
Abstract
Apoptosis is often deregulated in a number of human diseases. Heat-induced apoptosis is a model system for studying the consequences of protein misfolding and is mediated by the Bcl-2 family of proteins. This family consists of both pro-apoptotic and anti-apoptotic members that control mitochondrial integrity. The BH3-only pro-apoptotic members are strong inducers of apoptotic cell death. Protein damaging stress can activate a process of cellular destruction known as apoptosis. The pro-apoptotic BH3-only proteins and transcription factors activate this death pathway by inhibiting the anti-apoptotic Bcl-2 family proteins eliminating cancer cells in a short period of time.
General features of apoptosis
Protein damaging stress, such as exposure to elevated temperature, can activate a process of cellular destruction known as apoptosis (Mosser & Morimoto, Citation2004). Apoptosis is a regulated form of genetically programed cell death and has been recognized as an important part of cellular homeostasis in multicellular organisms (Kerr et al., Citation1972). It is an evolutionary conserved process that has a significant role in embryonic development and tissue homeostasis by eliminating unwanted, damaged, aged and misplaced cells (Borner, Citation2003; Hengartner, Citation2000; Meier et al., Citation2000). Regulated cell death was first discovered in 1842 as shrinkage necrosis, and later renamed apoptosis due to common morphological observations of dying cells (Kerr et al., Citation1972; Kerr, Citation1965; Meier & Vousden, Citation2007). In the first phase of apoptosis, known as the condensation phase, the cell undergoes chromosome condensation, shrinkage of the cytoplasm and nucleus and nuclear DNA fragmentation and membrane blebbing (Kerr et al., Citation1972; Taylor et al., Citation2008). The endoplasmic reticulum (ER), Golgi apparatus and mitochondria become disrupted (Taylor et al., Citation2008) and regulated proteolysis occurs (Kihlmark et al., Citation2001; Yoo et al., Citation2008). The second stage of apoptosis involves phagocytosis and destruction of the apoptotic bodies by phagocytes followed by lysosomal digestion (Kerr et al., Citation1972; Vaux & Strasser, Citation1996). Once triggered, the apoptotic process can occur rapidly within minutes or a few hours followed by the rapid clearance of the apoptotic bodies by phagocytic cells (Hengartner, Citation2000). The degradative process is mediated by a family of aspartate-specific proteases called caspases that cleave structural proteins and trigger the activation of endonucleases that are responsible for chromatin digestion (Vaux & Strasser, Citation1996).
An alternative pathway leading to cell death is known as necrosis. This process occurs upon ATP depletion and severe cellular injury such as toxic insults or physical damage of the cell (Edinger & Thompson, Citation2004). This phenomenon is characterized by membrane disruption linked to the release of cellular debris into the extracellular space. Necrotic cells undergo some DNA fragmentation as a result of lysosomal disruption however this process occurs without cellular energy consumption (Edinger & Thompson, Citation2004) ().
Figure 1. Apoptosis versus necrosis. Apoptosis (top) is an active and inherently controlled means of cell death. The physiological features of programed cell death include: cell shrinking, chromatin condensation, membrane blebbing and pinching off into apoptotic bodies that can be phagocytized by macrophages. Necrosis (bottom) is invariably caused by noxious stimuli as a result of an irreversible disturbance of cellular homeostatic mechanisms. Not involved in the control of cell populations, this form of cell death differs in that in comprises of cell swelling, loss of membrane integrity and organelles, degradation of DNA and eventual cell lysis into the extracellular environment.
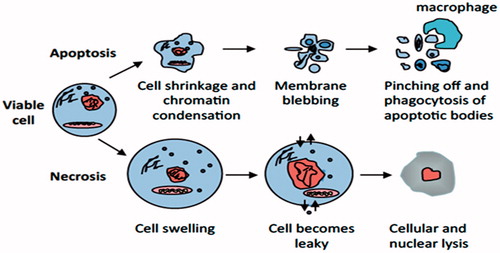
Throughout life, apoptosis is essential for the proper balance of rapidly renewing cells such as those of the hematopoietic system and intestinal epithelium, and for the eradication of irreparably damaged or potentially dangerous cells as seen in the immune system were unnecessary cells are removed after fighting an infection or become dangerous because their antigen receptors recognize self-tissue (Czabotar et al., Citation2014; Fadeel et al., Citation1999; Strasser et al., Citation2011). This physiological form of cell elimination is essential for embryonic development and for the removal of short-lived epithelial and immune cells as well as cells that have been damaged by adverse physiological conditions or viral infection. Moreover, apoptosis refers to physiological cell death caused by a variety of stimuli or conditions in multicellular organisms. As an example, apoptosis eliminates unnecessary interdigital cells during embryo formation. In humans, more than 60 billion cells are eliminated per day to maintain homeostasis as a balance to cell proliferation (Fadeel et al., Citation1999; Strasser et al., Citation2011).
Deregulated apoptosis is responsible for a large number of human diseases including neurological disorders and stroke where it occurs inappropriately and in cancer and autoimmune disorders in which it fails to occur (Czabotar et al., Citation2014; Fadeel et al., Citation1999). Different BCL-2 family members physiologically control apoptosis and interplay between different members, setting apoptotic threshold for novel cancer therapies and maintaining homeostasis. (Czabotar et al., Citation2014; Hardwick & Soane, Citation2013). Mutations in genes regulating apoptosis have been implicated in a number of human diseases including cancer and neurological disorders (Liadis et al., Citation2005; Rohn & Head, Citation2009). Abnormal resistance to apoptosis is considered a major factor for a variety of diseases such as infection, ischemia and chronic pathologies such as neurodegenerative diseases, neuromuscular diseases, and AIDS (Cory & Adams, Citation2002; Green & Evan, Citation2002; Hotchkiss et al., Citation2009; Mattson, Citation2000; Rathmell & Thompson, Citation2002; Thompson, Citation1995).
Regulation of apoptosis
Apoptosis can be initiated from signals at the cell surface and by the sensing of damage within the cell. Activation of the extrinsic pathway is signaled by the binding of death ligands to cell surface associated death receptors. The intrinsic pathway involves the transmission of signals generated by cellular damage to the mitochondrial membrane where apoptotic regulatory proteins control the release of caspase activating proteins into the cytosol. Caspases are proteases that contain a cysteine residue within their catalytic domain (Riedl & Shi, Citation2004; Wolf & Green, Citation1999). These proteases are normally present in healthy cells as inactive pro-caspases that are activated by induced dimerization or by catalytic cleavage. The caspases are divided into two groups, the initiator and effector caspases, based on their mode of activation. The first group is activated by recruitment to adaptor proteins and consists of the initiator caspases (caspase-8, 9 and 10). This activation is promoted either by apoptosome-mediated (intrinsic pathway) or DISC-mediated (extrinsic) formation of a platform for initiator caspase binding that results in their activation. The effector group of caspases (caspase-3, 6 and 7) is activated by proteolytic cleavage by the activated initiator caspases. Initiator caspase cleavage allows the formation of the active site within the effector caspase molecule. The effector caspases are responsible for the cleavage of over a 100 intracellular substrates, which results in the dismantling of the cell (Taylor et al., Citation2008). Recent studies have demonstrated the role of BCL-2 family selective and direct inhibitors in cancer therapeutics treatments by enhancing the efficacy in solid tumors via suppression of granulopoiesis both in vitro and in vivo and in chronic lymphocytic leukemia (Ashkenazi et al., Citation2017; Leverson et al., Citation2015).
Intrinsic and extrinsic apoptotic pathway
The intrinsic pathway, also known as the mitochondrial pathway, is activated from signals within the cells such as oncogenic stress, viral infection or cytotoxic insults that damage DNA or protein molecules (Cory et al., Citation2003) (). This pathway is triggered by stress-inducing stimuli that ultimately leads to the activation of the pro-apoptotic members of the Bcl-2 family BAX and BAK (Adams & Cory, Citation2007). Anti-apoptotic members of the Bcl-2 family regulate the intrinsic pathway by preventing BAX and Ba BAK activation. The BH3-only members of the Bcl-2 family are capable of sensing cellular stresses and act to inhibit the ability of the anti-apoptotic Bcl-2 proteins to repress BAX and BAK activation (indirect model). Some BH3-only proteins have also been demonstrated to directly activate BAX and BAK (direct model). BAX (and BAK) activation leads to their oligomerization in the mitochondrial outer membrane resulting in the formation of pores through which pro-apoptotic proteins can escape to the cytoplasm. BCL-2 member, BAX-mediated cytochrome C, have shown essential role in enhancing mitochondrial apoptosis via intrinsic pathway required in embryonic growth and cancer inhibition. Moreover, BAX overexpression induces mitochondrial fission that enhances cell destruction (Twig et al., Citation2008, Hardwick & Soane, Citation2013).
Figure 2. Intrinsic pathway of apoptosis. The intrinsic pathway shows an induction or post-translational activation of BH3-only protein upon induced stress, which results in the inactivation of some BCL-2 family members. This relieves inhibition of BAX and BAK activation, which in turn promotes apoptosis. BAX and/or BAK will be activated by BH3-only proteins. The pro-apoptotic Bcl-2 family members Bax and Bak oligomerize and undergo conformational changes in the mitochondrial membrane of stressed cells forming channels that permit the release of pro-apoptotic factor, cytochrome c, from mitochondria. Once released, activation of APAF1 into an apoptosome takes place followed by caspases-9 and-3 activation that ultimately causes the proteolytic dismantling of the dying cell.
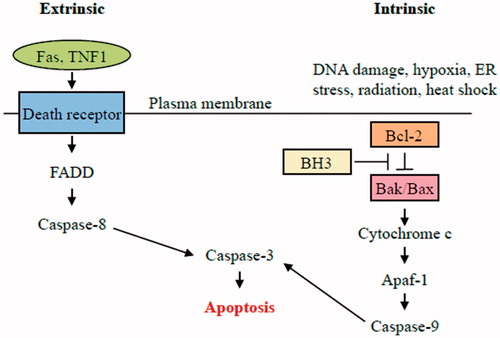
Cytochrome c released from mitochondria is responsible for apoptosome formation and caspase activation (Li et al., Citation1997). Cytochrome c binds to Apaf-1 (apoptotic protease activating factor 1) in the cytosol triggering conformational changes in the Apaf-1 molecule, which promote its oligomerization into the apoptosome. Apaf-1 consists of a caspase recruitment domain (CARD), which allows the Apaf-1 to interact with procaspase-9, a linker region (WD-40), which binds to cytochrome c, and a CED-4 homologous domain, which contains two nucleotide-binding sites. In addition to cytochrome c binding, apoptosome formation requires the binding of dATP or ATP. Once the apoptosome has formed, the CARD domain of Apaf-1 acts to recruit procaspase-9 by an interaction with a CARD domain located within the pro-domain of procaspase-9. Binding of procaspase-9 to the apoptosome allows caspase-9 to adopt a catalytically active conformation (Zou et al., Citation1999).
Extrinsic pathway (death receptor pathway) is activated by death ligands in response to binding to cell-surface death receptors on the cell. These receptors are members of the tumor necrosis factor receptor family (TNF) and includes the Fas receptor and the TNF receptor (Krammer, Citation2000). The death receptors contain intracellular death domains (DD) that serve as docking sites for DD domains within the adaptor protein FADD (Fas-associated death domain). A death effector domain (DED) on FADD can now attract and concentrate procaspase-8 on the activated receptor through an interaction with the DED domain in the pro-domain of the caspase-8. The formation of this complex, called the DISC (death-inducing signaling complex) results in pro-caspase-8 processing and activation. The active caspase-8 is now able to process and activate the effector caspase, caspase 3,6 and 7, which ultimately leads to the death of the cell. In some cells, a weak activation of the DISC leads to a delayed cell death that results from caspase-8 mediated cleavage of the BH3-only protein BID to form tBID (Peter & Krammer, Citation2003). tBID is able to translocate and insert into the outer mitochondrial membrane (OMM) where it can trigger BAX/BAK activation (Li et al., Citation1998). This mechanism allows crosstalk between the extrinsic and intrinsic pathways and permits the amplification of a weak signal at the DISC to efficient apoptosis induction through cytochrome c release and apoptosome formation (Youle & Strasser, Citation2008).
Bcl-2 protein family and the regulation of mitochondrial outer membrane permeability
Stress-induced cell death is under the control of the Bcl-2 family of apoptotic regulators (Chipuk et al., Citation2010) (). The pro-apoptotic Bcl-2 family members BAX and BAK oligomerize in the mitochondrial membrane of stressed cells forming channels that permit the release of cytochrome c (Roufayel & Kadry, Citation2017) (). Anti-apoptotic members (Bcl-2, Mcl-1, etc.) prevent the oligomerization of BAX and BAK. The BH3-only pro-apoptotic members (Noxa, Puma, Bad, BAK, Bid, etc.) act upstream of the anti-apoptotic members to inhibit their ability to block BAX and BAK activation. They do this by interactions between their BH3 domain and a hydrophobic pocket present on the anti-apoptotic Bcl-2 proteins. Regulation of the Bcl-2 family occurs through transcriptional, translational and post-translational (phosphorylation, proteolytic processing, and subcellular relocalization mechanisms.
Figure 3. Structure of the Bcl-2 protein family members. The BH1-3 domain forms a hydrophobic receptor domain able to sequester the BH3 domain-only proteins. BH1/2/3 domain of pro-survival Bcl-2 proteins acts as a receptor for the BH3 domain of other Bcl-2 family members.
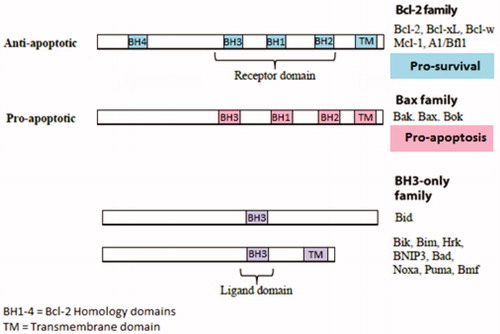
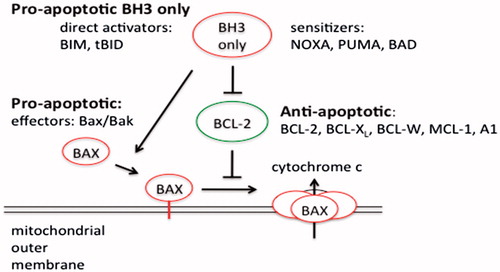
Figure 4. Schematic model of apoptosis regulation by Bcl-2 members protein family. Apoptosis is initiated by the oligomerization of Bax (or Bak) in the mitochondrial outer membrane, which causes the formation of a channel for the release of cytochrome c and other apoptogenic factors. The anti-apoptotic Bcl-2 family members inhibit Bax and Bak oligomerization. The pro-apoptotic BH3-only members of the Bcl-2 family are the direct sensors of cellular stress. They trigger cell death by either inhibiting the anti-apoptotic Bcl-2 members or in the case of BIM and tBID by directly activating BAX and BAK. The BH3-only proteins are regulated by a number of mechanism included increased gene expression or posttranslational modifications.
The Bcl-2 (B-cell lymphoma 2) family proteins are key regulators of stress-induced apoptosis and display either pro-apoptotic or anti-apoptotic activities. The Bcl-2 gene was discovered at the site of a t(14:18) chromosomal translocation in B-cell follicular lymphoma (Tsujimoto et al., Citation1984). The translocation places the Bcl-2 gene (18q21) under the influence a strong immunoglobulin enhancer (14q32) resulting in its elevated expression. However, unlike other oncogenes, Bcl-2 did not promote cell proliferation when overexpressed in a growth-factor dependent cell line but instead promoted cell survival when growth factors were removed (Vaux et al., Citation1988). Bcl-2 gene knockout in mice demonstrated that Bcl-2 function is essential for embryonic development and that animals lacking Bcl-2 showed a loss of mature B and T lymphocytes accompanied by abnormal kidney development (Kamada et al., Citation1995; Veis et al., Citation1993).
The Bcl-2 family proteins are expressed in all cells and consist of 12 distinct members each encoded by a single copy gene, many of which are expressed in different isoforms. These proteins contain at least one Bcl-2 domain known as the BH domain and have been grouped into three classes depending on their mode of action during apoptosis.
Anti-apoptotic Bcl-2 proteins
The anti-apoptotic members of the Bcl-2 family; Bcl-2, Bcl-xL, Bcl-W, Mcl-1, and A1 have 4 BH domains and play a role in maintaining the integrity of ER, mitochondrial and nuclear membranes. Most studies have concentrated on the effects that these proteins exert on the OMM. They contain a C-terminal hydrophobic domain, with the exception of A1, which functions to anchor them to the OMM (Er et al., Citation2006; Youle & Strasser, Citation2008). The BH1/2/3 domains form a hydrophobic groove that serves as a receptor for the BH3 domain of the pro-apoptotic Bcl-2 proteins. All of these anti-apoptotic proteins function to inhibit the release of cytochrome c from the mitochondria. They do this by preventing the oligomerization of the pro-apoptotic members BAX and BAK in the OMM (Chipuk et al., Citation2010).
Pro-apoptotic Bcl-2 effector proteins
The pro-apoptotic members include BAX, BAK and Bok. These proteins all contain three BH domains and have a similar 3D structure as the anti-apoptotic Bcl-2 proteins (Chipuk et al., Citation2010). They are the major inducers of mitochondrial outer membrane permeabilization (MOMP) resulting in cytochrome c release (Antignani & Youle, Citation2006). BAX and BAK exist in an inactive monomeric state in healthy cells. BAX is predominately cytosolic while BAK is associated with the OMM through insertion of its transmembrane domain. The transmembrane domain in BAX is buried within a hydrophobic cleft containing the BH1/2/3 domains. Apoptotic stimulation results in conformational changes in BAK and BAX that result in the formation of homo-oligomeric channel forming structures in the OMM. In the case of BAX activation, a priming step is required that results in the exposure of the transmembrane domain and its insertion into the OMM. Pore formation results in MOMP and the release of pro-apoptotic factors such as cytochrome c and SMAC/DIABLO into the cytoplasm (Du et al., Citation2000). Activated BAX and BAK are inhibited from forming channels by the anti-apoptotic Bcl-2 proteins. Oligomerization therefore requires the neutralization of these anti-apoptotic Bcl-2 proteins by the BH3-only proteins (Adams & Cory, Citation2007).
Pro-apoptotic BH3-only initiator proteins
The third group of the Bcl-2 family are the pro-apoptotic BH3-only proteins: Bid, Bad, Bim, Bik, NOXA, Puma, Hrk, BNIP1,2,3 and Bmf (Lomonosova & Chinnadurai, Citation2008). They have very diverse structures and are related only by the presence of a single BH3 domain. This domain consists of an α-helix that is capable of binding to the hydrophobic groove composed of the BH1/2/3 domains found on the anti-apoptotic Bcl-2 proteins. Various BH3-only members display specificity in their ability to interact with the various anti-apoptotic Bcl-2 proteins (Lomonosova & Chinnadurai, Citation2008) (). For example, NOXA binds only Mcl-1 and A1 while Bad binds to Bcl-2, Bcl-xl and Bcl-w (Chen et al., Citation2005; Kim et al., Citation2006). In contrast, the more promiscuous Bim, Bid and Puma interact with all of the anti-apoptotic Bcl-2 proteins and have also been demonstrated to activate Bax and BAK directly (Kuwana et al., Citation2005) (). The BH3-only proteins are differentially expressed in various cell types and are regulated at the transcriptional and post-transcriptional level. Individual members can be activated by distinct stress signals are believed to act as the sentinels of cell stress.
Figure 5. Schematic model showing the binding specificity of different BH3-only proteins. Bim, Puma and tBid (promiscuous members) are able to bind to all anti-apoptotic Bcl-2 family members. On the other hand, Bad and Noxa (selective members) bind only to certain anti-apoptotic Bcl-2 family members.
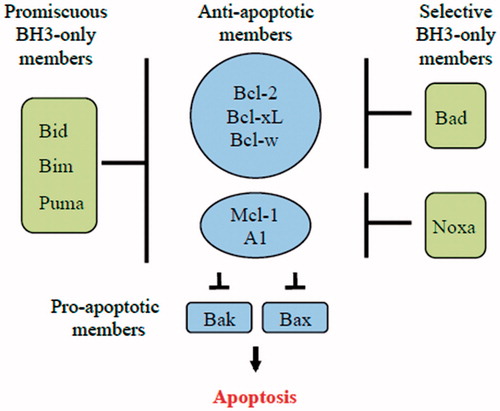
Figure 6. Comparison of the direct and indirect model of Bax and Bak activation. In the direct model, the BH3-only members (Bim, tBid and PUMA) act as activators and bind to Bax and Bak directly to induce pore formation in the OMM that permits the release of cytochrome c. On the other hand, the remaining BH3-only proteins act as sensitizers and bind to the Bcl-2 members, releasing bound Bim and tBid and allowing them to directly activate Bak and Bax. The indirect model shows that BH3-only proteins do not bind directly to Bax and Bak; however, they engage the anti-apoptotic proteins, causing the release of Bak and Bax. Some BH3-only proteins bind to specific anti-apoptotic Bcl-2 family proteins (selective) while others bind to all anti-apoptotic Bcl-2 proteins (promiscuous).
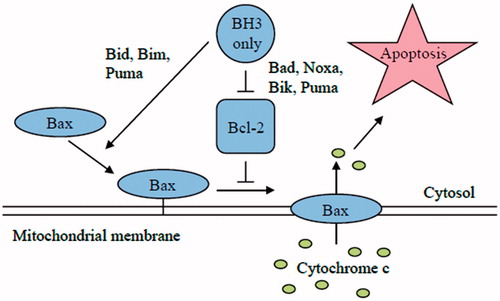
Two mechanisms have been proposed for the activation of BAX and BAK by the BH3-only proteins (Adams & Cory, Citation2007; Chipuk et al., Citation2010). In the indirect model, BAX and BAK activation occurs only when the anti-apoptotic Bcl-2 proteins are inhibited by the BH3-only proteins. BAX/BAK oligomerization ensues when the anti-apoptotic Bcl-2 proteins are no longer available to inhibit their oligomerization. The differential ability of certain BH3-only proteins to induce apoptosis when overexpressed can be explained by their differential ability to bind specific anti-apoptotic Bcl-2 proteins. Bim and Bid, which bind anti-apoptotic Bcl-2 proteins promiscuously, are potent inducers of apoptosis, while NOXA and Bad, which target only a subset of the anti-apoptotic Bcl-2 proteins, are weak inducers of apoptosis. However, co-expression of NOXA and Bad effectively triggers cell death as this combination can target all of the anti-apoptotic Bcl-2 proteins. In the direct model it is proposed that certain BH3-only proteins (Bid, Bim) are capable of directly interacting with BAX/BAK and inducing a conformational changes that leads to their activation and oligomerization. Recent structural studies have identified a binding domain on BAX for the Bid BH3-domain that is distinct from the BAX BH1/2/3 binding pocket (Adams & Cory, Citation2007; Chipuk et al., Citation2010). In this model, Bid and Bim are potent inducers of apoptosis, not simply because they are able to interact with all of the anti-apoptotic Bcl-2 proteins, but because of their ability to directly trigger BAX/BAK activation. Other BH3-only proteins serve to displace Bim/Bid from inhibitory complexes with the anti-apoptotic Bcl-2 proteins freeing them to directly activate BAX/BAK.
BH3-only protein NOXA (PMAIP1) and its regulation
The Noxa gene was first identified as ALT-deprived PMA-responsive gene (APR) in adult T-cell leukemia (ATL) and human embryonic blood cells (Hijikata et al., Citation1990). The same gene was later identified in a search for genes involved in DNA damage-induced apoptosis and named Noxa, which is Latin for damage (Oda et al., Citation2000). The HUGO approved name is PMAIP1 (phorbol-12-myristate-13-acetate-induced protein 1). The human Noxa gene is composed of three exons and two introns, which encodes a 54 amino acid protein containing one BH3 domain (Wang & Sun, Citation2008). The transcript encoding the NOXA protein consists of exon 1 and 3. Two splicing variants were shown to contain exon 2 and share only 19 amino acids with NOXA exon 1. However, both of these variants (NSV-1 and NSV-2) lack the BH3 domain and are only detected in cells treated with proteasomal inhibitors indicating their extremely short half-life. The function of these two variants is unknown. Therefore, only transcript 1, containing exons 1 and 3, encodes the functional NOXA protein.
Many cellular stresses such as DNA damage, hypoxia, glucose deprivation and proteasome inhibition showed an increase in Noxa expression (Ploner et al., Citation2008). Both tracriptional activation and posttranslational mechanisims of Noxa gene regulate its coresponding protein levels (Alves et al., Citation2006; Lowman et al., Citation2010). Noxa gene expression has been documented to be regulated by multiple transcription factors. A p53 response element in the Noxa promotor induced apoptosis in a p53 dependent pathway in mouse embryonic fibroblasts (MEF) (Oda et al., Citation2000). Another member of transcription factors belonging to the p53 family, p73, also showed increased Noxa protein levels in response to DNA damaging stress (Flinterman et al., Citation2005). Hypoxia stress also contributed in increased Noxa expression due to the hypoxia-responsive element present in Noxa promotor independent of p53 response element (Kim et al., Citation2004). Noxa expression have been regulated by E2F1 transcription factor binding to its corresponding E2F1-binding site mapped in the promotor region of the Noxa gene in a p53 independent pathway (Hershko & Ginsberg, Citation2004). In another study, Northern blotting showed Noxa mRNA induction upon γ-irradiation in the spleen and thymus but mRNA levels were also elevated in the absence of p53 in transverse colon of adult mice, revealing that p53-independent mechanisms also control Noxa gene transcription (Fei et al., Citation2002).
As a BH3-only member of the Bcl-2 family, NOXA functions in apoptosis induction by inhibiting the activity of specific anti-apoptotic Bcl-2 family members. Binding studies have demonstrated that NOXA is restricted in its interactions with association occurring only with Mcl-1 and A1 (Chen et al., Citation2005). Mutations within the BH3 domain of NOXA disrupt the interaction between NOXA and Mcl-1 (Chen et al., Citation2005). NOXA is localized to the mitochondria in healthy and dying cells. A mitochondrial targeting domain was mapped to a region C-terminal to the BH3 domain (Seo et al., Citation2003). Deletion of this region reduced its pro-apoptotic potential, however, given the small size of the protein (54 amino acids) and the close proximity of this domain to the BH3 domain it is possible that this deletion simply affected the proper folding of the BH3 domain. A recent study found that NOXA is localized to the cytoplasm in healthy cells but becomes mitochondrial associated in response to glucose stress (Lowman et al., Citation2010). Conversion of NOXA to the active mitochondrial-targeted state was dependent upon dephosphorylation of serine 13. This residue is constitutively phosphorylated by CDK5 in healthy growing cells but becomes dephosphorylated when cells are starved of glucose. The NOXA protein has a short half-life and treatments that inhibit proteasomal function result in NOXA accumulation and cell death (Baou et al., Citation2010; Qin et al., Citation2005). The proteasome inhibitor bortezomib can efficiently induce cell death in chronic lymphocytic leukemia cells and this has been shown to occur through a stabilization of Noxa protein levels as well through transcriptional induction of the Noxa gene. This upregulation was found to be independent of p53 since bortezomib treatment caused Noxa mRNA and protein induction in both p53 wild-type and p53-null melanoma cells (Fernandez et al., Citation2005).
BH3-only protein PUMA (bbc3) and its regulation
The p53 Up-regulated Modulator of Apoptosis protein known as PUMA was first discovered by two different laboratories back in 2001 (Nakano & Vousden, Citation2001; Yu et al., Citation2001). During the same year a third lab identified the bbc3 (Bcl-2 binding component 3) gene that corresponds to the PUMA cDNA (Han et al., Citation2001). This protein is by far the most powerful and effective member of the pro-apoptotic BH3-only protein family. Its gene encodes 4 different forms (α, β, γ and δ) of which only the α and β forms contain the BH3 domain and thus display the pro-apoptotic activity (Yu et al., Citation2001). The PUMA protein encodes 193 amino acids and is highly conserved among vertebrate species yet shows no significant homologies to any other known proteins aside from those with the BH3 domain (Hikisz and Kiliańska, Citation2012). Being mapped on chromosome 19q13.3, this gene is deleted in a large number of human cancers including B-cell malignancies, as well as neural, colorectal cancers (Yu et al., Citation2001). However, its presence has been reported to trigger and induce apoptosis effectively thus, eliminating cancer cell in a span of only a few hours (Jeffers et al., Citation2003; Yu et al., Citation2003).
Enhanced transcription, post-translational modifications and other distinct stimuli play a role in regulating the Bcl-2 family (Fricker et al., Citation2010). It is well documented that PUMA mRNA induction is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines (Yu & Zhang, Citation2008). Many pro-apoptotic activities of this protein have been reported in many cell lines (Han et al., Citation2001). Due to the complexity of this protein, PUMA is involved in many physiological and pathological processes, including the immune response, cancer, and neurodegenerative diseases as well as and viral infections (Jeffers et al., Citation2003). Many transcription factors such as TRB3, AP-1, c-Myc, p73, sp1, Fox03a, E2f1, CHOP including p53 coordinate the expression of PUMA mRNA and proteins during programed cell death (Nakano & Vousden, Citation2001; Jeffers et al., Citation2003; Hikisz & Kiliańska, Citation2012).
p53-dependent apoptosis
Being a direct transcriptional target of the tumor suppressor p53, the mutual interaction between p53 and PUMA is an efficient mechanism for preventing the growth and division of abnormal cells thus protecting cancer development (Happo et al., Citation2012; Strasser et al., Citation2011). In the presence of DNA damage, oxidative stress, deficiency of growth factors, or viral infection, p53 acts on PUMA induction (Wang & Sun, Citation2008; Hikisz & Kiliańska, Citation2012). On the other hand, any mutation or deletion of p53 function is associated with the lack of PUMA expression, therefore contributing to more than 50% of human cancers (Muller & Vousden, Citation2013). Furthermore, p53 acts as a sensor of cell stress, responsible for tumor growth inhibition by either cell cycle arrest followed by DNA repair or by causing apoptosis through activating the transcription of several pro-apoptotic genes, including PUMA. p53-dependent regulation of pro-apoptotic PUMA expression and subsequent apoptosis relies on the functioning of GSK-3 (Glycogen synthase kinase-3) and acetyltransferase Tip60, which control the choice between cell cycle arrest and apoptosis (Charvet et al., Citation2011).
p53-independent apoptosis
Up-regulation of PUMA expression can be regulated wither by p53-mediated activation or by other transcription factors (Yu & Zhang, Citation2008). The p53-independent apoptotic role of PUMA involves the removal of damaged cells during hypoxia, infection and cytokine or growth factor depletion indicating the severe apoptotic signals capable of damaging cells and tissues (Wu et al., Citation2007; Hikisz & Kiliańska, Citation2012). During such pathological conditions, the activity of other transcription factors, such as p73, Sp1 or Fox03a leads to the induction of PUMA mRNA expression and activity level depending on the cell types (Michalak et al., Citation2008; Yu & Zhang, Citation2008; Hikisz & Kiliańska, Citation2012). Although the mechanism remains unknown, the regulation of PUMA occurs mainly without the participation of p53 in compromised cells.
It has been documented that both p53-dependent and p53-independent inductions of apoptosis via PUMA are involved in the immune response after bacterial and viral infections (Wei et al., Citation2008). The immune response starts with increased T-cell proliferation but once the pathogen has been eliminated, the number of T cells needs to be controlled through apoptosis to decrease the immune response. PUMA plays a role in T cell apoptosis and is driven both by p53 and Fox03a (Fischer et al., Citation2008; Hikisz & Kiliańska, Citation2012). This ensures the proper functioning of the immune system to prevent pathological conditions, such as autoimmunity (Fischer et al., Citation2008). Puma expression levels are regulated by the tumor suppressor p53 via dependent and -independent mechanisms in human prostate cancer PC-3 cells (Shan et al., Citation2017). A recent study showed that PUMA overexpression not induced apoptosis in a p-53 silenced PC-3 cells (Yang et al., Citation2017). These results were also correlated with increased BAX expressions and caspase-3 activation independently of p53 induction. Therefore, PUMA overexpression could be a promising potential approach in cancer treatments.
Conclusion
Protein damaging stress can activate a process of cellular destruction known as apoptosis. Apoptosis is often deregulated in a number of human diseases. Stressed-induced apoptosis is a model system for studying the consequences of protein misfolding and is mediated by the Bcl-2 family of proteins. This family consists of both pro-apoptotic and anti-apoptotic members that control mitochondrial integrity. Apoptosis is controlled by pro and anti-apoptotic members of the Bcl-2 family. The BH3-only pro-apoptotic members NOXA and PUMA are strong inducers of apoptosis. They ultimately control cell fate by interacting with anti-apoptotic members and relieving their ability to suppress BAX/BAK oligomerization or, in the case of some (BIM, BID), by directly stimulating BAX/BAK activation. Both NOXA and MCL1 have short half-lives and therefore their abundance can be rapidly altered in stressed cells (Perciavalle & Opferman, Citation2013; Ploner et al., Citation2008). The pro-apoptotic BH3-only protein NOXA activates this death pathway by inhibiting the anti-apoptotic Bcl-2 family protein MCL-1. The other pro-apoptotic BH3-only protein PUMA shows changes in its mRNA and protein levels following exposure to stressed conditions. It act as sensors of diverse cytotoxic stress signals that act upstream of the anti-apoptotic members, inhibiting their ability to block BAK/BAX activation (Adams & Cory, Citation2007; Czabotar et al., Citation2014). Moreover, PUMA elevated levels trigger apoptosis and eliminate cancer cells in a short period of time. Aberrant apoptotic signaling contributes to numerous human disorders and diseases and abnormal resistance to apoptosis contributes to a wide range of acute diseases. It is accepted that diseases such as cancer, immunodeficiency syndromes and neurological disorders not only contribute to alterations in susceptibility to apoptosis, but can also enhance resistance to their conventional therapies.
Acknowledgements
This review was supported by the American University of the Middle East, Egaila, Kuwait.
Disclosure statement
The author report no conflicts of interest. The author alone is responsible for the content and writing of this article.
References
- Adams JM, Cory S. 2007. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26:1324–1337.
- Alves NL, Derks IA, Berk E, Spijker R, van Lier RA, Eldering E. 2006. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity 24:703–716.
- Antignani A, Youle RJ. 2006. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol 18:685–689.
- Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. 2017. From basicapoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 16:273–284.
- Baou M, Kohlhaas SL, Butterworth M, Vogler M, Dinsdale D, Walewska R, et al. 2010. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica 95:1510–1518.
- Borner C. 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol 39:615–647.
- Charvet C, Wissler M, Brauns-Schubert P, Wang S-J, Tang Y, Sigloch FC, Mellert H. 2011. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell 42:584–596.
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17:393–403.
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. 2010. The BCL-2 family reunion. Mol Cell 37:299–310.
- Cory S, Adams JM. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2:647–656.
- Cory S, Huang DC, Adams JM. 2003. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22:8590–8607.
- Czabotar PE, Lessene G, Strasser A, Adams JM. 2014. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15:49–63.
- Du C, Fang M, Li Y, Li L, Wang X. 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102:33–42.
- Edinger AL, Thompson CB. 2004. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16:663–669.
- Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM. 2006. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta 1757:1301–1311.
- Fadeel B, Orrenius S, Zhivotovsky B. 1999. Apoptosis in human disease: a new skin for the old ceremony? Biochem Biophys Res Commun 266:699–717.
- Fei P, Bernhard EJ, El-Deiry WS. 2002. Tissue-specific induction of p53 targets in vivo. Cancer Res 62:7316–7327.
- Fernandez Y, Verhaegen M, Miller TP, Rush JL, Steiner P, Opipari AW Jr, et al. 2005. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res 65:6294–6304.
- Fischer SF, Belz GT, Strasser A. 2008. BH3-only protein PUMA contributes to death of antigen-specific T cells during shutdown of an immune response to acute viral infection. Proc Natl Acad Sci USA 105:3035–3040.
- Flinterman M, Guelen L, Ezzati-Nik S, Killick R, Melino G, Tominaga K, et al. 2005. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem 280:5945–5959.
- Fricker M, O’Prey J, Tolkovsky AM, Ryan KM. 2010. Phosphorylation of PUMA modulates its apoptotic function by regulating protein stability. Cell Death Dis 1:e59.
- Green DR, Evan GI. 2002. A matter of life and death. Cancer Cell 1:19–30.
- Han J-w, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, et al. 2001. Expression of bbc3, a proapoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA 98:11218–11323.
- Happo L, Strasser A, Cory S. 2012. BH3-only proteins in apoptosis at a glance. J Cell Sci 125:1081–1987.
- Hardwick JM, Soane L. 2013. Multiple functions of BCL-2 family proteins. ColdSpring Harb Perspect Biol Cold Spring Harbor Lab 5:a008722.
- Hengartner MO. 2000. The biochemistry of apoptosis. Nature 407:770–776.
- Hershko T, Ginsberg D. 2004. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem 279:8627–8634.
- Hijikata M, Kato N, Sato T, Kagami Y, Shimotohno K. 1990. Molecular cloning and characterization of a cDNA for a novel phorbol-12-myristate-13-acetate-responsive gene that is highly expressed in an adult T-cell leukemia cell line. J Virol 64:4632–4639.
- Hikisz P, Kiliańska ZM, 2012. PUMA, a critical mediator of cell death-one decade on from its discovery. Cell Mol Biol Lett 17:646–669.
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. 2009. Cell death. New Engl J Med 361:1570–1583.
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang JLing, Brennan J, et al. 2003. PUMA is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328.
- Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, et al. 1995. bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res 55:354–359.
- Kerr JF. 1965. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol 90:419–435.
- Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257.
- Kihlmark M, Imreh G, Hallberg E. 2001. Sequential degradation of proteins from the nuclear envelope during apoptosis. J Cell Sci 114:3643–3653.
- Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. 2004. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med 199:113–124.
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. 2006. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8:1348–1358.
- Krammer PH. 2000. CD95’s deadly mission in the immune system. Nature 407:789–795.
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17:525–535.
- Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. 2015. Exploitingselective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 7:279ra40.
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489.
- Li H, Zhu H, Xu CJ, Yuan J. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501.
- Liadis N, Murakami K, Eweida M, Elford AR, Sheu L, Gaisano HY, et al. 2005. Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol 25:3620–3629.
- Lomonosova E, Chinnadurai G. 2008. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27(Suppl. 1):S2–S19.
- Lowman XH, McDonnell MA, Kosloske A, Odumade OA, Jenness C, Karim CB, et al. 2010. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell 40:823–833.
- Mattson MP. 2000. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1:120–129.
- Meier P, Finch A, Evan G. 2000. Apoptosis in development. Nature 407:796–801.
- Meier P, Vousden KH. 2007. Lucifer’s labyrinth-ten years of path finding in cell death. Mol Cell 28:746–754.
- Michalak EM, Villunger A, Adams JM, Strasser A. 2008. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 15:1019–1029.
- Mosser DD, Morimoto RI. 2004. Molecular chaperones and the stress of oncogenesis. Oncogene 23:2907–2918.
- Muller PAJ, Vousden KH. 2013. p53 mutations in cancer. Nat Cell Biol 15:2–8.
- Nakano K, Vousden KH. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694.
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058.
- Perciavalle RM, Opferman JT. 2013. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol 2013 23:22–29.
- Peter ME, Krammer PH. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10:26–35.
- Ploner C, Kofler R, Villunger A. 2008. Noxa: at the tip of the balance between life and death. Oncogene 27(Suppl. 1):S84–S92.
- Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. 2005. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 65:6282–6293.
- Rathmell JC, Thompson CB. 2002. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109:S97–S107.
- Riedl SJ, Shi Y. 2004. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5:897–907.
- Rohn TT, Head E. 2009. Caspases as therapeutic targets in Alzheimer’s disease: is it time to “cut” to the chase? Int J Clin Exp Pathol 2:108–118.
- Roufayel R, Kadry S. 2017. Expression of miR-23a by apoptotic regulators in human cancer: a review. Cancer Biol Ther 0:1–8.
- Seo YW, Shin JN, Ko KH, Cha JH, Park JY, Lee BR, et al. 2003. The molecular mechanism of Noxa-induced mitochondrial dysfunction in p53-mediated cell death. J Biol Chem 278:48292–48299.
- Shan Z, Liu Q, Li Y, Wu J, Sun D, Gao Z. 2017. PUMA decreases the growth of prostate cancer PC-3 cells independent of p53. Oncol Lett 13:1885–1890.
- Strasser A, Cory S, Adams JM. 2011. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J 30:3667–3683.
- Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241.
- Thompson CB. 1995. Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462.
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. 1984. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226:1097–1099.
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446.
- Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335:440–442.
- Vaux DL, Strasser A. 1996. The molecular biology of apoptosis. Proc Natl Acad Sci USA 93:2239–2244.
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. 1993. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75:229–240.
- Wang Z, Sun Y. 2008. Identification and characterization of two splicing variants of human Noxa. Anticancer Res 28:1667–1674.
- Wei J, O’Brien D, Vilgelm A, Piazuelo MB, Correa P, Washington MK, et al. 2008. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 134:1412–1423.
- Wolf BB, Green DR. 1999. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem 274:20049–20052.
- Wu B, Qiu W, Wang P, Yu H, Cheng T, Zambetti GP, et al. 2007. p53 independent induction of PUMA mediates intestinal apoptosis in response to ischaemia-reperfusion. Gut 56:645–654.
- Yang H, Xie Y, Yang D, Ren D. 2017. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget 8:25310–25322.
- Yoo H, Cha HJ, Lee J, Yu EO, Bae S, Jung JH, et al. 2008. Specific proteolysis of the A-kinase-anchoring protein 149 at the Asp582 residue by caspases during apoptosis. Oncol Rep 19:1577–1582.
- Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59.
- Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. 2003. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 100:1931–1936.
- Yu J, Zhang L. 2008. PUMA, a potent killer with or without p53. Oncogene 27:71–83.
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell Biol 7:673–682.
- Zou H, Li Y, Liu X, Wang X. 1999. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556.