cells this reduces proliferation. Nitrogenous bisphosphonates overcome some of the limitations of prenyltransferase inhibitors in this regard because they exhibit reduced effects on healthy cells through enriched distribution to the bone compartment and they prevent alternative prenylation through inhibiting an upstream enzyme, farnesyl diphosphate synthase (FDPS).
While FDPS is the molecular target of the clinical bisphosphonates, the molecular mechanism of action is often linked to downstream depletion of geranylgeranyl diphosphate. Therefore, bisphosphonate-based compounds that target other enzymes of isoprenoid biosynthesis (Wiemer et al., Citation2009) leading to geranylgeranyl diphosphate production may be viable therapies for GTPase-dependent disorders affecting the bone environment (Wiemer et al., Citation2011), and could be useful tools for understanding the biological consequences of geranylgeranyl diphosphate depletion in cells. This mini-review examines recent developments in our understanding of the relationship between geranylgeranyl diphosphate synthase (GGDPS) and cell survival and proliferation, which are affected by geranylgeranylated proteins in the membrane domain.
Expression and regulation of GGDPS
GGDPS is a ubiquitously-expressed 35-kDa enzyme found within the isoprenoid biosynthetic pathway (also known as the mevalonate pathway) (Wiemer et al., Citation2011) (). In human cells, GGDPS has been reported to form a large multimeric complex, which was initially characterized as an octomer (tetramer of dimers) (Kuzuguchi et al., Citation1999) but subsequently reported as a hexamer (trimer of dimers) (Kavanagh et al., Citation2006). Further biochemical studies appear to support the octomer model (Miyagi et al., Citation2007). Like FDPS, GGDPS is a prenyl synthase enzyme. Together, these enzymes produce the farnesyl diphosphate and geranylgeranyl diphosphate, respectively. GGDPS catalyzes the condensation of farnesyl diphosphate and isopentenyl diphosphate to geranylgeranyl diphosphate via an ionization-condensation-elimination mechanism (Poulter et al., Citation1978). Thus, depletion of either substrate may decrease GGDPS activity. GGDPS also has an additional binding site where geranylgeranyl diphosphate may bind and inhibit the enzymatic activity (Kavanagh et al., Citation2006, Guo et al., Citation2007). Farnesyl diphosphate and geranylgeranyl diphosphate, can subsequently be utilized as substrates by the prenyl transferase enzymes (farnesyl transferase and geranylgeranyl transferases I and II) as described above. Post-translational incorporation of the prenyl moiety increases membrane association of modified proteins, which is generally but not always required for normal activation and signaling.
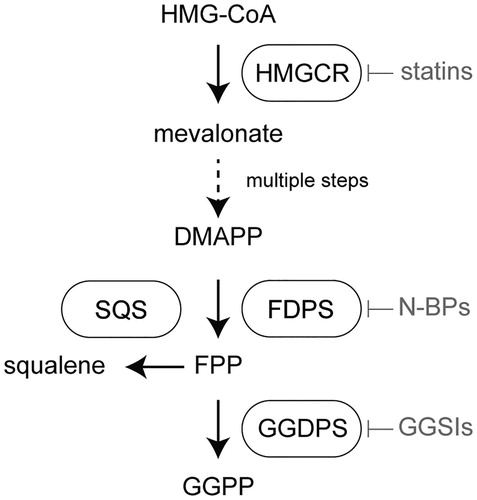
Figure 1. Location of geranylgeranyl diphosphate synthase (GGDPS) within the human isoprenoid biosynthetic pathway. The first and rate-limiting step of human isoprenoid synthesis is the conversion of HMG-CoA to mevalonate by HMG-CoA reductase (HMGCR). This enzyme is the molecular target of the statin drugs. Several steps downstream, farnesyl diphosphate synthase (FDPS) catalyzes the production of farnesyl diphosphate (FPP) from dimethylallyl diphosphate DMAPP and two equivalents of isopentenyl diphosphate (IPP). This enzyme is the molecular target of the nitrogenous bisphosphonates. Farnesyl diphosphate is a key branch point of isoprenoid metabolism, giving rise to squalene in a reaction catalyzed by squalene synthase (SQS) and geranylgeranyl diphosphate (GGPP) in the GGDPS reaction.
Many isoprenoid biosynthetic pathway enzymes are regulated at the transcriptional level to control isoprenoid production (). The classical model of this effect is observed when statin drugs are used to inhibit HMG-CoA reductase leading to the depletion of intracellular cholesterol (Brown and Goldstein, Citation1980). Cholesterol depletion triggers a restorative feedback response mediated by sterol regulatory element binding transcription factors, which upon translocation into the nucleus induce transcription of HMG-CoA reductase (Hua et al., Citation1993, Brown and Goldstein, Citation1997, Brown and Goldstein, Citation2009) (). It is unclear whether GGDPS expression is strongly dependent on sterol regulatory elements, though evidence suggests that its regulation by these factors is not as dramatic as the upstream enzymes such as HMG-CoA reductase. Nonetheless, inhibition of GGDPS leads to diversion of farnesyl diphosphate into sterol synthesis, which has an effect that is opposite to that of statins and nitrogenous bisphosphonates-decreased transcription of HMG-CoA reductase (Wiemer et al., Citation2011). Thus, in contrast to HMG-CoA reductase inhibition, cells appear to have a reduced ability to overcome the effects of GGDPS inhibition at the transcriptional level.
Figure 2. Key feedback mechanisms that control isoprenoid biosynthetic gene expression at the transcriptional level. (A) Sterol regulatory element-binding proteins (SREBPs) regulate transcription of HMG-CoA reductase and other enzymes of isoprenoid biosynthesis in response to cellular sterol levels, which indirectly affects synthesis of geranylgeranyl diphosphate. (B) Egr-1 regulates transcription of isoprenoid biosynthesis in response to activation of the ERK pathway, which directly regulates GGPDS expression at the transcriptional level.
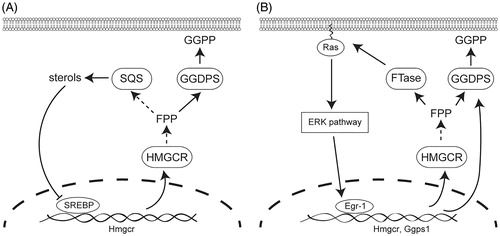
Sterol regulatory elements are not the only potential transcriptional regulators of GGDPS and other isoprenoid pathway enzymes. Activation of the extracellular signal-regulated kinase (ERK) pathway can activate the zinc-dependent transcription factor early growth response gene 1 (EGR1) to regulate expression of HMG-CoA reductase and other sterol pathway enzymes (Gokey et al., Citation2011) (). In contrast to transcription controlled by sterol regulatory elements, constitutively active EGR1 may also regulate GGDPS expression by upregulating transcription of the GGDPS gene, which forms a positive feedback loop (Shen et al., Citation2011). This pathway can be activated by insulin (Yu et al., Citation2011) and toxins such as cigarette smoke extract (Shen et al., Citation2011), which stimulate the ERK pathway and promote accumulation of EGR1 (Tao et al., Citation2013). But the relationship between EGR1 and GGDPS needs further investigation as inhibition of EGR1 transcription had no effect on GGDPS mRNA and protein levels in MCF-7 breast cancer cells (Tao et al., Citation2013).
Upregulation and/or dysregulation of the isoprenoid biosynthetic pathway enzymes including GGDPS has been indicated in oncogenic progression (Pandyra et al., Citation2015), demonstrating an important link between isoprenoids and cell survival and proliferation. For example, dysregulation of the mevalonate pathway by ectopic expression of HMG-CoA reductase promotes transformation of liver cells (Clendening et al., Citation2010). Additionally, mutant p53 can contribute to malignant cell morphology in mammary tissue in a way that is dependent upon the mevalonate pathway (Freed-Pastor et al., Citation2012). In cells depleted of mutant p53, the addition of mevalonate pathway metabolites could compensate for the loss of mutant p53 by restoring the invasive morphology. GGDPS itself is associated with development of liver cancer. In tissue from hepatocellular carcinoma patients, GGDPS mRNA expression was elevated in 85.29% of tumor tissue compared to surrounding cells (Yu et al., Citation2014). Correspondingly, they found that GGDPS protein expression was increased in tumor tissue compared to adjacent tissue in 11 of 15 patients.
While GGDPS expression is ubiquitous and its enzymatic product is often required for proliferation (Benford et al., Citation1999, van de Donk et al., Citation2003, Inoue et al., Citation2005, Brinkkoetter et al., Citation2006, Baulch-Brown et al., Citation2007, Sonnemann et al., Citation2007, Wiemer et al., Citation2007, Yanae et al., Citation2011, Tsubaki et al., Citation2013, Zafar et al., Citation2014), there is only limited evidence, as described in the preceding paragraph, to suggest a higher degree of GGDPS mutation in malignant cells relative to normal cells. Therefore, GGDPS inhibitors could be best classified as anti-metabolites targeting membrane domain proteins (Bennis et al., Citation1993, Mo and Elson, Citation2004). Even without malignant versus normal selectivity, therapeutic specificity could still be obtained by tissue-specific distribution such as that which directs bisphosphonates to localize to the bone environment due to the affinity of the substructure for calcium.
Geranylgeranyl diphosphate synthase inhibitors
Bisphosphonate-based drugs, such as alendronate (, compound 1), have been used therapeutically in humans for over half a century (Ebetino et al., Citation2011). In fact, the nitrogenous bisphosphonates that inhibit FDPS gained therapeutic use prior to determination of their molecular mechanism of action. These agents were selected in part because of the usefulness of the bisphosphonate substructure for binding calcium ions. It was later revealed that the nitrogenous bisphosphonates act as potent inhibitors of FDPS. However, the cellular effects of bisphosphonates on osteoclast-mediated bone resorption are not due to depletion of the immediate enzymatic product, farnesyl diphosphate, but rather to depletion of the subsequent molecule in the pathway, geranylgeranyl diphosphate (van beek et al., Citation1999a). Around the same time, it was demonstrated that the bisphosphonate substructure is useful for targeting inhibitors such as compound 2 to squalene synthase (Ciosek et al., Citation1993). As bisphosphonates can be viewed as analogs of diphosphates, one could readily hypothesize that other enzymes that metabolize diphosphate, such as GGDPS, may also be targeted by bisphosphonate-based drugs.
Figure 3. Structural evolution of GGDPS inhibitors. Small nitrogen-containing bisphosphonates are inhibitors of farnesyl diphosphate synthase (FDPS). Lipophilic bisphosphonates can inhibit the enzymes of farnesyl diphosphate metabolism, including GGDPS and squalene synthase. Branched bisalkyl bisphosphonates retain specificity for GGDPS. Non-bisphosphonate inhibitors of GGDPS have also been identified.
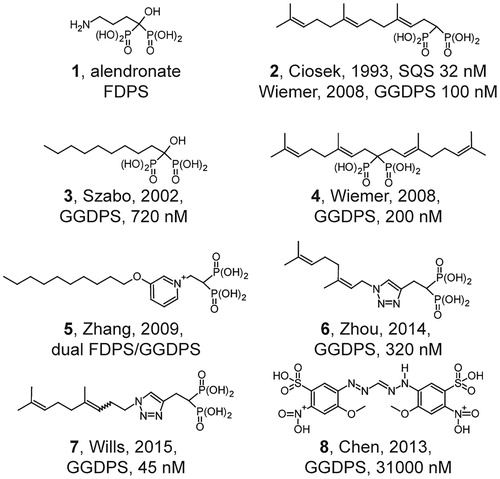
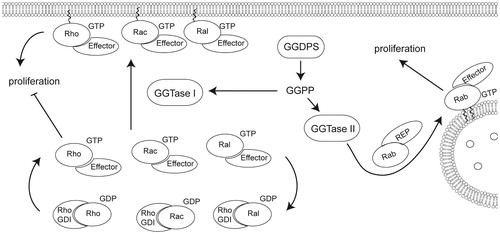
Figure 4. Geranylgeranylation of small GTPases including Rac, Rho, Rab, and Cdc42 promotes localization to the membrane domain, affecting proliferation. Geranylgeranyl transferase I catalyzes the addition of geranylgeranyl diphosphate to the C-terminus of Rho, Rac, or Cdc42 while geranylgeranyl transferase II catalyzes the addition of one or two geranylgeranyl moieties to Rab GTPases. Rab GTPases are primarily involved in vesicle trafficking, though disruption of Rab geranylgeranylation can affect cell viability and proliferation. Rho, Rac, and Cdc42 are involved in proliferation in addition to adhesion and migration regulation.
Initial studies on GGDPS demonstrated that lipophilic bisphosphonates such as compound 3 inhibit the enzyme, while the nitrogenous bisphosphonate drugs do not (Bergstrom et al., Citation2000, Szabo et al., Citation2002). Given this knowledge, it was hypothesized that bisphosphonates bearing isoprenoid substituents may also function as GGDPS inhibitors. We synthesized and characterized an initial library of bis-prenyl bisphosphonates (Shull et al., Citation2006) including digeranyl bisphosphonate (4), demonstrating potent inhibition of cellular geranylgeranylation with no effect on cellular farnesylation. Subsequent studies revealed nanomolar inhibition of GGDPS with no inhibition of FDPS (Wiemer et al., Citation2007, Citation2008).
Lipophilic bisphosphonates bind up to three sites in the GGDPS enzyme (Guo et al., Citation2007), including both the substrate binding pockets of isopentenyl diphosphate and farnesyl diphosphate, as well as a geranylgeranyl diphosphate product/inhibitory binding site. Further studies comparing bis-prenyl and mono-prenyl bisphosphonates showed potent GGDPS inhibition of both types of compounds (Wiemer et al., Citation2008). Structural analysis demonstrated that branched bisphosphonates such as digeranyl bisphosphonate (DGBP) bind to the enzyme with the bisphosphonate in complex with two magnesium ions and the dual alkyl chains extending into the farnesyl diphosphate substrate and the geranylgeranyl diphosphate product binding sites (Chen et al., Citation2008).
Shorter-chain mono-alkyl nitrogenous bisphosphonates (e.g. compound 5) allow for dual inhibition of FDPS and GGDPS (Zhang et al., Citation2009). Compound 5 demonstrated good anti-tumor activity in a mouse model of breast cancer. Additionally, the authors argued that these GGDPS inhibitors may be effective anti-malarial compounds, due to their two-fold effect (Zhang et al., Citation2013): (1) Inhibition of the parasite GGDPS enzyme causes direct inhibition of parasite growth, and (2) inhibition of human FDPS causes activation of a human immune response through indirect activation of gamma delta T cells. Both mono-aromatic non-nitrogenous bisphosphonates (Barney et al., Citation2010) and triazole-based compounds such as compound 6 appear to retain specificity for inhibition of GGDPS over FDPS, in contrast to mono-alkyl nitrogenous bisphosphonates which appear to inhibit both FDPS and GGDPS (Zhou et al., Citation2014). In fact, a recently identified triazole-based compound (7) is the most potent GGDPS inhibitor described to date (Wills et al., Citation2015).
While most reports have examined modifications to the bisphosphonate substructure, non-bisphosphonate inhibitors of GGDPS have also been recently described (e.g. compound 8) (Chen et al., Citation2013). Although these compounds are significantly less potent inhibitors of GGDPS, they retain specificity over FDPS. Lack of the bisphosphonate would be expected to increase peripheral distribution, which could increase the relevance for treatment of non-bone disorders that are dependent upon geranylgeranylation. However, this strategy may also result in increased toxicity towards healthy peripheral tissues. Taken together, a diverse group of bisphosphonate and non-bisphosphonate based GGDPS inhibitors has been developed, yielding a variety of options with respect to target specificity and cellular availability that could determine the consequences of geranylgeranyl diphosphate depletion.
GGDPS inhibitors as anti-proliferative agents targeting small GTPases
Several groups have examined the anti-proliferative activity of compounds that inhibit the enzymes of the isoprenoid pathway. The major premise is that protein prenylation is vital to cell growth and survival and that blocking prenylation through isoprenoid depletion would negatively impact cell health . While it is straightforward to broadly determine whether prenylation is required for proliferation in a given system, it is more difficult to determine which specific prenylated proteins mediate the effect. This problem is especially difficult with respect to geranylgeranylation, because approximately two thirds of the over 150 prenylated proteins are predicted to be geranylgeranylated rather than farnesylated (Reid et al., Citation2004, Maurer-Stroh et al., Citation2007). Many of these proteins are small GTPases of the Ras superfamily, which is typically divided into five families based on sequence and functional homology (Ras, Rho, Rab, Ran and Arf) (Wennerberg et al., Citation2005). While the Ras family GTPases are known to regulate proliferation, these proteins are predominately farnesylated and usually geranylgeranylated only when their farnesylation is blocked. In contrast, the Rho and Rab GTPases, while predominately geranylgeranylated, are traditionally thought to function mainly in cytoskeletal reorganization and vesicle trafficking, respectively. Their roles in proliferation are less clear. Here, we examine the connection between geranylgeranylated small GTPases and the anti-proliferation phenotype caused by geranylgeranyl diphosphate depletion in order to determine whether GGDPS inhibitors would reduce proliferation by affecting a particular GTPase or GTPase family.
In some cases, the anti-proliferative effect caused by isoprenoid depletion using HMG-CoA reductase inhibitors (statin drugs) or FDPS inhibitors (bisphosphonate drugs) is dependent upon reduced prenylation of oncogenic small GTPases. The primary rationale for developing direct prenylation inhibitors (e.g. farnesyl transferase inhibitors) as anti-cancer agents stemmed from the finding that H- N- and K-Ras, which are among the most commonly mutated oncogenes, are mostly farnesylated (Cox et al., Citation2015). Isoprenoid biosynthesis inhibitors or farnesyl transferase inhibitors can block Ras prenylation, in turn reducing Ras signaling and preventing cellular growth. For example, in some human leukemia cells the HMG-CoA reductase inhibitor lovastatin potentiates the cell toxicity of a checkpoint inhibitor, which is abrogated by ectopic expression of constitutively active Ras leading to elevated downstream ERK signaling (Dai et al., Citation2007).
At times some Ras isoforms can be geranylgeranylated when their farnesylation is blocked (Whyte et al., Citation1997), suggesting that farnesyl transferase is not an ideal target for disrupting Ras prenylation. Some groups have proposed combining farnesyl transferase inhibitors with geranylgeranyl transferase inhibitors to achieve complete Ras blockade (Lobell et al., Citation2001). However, another potential route to overcome this limitation is to utilize a prenyl synthase inhibitor which acts upstream to deplete both farnesyl and geranylgeranyl diphosphate, and block both routes of prenylation. For example, exposure to a lipophilic bisphosphonate decreases the plasma membrane levels of K-Ras and H-Ras and leads to caspase-dependent apoptosis (Xia et al., Citation2014). Targeting only farnesyl transferase lacked clinical efficacy, therefore targeting multiple isoprenoid enzymes may be more advantageous (Cox et al., Citation2015), provided that unwanted toxicity to healthy cells can be avoided through other means such as tumor-specific delivery.
Although Ras is mutated in a number of cancers like pancreatic and lung adenocarcinomas (McCormick, Citation2015, Padavano et al., Citation2015), the majority of prenylated proteins are not mutated in cancer (Alan and Lundquist, Citation2013). Notable exceptions are that dominant negative RhoA can be found in angioimmunoblastic T-cell lymphoma and mutated RhoH can be found in non-Hodgkin lymphoma and multiple myeloma (Preudhomme et al., Citation2000, Nagata et al., Citation2016). However, even these non-mutated prenylated proteins can still be targets for cancer therapy because of their role in cell proliferation. Targeting prenylation requires that both the protein and its prenylated state are important in proliferation. For instance, Rac, which is a substrate of geranylgeranyl transferase I, has been shown to be involved in proliferation and decreasing prenylation decreases proliferation (Baba et al., Citation2008). Despite these advantages, it is challenging to identify the specific protein involved in the pathway because many GTPases share effector and regulatory proteins and the exogenous expression of one GTPase may influence the activity of other GTPases.
Taking those caveats into account, most studies have implicated geranylgeranylation of Rho family proteins, specifically RhoA, as a mediator of the anti-proliferative effects of geranylgeranyl diphosphate depletion. For example, depletion of geranylgeranyl diphosphate interrupts the transition from G1 to S phase in thyroid cells by preventing a decrease in p27 expression (Hirai et al., Citation1997). The phenotype is rescued by co-incubation with exogenous geranylgeranyl diphosphate. However, in the presence of botulinum C3 exoenzyme (which specifically inhibits Rho proteins), geranylgeranyl diphosphate is unable to rescue the phenotype. Thus, the authors concluded that geranylgeranylation of Rho is required for proliferation of these cells.
In vascular smooth muscle cells, statins induce an apoptotic effect, which is rescued by co-incubation with either farnesyl or geranylgeranyl diphosphate, indicating a role for geranylgeranylation in the apoptotic mechanism. Overexpression of constitutively active RhoA (L63) reduces the effect of the statin on apoptotic DNA laddering (Blanco-Colio et al., Citation2002). In endothelial cells, statins potentiate apoptosis induced by tumor necrosis factor-α. This effect is due to reduced RhoA geranylgeranylation, because the phenotype can be mimicked by expression of a dominant negative RhoA construct (N19). More importantly, the effect of the statin can be rescued by expression of constitutively active RhoA (L63) (Tang et al., Citation2006). In osteosarcoma cells, statin treatment induces caspase-dependent apoptosis. Here, the authors demonstrated that atorvastatin increases caspase activity, which is rescued by constitutively active RhoA (V14) but not wild-type RhoA (Fromigue et al., Citation2006). In a well-designed study, Allal et al. generated plasmids coding for farnesylated-RhoA and farnesylated-RhoB in which the CAAX domain is mutated to a farnesylation sequence (CVLS) as opposed to the normal geranylgeranylation sequence (CVLV). They found that farnesylated-RhoB and not farnesylated RhoA evades G0/G1 cell cycle arrest induced by geranylgeranyl transferase inhibition (Allal et al., Citation2002). Together, these studies provide mechanistic evidence for a requirement of not only Rho but also of Rho geranylgeranylation for cell proliferation and survival.
While disruption of RhoA geranylgeranylation resulting in mislocalization has negative consequences on proliferation and survival, it is unclear in some cases whether disruption of geranylgeranylation inhibits or activates RhoA signaling. For example, in HepG2 cells, a decrease in RhoA activity is implicated in mediating apoptosis via lipophilic statins (Maeda et al., Citation2010). These effects are rescued by addition of geranylgeranyl diphosphate. Similarly, in colorectal cancer cells, treatment with a statin induces apoptosis, which is rescued by exogenous geranylgeranyl diphosphate but not farnesyl diphosphate (Zhu et al., Citation2013). In contrast to the HepG2 cells, partial rescue of the statin-induced phenotype is observed not only with RhoA silencing, but also with Rac1 silencing, Rac1 inhibitors, and expression of dominant negative Rac1 (N17). This led the authors to suggest not only that both RhoA and Rac1 mediate the effects of statins in this system, but also that statins elevate (rather than reduce) Rho and Rac signaling activity, which leads to apoptosis.
These results agree with a prior study that observed GTPase activation in macrophages that had been treated with nitrogenous bisphosphonates. In the macrophage system, increases are observed in RhoA, Rac1, and Cdc42 GTP binding in response to isoprenoid depletion. The authors propose that mislocalization of the improperly prenylated proteins may restrict their ability to interact with their regulatory partners. Furthermore, in this system Rac1 silencing was able to block the increase in p38 phosphorylation that was also observed (Dunford et al., Citation2006).
The Ras family members RalA and RalB have also been implicated in the phenotypes caused by inhibition of geranylgeranylation (Falsetti et al., Citation2007). Here, the authors examined the mechanism of growth inhibition that was induced by geranylgeranyl transferase type I inhibitors in pancreatic cancer cells. Treatment with the geranylgeranyl transferase inhibitors causes anchorage-independent growth and induces apoptosis. These studies used RalA and RalB constructs in which the CAAX prenylation signal had been altered to allow these naturally geranylgeranylated proteins to be modified instead by farnesylation. Interestingly, the farnesylated RalB construct rescued the apoptotic phenotype while expression of the farnesylated RalA construct rescued the anchorage-independent growth phenotype.
Rap1 is another geranylgeranylated GTPase that may play a role in tumor malignancy. While mechanistic studies demonstrating functional significance of Rap1 prenylation to the anti-proliferative phenotypes of isoprenoid biosynthesis inhibitors are lacking, the development of antibodies to the non-prenylated form of Rap1 have facilitated the in vitro study of geranylgeranylation by eliminating the need for preparing cytoplasmic and membrane fractions for Western blot analysis. Rap1 is routinely used as a biomarker for cellular geranylgeranylation, and is especially useful for in tissue studies in which tissue for analysis is limited, precluding analysis by subcellular fractionation. For example, Rap1 geranylgeranylation was detected to be impaired by GGDPS inhibitors at metastatic sites in the adrenal gland and mesenteric metastatic sites, while it was not altered in non-tumorous adrenal glands. Corresponding with the inhibition of Rap1 prenylation, mice treated with the GGDPS inhibitor also show reduced tumor burden (Reilly et al., Citation2015).
An important advantage of GGDPS inhibition is the ability to impair geranylgeranylation of proteins that are modified by both geranylgeranyl transferase type I, which I modifies Rho family proteins like Rho, Rac, cdc42 and type II, which modifies members of the Rab family (Stigter et al., Citation2012). These proteins have been traditionally associated with regulation of membrane trafficking; however, there is recent evidence implicating Rab prenylation in the anti-proliferative effect of isoprenoid depletion. For example, it has been reported that overexpression of Rab27B can promote tumor growth (Hendrix et al., Citation2010). Mutants of Rab27B that are deficient in their prenylation lack this effect.
The impact of Rab GTPases is also seen in inhibitor studies. Both zoledronate and a geranylgeranyl transferase inhibitor decrease the membrane to cytoplasmic ratios of RhoA, Cdc42 and Rab6. Most notably, the effects of zoledronate are similar to that of a geranylgeranyl transferase II inhibitor suggesting that the effects of zoledronate likely depend on the geranylgeranylation of Rab proteins including Rab6. Zoledronate ultimately induces apoptosis in some cells and S-phase cell cycle arrest in others. An inhibitor of geranylgeranyl transferase II also demonstrated a similar effect, and the authors suggested that zoledronate-mediated apoptosis was driven by loss of Rab geranylgeranylation (Okamoto et al., Citation2014).
In addition to roles in cell proliferation regulation, Rab geranylgeranylation has been implicated in regulation of autophagy. Geranylgeranyl diphosphate depletion in response to GGDPS inhibition increases expression of LC3-II (Wasko et al., Citation2011, Dykstra et al., Citation2015), indicative of autophagy (Tanida et al., Citation2008). Co-incubation with exogenous geranylgeranyl diphosphate prevented accumulation of LC3-II and impairment of Rab geranylgeranylation caused by isoprenoid depletion. Dykstra found that geranylgeranyl transferase I inhibitor (GGTI-2133) is not able to induce LC3-II accumulation despite effective inhibition of geranylgeranylation (Dykstra et al., Citation2015). Moreover, knockdown of GGDPS induces autophagy and delays tumor growth in vivo (Jiang et al., Citation2014).
Conclusion
While many papers have shown that depletion of isoprenoids or small GTPases has a negative impact on proliferation and viability, fewer have investigated whether the anti-proliferative effects of isoprenoid depletion can be rescued by molecular manipulation of a prenylated small GTPase. The more successful approaches have been to rescue proliferation with either an alternatively prenylated construct or a constitutively active construct, or fail to rescue with exogenous isoprenoid plus a dominant negative construct or inhibitor. In some cases, GTPase mislocalization causes activation rather than inactivation, and the phenotype can be directly rescued with a GTPase inhibitor. The studies described herein show that although there are dozens of predicted geranylgeranylated proteins, the anti-proliferative mechanism of geranylgeranyl diphosphate depletion can at times rest on mislocalization of a single geranylgeranylated small GTPase. RhoA geranylgeranylation has been the most frequently described critical lynchpin, but the phenomenon varies by model system, with Rac, Ral, and even Rab geranylgeranylation mediating the anti-proliferative effects in some models. By extension, inhibitors of GGDPS or the geranylgeranyl transferases may be most relevant to indications that depend upon the function of RhoA or its related proteins. Future studies on the consequences of disrupting geranylgeranylation would be aided by molecular approaches that identify the specific GTPase(s) involved.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Compound 4 is covered by a patent that is owned by the University of Iowa. A.J.W. owns shares in Terpenoid Therapeutics, Inc., which has licensed the patent.
References
- Adnane J, Bizouarn FA, Qian Y, Hamilton AD, Sebti SM. 1998. P21wafi/cipi is upregulated by the geranylgeranyltransferase i inhibitor ggti-298 through a transforming growth factor b- and sp1-responsive element: involvement of the small gtpase rhoa. Mol Cell Biol 18:6962–6970.
- Alan JK, Lundquist EA. 2013. Mutationally activated rho gtpases in cancer. Small GTPases 4:159–163.
- Allal C, Favre G, Couderc B, Salicio S, Sixou S, Hamilton AD, et al. 2000. Rhoa prenylation is required for promotion of cell growth and transformation and cytoskeleton organization but not for induction of serum response element transcription. J Biol Chem 275:31001–31008.
- Allal C, Pradines A, Hamilton AD, Sebti SM, Favre G. 2002. Farnesylated rhob prevents cell cycle arrest and actin cytoskeleton disruption caused by the geranylgeranyltransferase i inhibitor ggti-298. Cell Cycle 1:430–437.
- Amin D, Cornell SA, Gustafson SK, Needle SJ, Ullrich JW, Bilder GE, Perrone MH. 1992. Bisphosphonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis. J Lipid Res 33:1657–1663.
- Baba TT, Nemoto TK, Miyazaki T, Oida S. 2008. Simvastatin suppresses the differentiation of c2c12 myoblast cells via a rac pathway. J Muscle Res Cell Motil 29:127–134.
- Barney RJ, Wasko BM, Dudakovic A, Hohl RJ, Wiemer DF. 2010. Synthesis and biological evaluation of a series of aromatic bisphosphonates. Bioorg Med Chem 18:7212–7220.
- Baulch-Brown C, Molloy TJ, Yeh SL, Ma D, Spencer A. 2007. Inhibitors of the mevalonate pathway as potential therapeutic agents in multiple myeloma. Leuk Res 31:341–352.
- Benford HL, Frith JC, Auriola S, Monkkonen J, Rogers MJ. 1999. Farnesol and geranylgeraniol prevent activation of caspases by aminobisphosphonates: biochemical evidence for two distinct pharmacological classes of bisphosphonate drugs. Mol Pharmacol 56:131–140.
- Bennis F, Favre G, Le Gaillard F, Soula G. 1993. Importance of mevalonate-derived products in the control of hmg-coa reductase activity and growth of human lung adenocarcinoma cell line a549. Int J Cancer 55:640–645.
- Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G. 2000. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch Biochem Biophys 373:231–241.
- Berndt N, Hamilton AD, Sebti SM. 2011. Targeting protein prenylation for cancer therapy. Nat Rev Cancer 11:775–791.
- Blanco-Colio LM, Villa A, Ortego M, Hernandez-Presa MA, Pascual A, Plaza JJ, Egido J. 2002. 3-hydroxy-3-methyl-glutaryl coenzyme a reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of bcl-2 expression and rho a prenylation. Atherosclerosis 161:17–26.
- Brinkkoetter PT, Gottmann U, Schulte J, Van Der Woude FJ, Braun C, Yard BA. 2006. Atorvastatin interferes with activation of human cd4(+) t cells via inhibition of small guanosine triphosphatase (gtpase) activity and caspase-independent apoptosis. Clin Exp Immunol 146:524–532.
- Brown MS, Goldstein JL. 1980. Multivalent feedback regulation of hmg coa reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res 21:505–517.
- Brown MS, Goldstein JL. 1997. The srebp pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340.
- Brown MS, Goldstein JL. 2009. Cholesterol feedback: from schoenheimer’s bottle to scap’s meladl. J Lipid Res 50(Suppl.):S15–S27.
- Chen CK-M, Hudock MP, Zhang Y, Guo RT, Cao R, No JH, et al. 2008. Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: a crystallographic and computational investigation. J Med Chem 51:5594–5607.
- Chen SH, Lin SW, Lin SR, Liang PH, Yang JM. 2013. Moiety-linkage map reveals selective nonbisphosphonate inhibitors of human geranylgeranyl diphosphate synthase. J Chem Inf Model 53:2299–2311.
- Ciosek CP Jr, Magnin DR, Harrity TW, Logan JV, Dickson JK Jr, Gordon EM, et al. 1993. Lipophilic 1,1-bisphosphonates are potent squalene synthase inhibitors and orally active cholesterol lowering agents in vivo. J Biol Chem 268:24832–24837.
- Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. 2010. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA 107:15051–15056.
- Clezardin P, Benzaid I, Croucher PI. 2011. Bisphosphonates in preclinical bone oncology. Bone 49:66–70.
- Cox AD, Der CJ, Philips MR. 2015. Targeting ras membrane association: back to the future for anti-ras drug discovery? Clin Cancer Res 21:1819–1827.
- Dai Y, Khanna P, Chen S, Pei XY, Dent P, Grant S. 2007. Statins synergistically potentiate 7-hydroxystaurosporine (ucn-01) lethality in human leukemia and myeloma cells by disrupting ras farnesylation and activation. Blood 109:4415–4423.
- Denoyelle C, Hong L, Vannier JP, Soria J, Soria C. 2003. New insights into the actions of bisphosphonate zoledronic acid in breast cancer cells by dual rhoa-dependent and -independent effects. Br J Cancer 88:1631–1640.
- Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, Coxon FP. 2006. Inhibition of protein prenylation by bisphosphonates causes sustained activation of rac, cdc42, and rho gtpases. J Bone Miner Res 21:684–694.
- Dykstra KM, Allen C, Born EJ, Tong H, Holstein SA. 2015. Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget 6:41535–41549.
- Ebetino FH, Hogan AM, Sun S, Tsoumpra MK, Duan X, Triffitt JT, et al. 2011. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 49:20–33.
- Falsetti SC, Wang DA, Peng H, Carrico D, Cox AD, Der CJ, et al. 2007. Geranylgeranyltransferase i inhibitors target ralb to inhibit anchorage-dependent growth and induce apoptosis and rala to inhibit anchorage-independent growth. Mol Cell Biol 27:8003–8014.
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. 2012. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148:244–258.
- Fromigue O, Hay E, Modrowski D, Bouvet S, Jacquel A, Auberger P, Marie PJ. 2006. Rhoa gtpase inactivation by statins induces osteosarcoma cell apoptosis by inhibiting p42/p44-mapks-bcl-2 signaling independently of bmp-2 and cell differentiation. Cell Death Differ 13:1845–1856.
- Ghosh PM, Ghosh-Choudhury N, Moyer ML, Mott GE, Thomas CA, Foster BA, et al. 1999. Role of rhoa activation in the growth and morphology of a murine prostate tumor cell line. Oncogene 18:4120–4130.
- Gnant M. 2012. Adjuvant bisphosphonates: a new standard of care? Curr Opin Oncol 24:635–642.
- Gokey NG, Lopez-Anido C, Gillian-Daniel AL, Svaren J. 2011. Early growth response 1 (egr1) regulates cholesterol biosynthetic gene expression. J Biol Chem 286:29501–29510.
- Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, et al. 2007. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA 104:10022–10027.
- Hendrix A, Maynard D, Pauwels P, Braems G, Denys H, Van Den Broecke R, et al. 2010. Effect of the secretory small gtpase rab27b on breast cancer growth, invasion, and metastasis. J Natl Cancer Inst 102:866–880.
- Hirai A, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Tatsuno I, et al. 1997. Geranylgeranylated rho small gtpase(s) are essential for the degradation of p27kip1 and facilitate the progression from g1 to s phase in growth-stimulated rat frtl-5 cells. J Biol Chem 272:13–16.
- Hottman DA, Li L. 2014. Protein prenylation and synaptic plasticity: implications for Alzheimer’s disease. Mol Neurobiol 50:177–185.
- Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. 1993. Srebp-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA 90:11603–11607.
- Ibrahim A, Scher N, Williams G, Sridhara R, Li N, Chen G, et al. 2003. Approval summary for zoledronic acid for treatment of multiple myeloma and cancer bone metastases. Clin Cancer Res 9:2394–2399.
- Inoue R, Matsuki NA, Jing G, Kanematsu T, Abe K, Hirata M. 2005. The inhibitory effect of alendronate, a nitrogen-containing bisphosphonate on the pi3k-akt-nfkappab pathway in osteosarcoma cells. Br J Pharmacol 146:633–641.
- Jiang P, Mukthavaram R, Chao Y, Nomura N, Bharati IS, Fogal V, et al. 2014. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br J Cancer 111:1562–1571.
- Kavanagh KL, Dunford JE, Bunkoczi G, Russell RG, Oppermann U. 2006. The crystal structure of human geranylgeranyl pyrophosphate synthase reveals a novel hexameric arrangement and inhibitory product binding. J Biol Chem 281:22004–22012.
- Kuzuguchi T, Morita Y, Sagami I, Sagami H, Ogura K. 1999. Human geranylgeranyl diphosphate synthase. Cdna cloning and expression. J Biol Chem 274:5888–5894.
- Li X, Liu L, Tupper JC, Bannerman DD, Winn RK, Sebti SM, et al. 2002. Inhibition of protein geranylgeranylation and rhoa/rhoa kinase pathway induces apoptosis in human endothelial cells. J Biol Chem 277:15309–15316.
- Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, et al. 2001. Evaluation of farnesyl:Protein transferase and geranylgeranyl:Protein transferase inhibitor combinations in preclinical models. Cancer Res 61:8758–8768.
- Maeda A, Yano T, Itoh Y, Kakumori M, Kubota T, Egashira N, Oishi R. 2010. Down-regulation of rhoa is involved in the cytotoxic action of lipophilic statins in hepg2 cells. Atherosclerosis 208:112–118.
- Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. 2007. Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput Biol 3:e66.
- McCormick F. 2015. The potential of targeting ras proteins in lung cancer. Expert Opin Ther Targets 19:451–454.
- Miyagi Y, Matsumura Y, Sagami H. 2007. Human geranylgeranyl diphosphate synthase is an octamer in solution. J Biochem 142:377–381.
- Mo H, Elson CE. 2004. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med (Maywood) 229:567–585.
- Nagata Y, Kontani K, Enami T, Kataoka K, Ishii R, Totoki Y, et al. 2016. Variegated rhoa mutations in adult t-cell leukemia/lymphoma. Blood 127:596–604.
- Okamoto S, Jiang Y, Kawamura K, Shingyoji M, Tada Y, Sekine I, et al. 2014. Zoledronic acid induces apoptosis and s-phase arrest in mesothelioma through inhibiting rab family proteins and topoisomerase ii actions. Cell Death Dis 5:e1517.
- Padavano J, Henkhaus RS, Chen H, Skovan BA, Cui H, Ignatenko NA. 2015. Mutant k-ras promotes invasion and metastasis in pancreatic cancer through gtpase signaling pathways. Cancer Growth Metastasis 8:95–113.
- Palsuledesai CC, Distefano MD. 2015. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem Biol 10:51–62.
- Pandyra AA, Mullen PJ, Goard CA, Ericson E, Sharma P, Kalkat M, et al. 2015. Genome-wide rnai analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 6:26909–26921.
- Poulter CD, Argyle JC, Mash EA. 1978. Farnesyl pyrophosphate synthetase. Mechanistic studies of the 1'-4 coupling reaction with 2-fluorogeranyl pyrophosphate. J Biol Chem 253:7227–7233.
- Preudhomme C, Roumier C, Hildebrand MP, Dallery-Prudhomme E, Lantoine D, Lai JL, et al. 2000. Nonrandom 4p13 rearrangements of the rhoh/ttf gene, encoding a gtp-binding protein, in non-hodgkin's lymphoma and multiple myeloma. Oncogene 19:2023–2032.
- Qiu RG, Chen J, Mccormick F, Symons M. 1995. A role for rho in ras transformation. Proc Natl Acad Sci USA 92:11781–11785.
- Reid TS, Terry KL, Casey PJ, Beese LS. 2004. Crystallographic analysis of caax prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol 343:417–433.
- Reilly JE, Neighbors JD, Tong H, Henry MD, Hohl RJ. 2015. Targeting geranylgeranylation reduces adrenal gland tumor burden in a murine model of prostate cancer metastasis. Clin Exp Metastasis 32:555–566.
- Russell RGG, Watts NB, Ebetino FH, Rogers MJ. 2008. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 19:733–759.
- Shen N, Shao Y, Lai SS, Qiao L, Yang RL, Xue B, et al. 2011. Ggpps, a new egr-1 target gene, reactivates erk 1/2 signaling through increasing ras prenylation. Am J Pathol 179:2740–2750.
- Shipman CM, Croucher PI, Russell RG, Helfrich MH, Rogers MJ. 1998. The bisphosphonate incadronate (ym 175) causes apoptosis of human myeloma cels in vitro by inhibiting the mevalonate pathway. Cancer Res 58:5294–5297.
- Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF. 2006. Synthesis and biological activity of isoprenoid bisphosphonates. Bioorg Med Chem 14:4130–4136.
- Sonnemann J, Bumbul B, Beck JF. 2007. Synergistic activity of the histone deacetylase inhibitor suberoylanilide hydroxamic acid and the bisphosphonate zoledronic acid against prostate cancer cells in vitro. Mol Cancer Ther 6:2976–2984.
- Stigter EA, Guo Z, Bon RS, Wu YW, Choidas A, Wolf A, et al. 2012. Development of selective, potent rabggtase inhibitors. J Med Chem 55:8330–8340.
- Szabo CM, Matsumura Y, Fukura S, Martin MB, Sanders JM, Sengupta S, et al. 2002. Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates and diphosphates: a potential route to new bone antiresorption and antiparasitic agents. J Med Chem 45:2185–2196.
- Tang D, Park HJ, Georgescu SP, Sebti SM, Hamilton AD, Galper JB. 2006. Simvastatin potentiates tumor necrosis factor alpha-mediated apoptosis of human vascular endothelial cells via the inhibition of the geranylgeranylation of rhoa. Life Sci 79:1484–1492.
- Tanida I, Ueno T, Kominami E. 2008. Lc3 and autophagy. Methods Mol Biol 445:77–88.
- Tao W, Shi JF, Zhang Q, Xue B, Sun YJ, Li CJ. 2013. Egr-1 enhances drug resistance of breast cancer by modulating mdr1 expression in a ggpps-independent manner. Biomed Pharmacother 67:197–202.
- Terpos E, Berenson J, Raje N, Roodman GD. 2014. Management of bone disease in multiple myeloma. Expert Rev Hematol 7:113–125.
- Tsubaki M, Itoh T, Satou T, Imano M, Komai M, Ogawa N, et al. 2013. Nitrogen-containing bisphosphonates induce apoptosis of hematopoietic tumor cells via inhibition of ras signaling pathways and bim-mediated activation of the intrinsic apoptotic pathway. Biochem Pharmacol 85:163–172.
- Van Beek E, Lowik C, Van Der Pluijm G, Papapoulos S. 1999a. The role of geranylgeranylation in bone resorption and its suppression by bisphosphonates in fetal bone explants in vitro: a clue to the mechanism of action of nitrogen-containing bisphosphonates. J Bone Miner Res 14:722–729.
- Van Beek E, Pieterman E, Cohen L, Lowik C, Papapoulos S. 1999b. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun 264:108–111.
- Van De Donk NW, Kamphuis MM, Van Kessel B, Lokhorst HM, Bloem AC. 2003. Inhibition of protein geranylgeranylation induces apoptosis in myeloma plasma cells by reducing mcl-1 protein levels. Blood 102:3354–3362.
- Wasko BM, Dudakovic A, Hohl RJ. 2011. Bisphosphonates induce autophagy by depleting geranylgeranyl diphosphate. J Pharmacol Exp Ther 337:540–546.
- Wennerberg K, Rossman KL, Der CJ. 2005. The ras superfamily at a glance. J Cell Sci 118:843–846.
- Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. 1997. K- and n-ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 272:14459–14464.
- Wiemer AJ, Hohl RJ, Wiemer DF. 2009. The intermediate enzymes of isoprenoid metabolism as anticancer targets. Anticancer Agents Med Chem 9:526–542.
- Wiemer AJ, Tong H, Swanson KM, Hohl RJ. 2007. Digeranyl bisphosphonate inhibits geranylgeranyl pyrophosphate synthase. Biochem Biophys Res Commun 353:921–925.
- Wiemer AJ, Wiemer DF, Hohl RJ. 2011. Geranylgeranyl diphosphate synthase: an emerging therapeutic target. Clin Pharmacol Ther 90:804–812.
- Wiemer AJ, Yu JS, Lamb KM, Hohl RJ, Wiemer DF. 2008. Mono- and dialkyl isoprenoid bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg Med Chem 16:390–399.
- Wills VS, Allen C, Holstein SA, Wiemer DF. 2015. Potent triazole bisphosphonate inhibitor of geranylgeranyl diphosphate synthase. ACS Med Chem Lett 6:1195–1198.
- Xia Y, Liu YL, Xie Y, Zhu W, Guerra F, Shen S, et al. 2014. A combination therapy for kras-driven lung adenocarcinomas using lipophilic bisphosphonates and rapamycin. Sci Transl Med 6:263ra161.
- Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, Yamazoe Y, Nishida S. 2011. Statin-induced apoptosis via the suppression of erk1/2 and akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res 30:74.
- Yoshida T, Clark MF, Stern PH. 2009. The small gtpase rhoa is crucial for mc3t3-e1 osteoblastic cell survival. J Cell Biochem 106:896–902.
- Yu DC, Liu J, Chen J, Shao JJ, Shen X, Xia HG, et al. 2014. Ggpps1 predicts the biological character of hepatocellular carcinoma in patients with cirrhosis. BMC Cancer 14:248.
- Yu X, Shen N, Zhang ML, Pan FY, Wang C, Jia WP, et al. 2011. Egr-1 decreases adipocyte insulin sensitivity by tilting pi3k/akt and mapk signal balance in mice. EMBO J 30:3754–3765.
- Zafar S, Coates DE, Cullinan MP, Drummond BK, Milne T, Seymour GJ. 2014. Zoledronic acid and geranylgeraniol regulate cellular behaviour and angiogenic gene expression in human gingival fibroblasts. J Oral Pathol Med 43:711–721.
- Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, et al. 2009. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: an x-ray and nmr investigation. J Am Chem Soc 131:5153–5162.
- Zhang Y, Zhu W, Liu YL, Wang H, Wang K, Li K, et al. 2013. Chemo-immunotherapeutic anti-malarials targeting isoprenoid biosynthesis. ACS Med Chem Lett 4:423–427.
- Zhou X, Ferree SD, Wills VS, Born EJ, Tong H, Wiemer DF, Holstein SA. 2014. Geranyl and neryl triazole bisphosphonates as inhibitors of geranylgeranyl diphosphate synthase. Bioorg Med Chem 22:2791–2798.
- Zhu Y, Casey PJ, Kumar AP, Pervaiz S. 2013. Deciphering the signaling networks underlying simvastatin-induced apoptosis in human cancer cells: evidence for non-canonical activation of rhoa and rac1 gtpases. Cell Death Dis 4:e568.