Abstract
The human disease protein, Bestrophin-1, associated with vitelliform macular dystrophy, has recently been shown to be an integral membrane anion channel-forming protein. In this study we have recovered all bestrophin homologues from the NCBI database and analyzed their sequences using bioinformatic approaches. Eukaryotic homologues were found in animals and fungi but not in plants or protozoans, and prokaryotic homologues distantly related to the eukaryotic proteins, were identified in certain Gram-negative bacterial kingdoms but not in Gram-positive bacteria or archaea. Our analyses suggest a uniform 4 TMS topology for most of these homologues with regions of conservation overlapping and preceding the odd numbered TMSs and overlapping and following the even numbered TMSs. Well-conserved motifs were identified in both the eukaryotic and the prokaryotic homologues, and these proved to overlap, suggesting common structural and functional properties. Phylogenetic analyses revealed that the eukaryotic proteins cluster according to organismal type, and that the prokaryotic proteins sometimes (but not always) do so. This suggests that eukaryotic paralogues arose exclusively by recent gene duplication events although both early and late gene duplication events occurred in prokaryotes.
Introduction
In 1998, the mutant gene responsible for the human autosomal dominant disease vitelliform macular degeneration Type 2 (VMD2) or Best's macular dystrophy was identified (Petrukhin et al. [Citation1998]). Subsequently, many mutations giving rise to a spectrum of disease phenotypes were identified (Allikmets et al. [Citation1999]; Bakall et al. [Citation1999]; Caldwell et al. [Citation1999]; Marchant et al. [Citation2001]). The genetic lesions cause loss of central vision and defects in the retinal pigment epithelium (Bakall et al. [Citation1999]; Marmorstein et al. [Citation2002]; Petrukhin et al. [Citation1998]). Bestrophin-1 (the VMD2 gene product), also called the Best disease protein, is an integral membrane protein of 585 amino acyl residues (aas). In a hydropathy plot, the VMD2 gene product exhibits two strongly hydrophobic peaks at residue positions 31–50 and 72–90, two central moderately hydrophobic peaks at positions 131–148 and 185–210, and two more strongly hydrophobic peaks at positions 229–253 and 271–288. The remainder of the protein (residues 290–585) is strongly hydrophilic (see fig. 8 in Tsunenari et al. [Citation2003]). Analyses suggest that the N- and C-termini are in the cytoplasm. Tsunenari et al. ([Citation2003]) proposed a 4 TMS topological model with the polypeptide chain dipping into the membrane between TMSs 3 and 4 and with cytosolic N- and C-termini. Alternatively, Qu et al. ([Citation2003]) have proposed a 6 TMS model with the moderately hydrophobic regions being transmembrane.
Multiple members of the Bestrophin family (TC #1.A.46) are found in mammals, insects and worms (Stohr et al. [Citation2002]; Tsunenari et al. [Citation2003]). Sun et al. ([Citation2002]) demonstrated that the wild-type Bestrophin protein forms a functional chloride channel. Different functionally characterized bestrophin homologues (two from Homo sapiens, one from Mus musculus, two from Xenopus laevis, one from Drosophila melanogaster and one from Caenorhabditis elegans) each produce chloride conductances with distinctive I–V relationships and ion selectivities (Boese et al. [Citation2004]; Qu et al. [Citation2003]; Sun et al. [Citation2002]; Tavsanli et al. [Citation2001]; Tsunenari et al. [Citation2003]). For example, the Xenopus homologue transports anions with the selectivity order I−>Br−>Cl− >> aspartate (Qu et al. [Citation2003]). At least some of these anion channels are Ca2 + activated (Boese et al. [Citation2004]; Qu et al. [Citation2003], [Citation2004]). A dominant negative phenotype of non-functional mutant channel proteins in the presence of the wild-type protein suggests that they form oligomeric (tetrameric or pentameric) channels (Qu et al. [Citation2003], [Citation2004]). Bestrophin, a phosphoprotein, also interacts, both physically and functionally, with other proteins such as the protein phosphatase 2A via its hydrophilic C-terminal domain (Marmorstein et al. [Citation2002]).
We have searched the databases for bestrophin homologues and found them in animals and fungi, but not in plants and protozoans. We also found them in certain Gram-negative bacteria but not in Gram-positive bacteria or archaea. These homologues are generally encoded in large genome organisms but not in pathogenic bacteria with reduced genome sizes. Most homologues appear to exhibit a probable 4 TMS topology with regions of good conservation overlapping and preceding the odd numbered TMSs and overlapping and following the even numbered TMSs. Analysis of the multiple alignments allowed identification of well-conserved sequence motifs. Phylogenetic analyses revealed the relationships of these proteins to each other, showing that the eukaryotic proteins (and sometimes the prokaryotic proteins) cluster according to organismal type. This last observation suggests that gene duplication events giving rise to multiple paralogues in eukaryotes occurred late, after segregation of the organismal types. Additionally, the gene duplication events giving rise to multiple paralogues in prokaryotes probably occurred both early and late during family evolution, although the possibility that horizontal transfer of genetic information has occurred repeatedly cannot be ruled out. Our studies provide the first detailed analysis of the Bestrophin family of anion channel proteins.
Materials and methods
Computer methods
With the protein sequence of Bestrophin-1 (the VMD2 gene product) of Homo sapiens (Hsa1 in ) as query, the PSI-Blast search tool was used to identify proteins of similar sequences (Altschul et al. [Citation1997]). These sequences were retrieved from the NCBI database (e-value ≤10−4). Redundant sequences were eliminated using an unpublished program (S. Singhi & M.H. Saier unpublished).
Multiple sequence alignments were generated using the Clustal X program, version 1.83 (Thompson et al. [Citation1997]) as well as the TREE program (Feng & Doolittle [Citation1990]). The Clustal X multiple alignments can be found on our website: http://www.biology.ucsd.edu/∼msaier/supmat/Bestrophin/index.html. Figure S1 is the multiple alignment of all eukaryotic Bestrophin homologues (the basis for and ), Figure S2 is an alignment of all but the most divergent sequences, and Figure S3 is the multiple alignment of all prokaryotic homologues (the basis for and ). The BLOSUM30 scoring matrix was used with both programs. Neighbor joining trees were constructed using the above two programs. Parsimony trees were generated using the Phylip program, version 3.62 (Felsenstein [Citation1989]) as well as the PAUP program, version 4.0b10 for Macintosh (Swofford [Citation2003]). All trees were drawn using the TREEVIEW program (Page [Citation1996]; Zhai et al.[Citation2002]). When using Clustal X, the neighbor joining tree (Phylip format) and the default Guide Tree gave virtually identical results. All four tree-construction methods gave similar branching orders. Only the Clustal X-trees are presented.
Statistical analyses of binary sequence comparisons were performed with the GAP program (Devereux et al. [Citation1984]). The standard for establishing homology between two proteins is 9 SDs for regions of at least 60 residues that are compared with the GAP program, using 500 random shuffles, a gap opening penalty of eight and a gap extension penalty of two (Saier [Citation1994]). Sequence comparisons between multiple homologues were performed using the IC program (Zhai & Saier [Citation2002]). The TMS-SPLIT program (Zhou et al. [Citation2003]) was used to generate fragmented protein sequences used for analysis of potential internal duplications using the IC program (Zhai & Saier [Citation2002]), the GAP program (Devereux et al. [Citation1984]) and the TMS-ALIGN program (Zhou et al. [Citation2003]).
The TMHMM (Krogh et al. [Citation2001]), HMMTOP (Tusnady & Simon [Citation1998]), Phobius (Kall et al. [Citation2004]) and WHAT (Zhai & Saier [Citation2001b]) programs were used to estimate the topologies of individual membrane proteins (von Heijne [Citation1986], [Citation1991]). In and , the average values obtained using these four methods are reported. The values obtained with the four programs can be found in and on our website. The Tmap program (Persson & Argos [Citation1994], [Citation1996]), using the Clustal X multiple alignment, was also used for topological prediction. The WHAT program was used to predict secondary structure and display regions of relative amphipathicity. The AveHAS program (Zhai & Saier [Citation2001a]) was used for plotting the average hydropathy, similarity and amphipathicity as a function of alignment position for the eukaryotic and prokaryotic homologues after aligning the sequences with the Clustal X program. Conserved consensus motifs, derived from the Clustal X multiple alignments, characterize and serve as fingerprints for the two Bestrophin subfamilies, the eukaryotic and prokaryotic subfamilies. In the motifs presented in the text, asterisks indicate fully conserved residues; residues present in a majority of the proteins at a particular position are indicated either as single residues or as groups of similar residues in parentheses; Hy indicates occurrences of hydrophobic residues, non-conserved positions are indicated with an X.
Table S1. Eukaryotic Members of the Bestrophin Family.1
Table S2. Prokaryotic Members of the Bestrophin Family.
Results
Eukaryotic homologues
Seventy-three bestrophin homologues were identified in eukaryotes, and using four different programs, most were predicted to have 4 TMSs (). Four of these homologues were from fungi and 69 were from animals; C. elegans contained 26 paralogues, but Drosophila melanogaster and mammals have just four. Only two paralogues from either Anopheles gambiae or Xenopus laevis were identified. Of the four fungal homologues, two were from Aspergillus nidulans, and one each was from Neurospora crassa and Magnaporthe grisea. None was found in yeast, plants or protozoans although complete genome sequences are available for representative organisms in each of these eukaryotic kingdoms.
Table I. Eukaryotic Members of the Bestrophin Family1.
One of the paralogues from C. elegans (Cel5) had a unique hydrophilic N-terminal extension of about 900 residues preceding putative TMS 1, and another (Cel19) had two putative 4 TMS internal repeat elements that exhibited 39% identity and 47% similarity with each other. In all of these eukaryotic homologues, putative TMSs 1 and 2 proved to be fairly well conserved, and good conservation was also observed in the hydrophilic regions just preceding TMSs 1 and just following TMSs 2. This fact is revealed by comparison of the average hydropathy and average similarity plots shown in . In this figure, the first pair of conserved hydrophobic peaks are labeled 1 and 2, the second pair of conserved hydrophobic peaks are labeled 3 and 4, and the two centrally located moderately hydrophobic peaks are labeled M1 and M2. Note that the large non-conserved peak of hydrophobicity between peaks M2 and 3 is present only in one protein, Cel10 (see the multiple alignment presented in suppl. Figure S1 on our website).
Figure 1. Average hydropathy (black) and similarity (gray) plots for the eukaryotic bestrophin homologues listed in . The plots are based on the multiple alignment generated with the CLUSTAL X program (see Figure S1 on our website). The AveHAS program (Zhai & Saier [Citation2001a]) was used to derive the plots with a sliding window of 19 residue positions. Peaks 1–4, the strongly conserved hydrophobic peaks, are numbered above the peaks of hydropathy and correspond to putative TMSs 1-4. The two centrally located moderately hydrophobic peaks are labeled M1 and M2. An expanded version of the average hydropathy and average similarity plots for alignment positions 900–1600 can be found on our website (Figure S4).
![Figure 1. Average hydropathy (black) and similarity (gray) plots for the eukaryotic bestrophin homologues listed in Table I. The plots are based on the multiple alignment generated with the CLUSTAL X program (see Figure S1 on our website). The AveHAS program (Zhai & Saier [Citation2001a]) was used to derive the plots with a sliding window of 19 residue positions. Peaks 1–4, the strongly conserved hydrophobic peaks, are numbered above the peaks of hydropathy and correspond to putative TMSs 1-4. The two centrally located moderately hydrophobic peaks are labeled M1 and M2. An expanded version of the average hydropathy and average similarity plots for alignment positions 900–1600 can be found on our website (Figure S4).](/cms/asset/d72603b6-fb61-43f4-b003-3980c7b4f79a/imbc_a_112954_uf0001_b.jpg)
The pattern of conservation observed for the first pair of putative TMSs (peaks 1 and 2 in ) was also observed for the second pair of strongly hydrophobic TMSs (peaks 3 and 4 in ). Thus, good conservation was observed in the hydrophilic regions just preceding TMSs 3 and just following TMSs 4. In spite of this pattern of similarity between these two putative hairpin structures, we could not detect sufficient sequence similarity to establish homology (see Discussion section).
Sequence analyses of eukaryotic homologues
The multiple alignment generated from the 73 eukaryotic homologues upon which was based, revealed that several apparently truncated proteins, as well as several apparent fusions and insertions introduced large gaps in the alignment, making interpretation difficult. The anomalous proteins as well as the four sequence divergent fungal homologues were therefore removed. With 66 animal proteins of the original 73 sequences remaining in the alignment, poor conservation was still observed (compare Figures S1 and S2 on our website: http://www.biology.ucsd.edu/∼msaier/supmat/Bestrophin/index.html). Only a few fully conserved residues were present. In the region of the first two TMSs, several positions were well (but not fully) conserved, e.g., (RK)GS(LIV)(WY)K at alignment positions 904–909 in TMS 1 and P(LIV)S(Hy)3GF(FY)(LIV)(ST)Hy3XRW within TMS 2 at alignment positions 965–981 [X = any residue; Hy = any hydrophobic residue; alternative possibilities for a single alignment position are indicated in parentheses.]. TMSs 3 and 4 proved to be better conserved. In front of and overlapping TMS 3 was a well-conserved region with four fully conserved residues (marked by asterisks) as follows: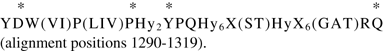 Within TMS 4 and to the right of it, was the consensus sequence:
Within TMS 4 and to the right of it, was the consensus sequence: This is the best-conserved region in these proteins with six fully conserved residues.
This is the best-conserved region in these proteins with six fully conserved residues.
Prokaryotic homologues
Thirty prokaryotic homologues were identified, and all proved to be from certain Gram-negative bacterial kingdoms (). These kingdoms included the cyanobacteria, the cytophaga and the α-, β-, γ- and δ-proteobacteria. Prokaryotic homologues are substantially smaller than the eukaryotic homologues with an average size of about 300 residues. Almost all of them are predicted to have 4 TMSs (). Comparison scores of up to 17 SD were obtained when prokaryotic and eukaryotic proteins were compared establishing that they are all homologous and belong to a single family.
Table II. Prokaryotic Members of the Bestrophin Family.
Sequence analyses of prokaryotic homologues
The average hydropathy and similarity plots for the prokaryotic proteins, based on the multiple alignment of the bacterial bestrophin homologues shown in Figure S3 of our website, are shown in . The plots, although much simpler, exhibit certain similarities with the ones shown in for the eukaryotic homologues. Thus, all four putative TMSs, corresponding to the four peaks of strong hydrophobicity, are well conserved, and the regions immediately preceding TMSs 1 and 3 and following TMSs 2 and 4 are well conserved. The two centrally located moderately hydrophobic peaks are less well conserved and do not exhibit sufficient degrees of hydrophobicity to suggest that they are transmembrane (see also ).
Figure 2. Average hydropathy (black) and similarity (gray) plots for the prokaryotic bestrophin homologues listed in . The plots are based on the multiple alignment generated with the Clustal X program (see Figure S3 on our website). The AveHAS program (Zhai & Saier [Citation2001a]) was used to derive the plots with a sliding window of 19 residue positions. Peaks 1-4, the strongly conserved hydrophobic peaks, correspond to putative TMSs 1-4. They are numbered above the peaks of hydropathy. The two central moderately hydrophobic peaks are labeled M1 and M2 as indicated in .
![Figure 2. Average hydropathy (black) and similarity (gray) plots for the prokaryotic bestrophin homologues listed in Table II. The plots are based on the multiple alignment generated with the Clustal X program (see Figure S3 on our website). The AveHAS program (Zhai & Saier [Citation2001a]) was used to derive the plots with a sliding window of 19 residue positions. Peaks 1-4, the strongly conserved hydrophobic peaks, correspond to putative TMSs 1-4. They are numbered above the peaks of hydropathy. The two central moderately hydrophobic peaks are labeled M1 and M2 as indicated in Figure 1.](/cms/asset/b34233ec-78f7-4eda-bd9b-8ba37d0433e0/imbc_a_112954_uf0002_b.jpg)
The prokaryotic and eukaryotic homologues proved to share consensus motif elements. Thus, in front of TMS 1 of the prokaryotic homologues is the sequence motif: GS(LIV)2X2(LIV)2 (positions 151–158 in the multiple alignment), similar to, but less well conserved than the corresponding motif in the animal homologues. To the right of TMS 2 is the well-conserved sequence motif:![]() In front of TMS 3 is a well-conserved region:
In front of TMS 3 is a well-conserved region:![]() Finally, TMS 4 is terminated by a short well-conserved motif:
Finally, TMS 4 is terminated by a short well-conserved motif: ![]()
Predicted uniform topology of all bestrophin homologues
Four programs for topological prediction of individual proteins (WHAT, TMHMM, HMMTOP and Phobius) and two programs for analyzing multiple alignments (AveHas and Tmap) were applied to both the eukaryotic and prokaryotic subfamilies of the Bestrophin family in order to estimate the most reasonable topology (see Methods section). Using all six programs, the consensus prediction was that peaks 1, 2, 3 and 4 in and are transmembrane α-helical spanners while peaks M1 and M2 are not. The Tmap results were particularly clear-cut and can be found on our website (Figures S5 and S6 for the eukaryotic and prokaryotic subfamilies, respectively). Thus, we conclude that most Bestrophin homologues consist of a basic 4 TMS unit.
Eukaryotic homologue phylogeny
The phylogenetic tree for the eukaryotic proteins is shown in . The tree reveals strict clustering according to organismal type. Thus, all fungal proteins cluster loosely together, all insect proteins cluster together, all vertebrate proteins cluster together, and the worm proteins comprise the rest of the tree. Of the four Drosophila paralogues (Dme1, Dme2, Dme3 and Dme4), two (Aga1 and Aga2) are present in the mosquito; the other two may be revealed when the fully sequenced mosquito genome becomes available. Within the mammalian branch of the tree, all four human paralogues (Hsa1, Hsa2, Hsa3 and Hsa4) have counterparts in rats (Rno1, Rno2, Rno3 and Rno4) and mice (Mmu1, Mmu2, Mmu3 and Mmu4). It is apparent that the different groups of mammalian orthologues have diverged in sequence at different rates with orthologues 2 and 4 diverging less rapidly than orthologues 1 and 3 (see ). In these cases, branch length distance between two orthologues is approximately proportional to the numbers of non-conserved amino acids between those two proteins.
Figure 3. Phylogenetic tree for the eukaryotic bestrophin homologues. The CLUSTAL X program (Jeanmougin et al. [Citation1998]; Thompson et al. [Citation1997]) was used to generate the multiple alignment (Figure S1 on our website) upon which the tree (drawn using the TreeView program; Page, [Citation1996]) was based. Abbreviations of the proteins are as indicated in . Bootstrap values (percentage) from one thousand replications are presented at the nodes. Nodes where no values are indicated have a bootstrap value of 100%.
![Figure 3. Phylogenetic tree for the eukaryotic bestrophin homologues. The CLUSTAL X program (Jeanmougin et al. [Citation1998]; Thompson et al. [Citation1997]) was used to generate the multiple alignment (Figure S1 on our website) upon which the tree (drawn using the TreeView program; Page, [Citation1996]) was based. Abbreviations of the proteins are as indicated in Table I. Bootstrap values (percentage) from one thousand replications are presented at the nodes. Nodes where no values are indicated have a bootstrap value of 100%.](/cms/asset/4999dc32-ba3b-440e-9f18-c206e2b1f680/imbc_a_112954_uf0003_b.jpg)
Many of the worm homologues are found in both C. elegans and C. briggsae. The tree reveals that the different worm orthologous pairs diverged in sequence at very different rates. Thus, orthologous pair 9 (Cel9 and Cbr9) diverged most rapidly, with pairs 7 and 5 coming in second place. Orthologous pairs 4 and 14 have hardly diverged in sequence at all since C. elegans and C. briggsae diverged from each other. These different divergence rates in both the worm and mammalian kingdoms suggest that different paralogues in both kingdoms have been under different pressures to either diverge from or retain their original sequence. In conclusion, it appears that the gene duplication events that have given rise to paralogous eukaryotic bestrophin sequences have occurred within each of the organismal groups (insects, worms, vertebrates and fungi), and that each of the resultant paralogues has diverged in sequence at its own characteristic rate.
Prokaryotic homologue phylogeny
The tree of prokaryotic homologues is shown in . Phylogenetic clustering is often but not always in accordance with organismal phylogeny. For example, cluster 1 proteins are derived exclusively from γ-proteobacteria. Two paralogues in Pseudomonas syringae (Psy1 and Psy2) are found in this cluster, and they arose by a relatively recent gene duplication event. Cluster 2 consists of distantly related proteins from β-, γ- and δ-proteobacterial species. The two paralogues from Burkholderia fungorum (Bfu1 and Bfu2) both fall into this cluster although they are distantly related. Bfu1 is probably orthologous to Rso. Cluster 3 consists of closely related proteins from enteric γ-proteobacteria, but cluster 4 consists of distantly related proteins from the α-, β- and γ-proteobacterial types. Within this cluster, Pfl1 and Ppu are probably orthologues. The paralogue of Pfl1, Pfl2, is present in cluster 1. Cluster 5 is really not a cluster, but two very distantly related cytophagal paralogues. Finally, cluster 6 proteins all derive from cyanobacteria. Thus, four of the six clusters are derived from a single bacterial kingdom or subkingdom, and even the two more diverse clusters are derived exclusively from proteobacteria.
Figure 4. Phylogenetic tree for the prokaryotic bestrophin homologues. The CLUSTAL X program (Jeanmougin et al. [Citation1998]; Thompson et al. [Citation1997]) was used to generate the multiple alignment (Figure S3 on our website) upon which the tree (drawn using the TreeView program; Page, [Citation1996]) was based. Abbreviations of the proteins are as indicated in . Bootstrap values (percentage) from one thousand replications are presented at the nodes.
![Figure 4. Phylogenetic tree for the prokaryotic bestrophin homologues. The CLUSTAL X program (Jeanmougin et al. [Citation1998]; Thompson et al. [Citation1997]) was used to generate the multiple alignment (Figure S3 on our website) upon which the tree (drawn using the TreeView program; Page, [Citation1996]) was based. Abbreviations of the proteins are as indicated in Table II. Bootstrap values (percentage) from one thousand replications are presented at the nodes.](/cms/asset/eff3d8e8-a75f-40f8-a607-64dd293b94d1/imbc_a_112954_uf0004_b.jpg)
We were not able to identify bestrophin homologues in several prokaryotic kingdoms including Gram-positive bacteria, mycoplasmas, spirochetes, Chlamydia, and archaea. It appears that most Bestrophin homologues occur in large genome prokaryotes that are at least capable of a free-living existence (). They are not found in bacterial pathogens with reduced genome sizes that have adapted to and are restricted to an animal host. It therefore seems likely that these proteins function in stress adaptation, and that their loss in small genome pathogens results in part from adaptation to a homeostatic environment (Booth & Louis [Citation1999]; Pivetti et al. [Citation2003]). However, this observation does not explain their apparent absence in Gram-positive bacteria and archaea that include sequenced members with relatively large genomes.
Discussion
We have described a family of anion channel proteins characterized only in animals but also found in fungi and Gram-negative bacteria. The restricted distribution of these proteins is noteworthy. In eukaryotes, they were identified in animals and a few fungi, but not in plants, protozoans or yeast. In prokaryotes, we found them in several subdivisions of the proteobacteria as well as in the cytophagal and cyanobacterial kingdoms, but we did not find them in archaea, Gram-positive bacteria, mycoplasmas, spirochetes, chlamydia or primitive bacteria such as Aquifex aeolicus, Deinococcus radiodurans, or Thermotoga maritima. In part, this may be due to the fact that almost all prokaryotic bestrophin homologues are present in large genome bacteria that are capable of living free in nature although many can form symbiotic or pathogenic relationships with eukaryotes or other prokaryotes. This fact suggests a role in adaptation to stress such as to osmotic or pH stress. Such a possibility is consistent with previously published data for various other families of channel proteins found in bacteria (Booth & Louis [Citation1999]; Kuo et al. [Citation2003]; Pivetti et al. [Citation2003]). If this interpretation is correct, the presence of two bestrophin paralogues in each of several bacteria () may have survival value in rendering these organisms resistant to certain stress conditions.
The eukaryotic bestrophin phylogenetic tree () revealed strict clustering according to organismal type. Thus, all insect proteins cluster together as do the vertebrate proteins, and the fungal proteins similarly form their own cluster. Moreover, the tendency to generate paralogues is kingdom specific. Sequenced fungi, for example, have maximally two paralogues while insects and vertebrates may have four paralogues each. Surprisingly, worms have proliferated far more, possibly 27 or more paralogues. It will be interesting to learn why worms need so many sequence divergent bestrophin paralogues. The physiological functions of these worm channel proteins have yet to be investigated but could be cell type or developmental stage specific.
The prokaryotic bestrophin phylogenetic tree () revealed loose clustering that usually, but not always correlated with organismal type. Thus, four of the six clusters included proteins that were derived from a single bacterial kingdom. The other two clusters were derived from α-, β- and γ-proteobacteria (cluster 4) and β-, γ- and δ-proteobacteria (cluster 2), respectively. Furthermore, clusters 1 and 3 consisted exclusively of γ-proteobacterial proteins, and one γ-proteobacterium, Pseudomonas fluorescence, had two bestrophin paralogues (Pfl1 and Pfl2) which segregated into clusters 1 and 4. Thus, γ-proteobacterial proteins are found in four phylogenetic clusters, while β-proteobacterial proteins are found in two of these clusters. It is therefore most likely that early gene duplication events gave rise to distantly related homologues early during the evolution of the bacterial bestrophin family. Based on previously reported observations, we consider it less likely that horizontal gene transfer is responsible for the sequence divergent paralogues within a single bacterium or group of bacteria (Saier [Citation2003b]).
The eukaryotic bestrophin phylogenetic tree () revealed that in both worms and vertebrates, the rates of sequence divergence between orthologues varies widely. Some orthologues have undergone rapid sequence divergence since the species diverged from each other while other orthologues have hardly diverged at all. This may have resulted from (1) a need to retain a specific function dependent on sequence (thus minimizing sequence divergence) and (2) the need to adapt to changing conditions that have diverged as the species have diverged (thus maximizing sequence divergence). In any case, it seems clear that different rates of sequence divergence must reflect functional requirements, and that the different paralogues therefore serve different functions.
The average hydropathy and similarity plots ( and ) clearly suggested that bestrophins are 4 (rather than 6) TMS proteins (see Introduction section). This is particularly evident from the bacterial bestrophin plot () where the two central peaks of moderate hydrophobicity (M1 and M2) are actually quite hydrophilic. Moreover, we consider it possible that the putative 4 TMS bestrophin homologues arose by an internal gene duplication event where a primordial 2 TMS element generated the present day 4 TMS proteins. Such events have been documented for numerous families of transport proteins (Saier [Citation2003a]). However, in spite of a fair degree of sequence conservation in the regions of the TMSs, no sequence or motif similarity between the two halves of these proteins could be detected. Possibly the two halves have evolved to perform dissimilar functions.
One of the bestrophin homologues, a protein from C. elegans (Cel19) proved to possess 8 putative TMSs and apparently arose by intragenic duplication of the standard 4 TMS-encoding gene. Assuming that both halves are functional, this observation could be explained if one assumes that channel formation requires oligomerization of the 4 TMS unit. This suggestion agrees with the studies of Qu et al. ([Citation2003], [Citation2004]) showing that mutant bestrophin homologues can exhibit a dominant negative phenotype over the wild-type protein. These two independent lines of evidence agree with the concept that bestrophins form oligomeric structures.
This report serves to characterize a large and novel family of putative anion selective channel proteins, distributed widely in nature, but only in restricted organismal kingdoms. With the sole exception of the human VMD2 gene product (see Introduction), the physiological functions of these proteins remain a mystery in spite of fairly extensive electrophysiological data for several of the animal proteins. We have suggested that the functions of the bacterial homologues concern stress responses, and the same can be proposed for the fungal homologues. The motif, phylogenetic and distribution analyses reported here as well as our functional predictions should provide guides for future research. This paper was first published online on prEview on 29 June 2005.
This work was supported by NIH grant GM64368. We thank Mary Beth Hiller for her assistance in the preparation of this manuscript.
References
- Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S, Gerrard B, Li W, Metzker ML, Wadelius C, et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet 1999; 104: 449–53
- Altschul SF, Madden TL, Schaeffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402
- Bakall B, Marknell T, Ingvast S, Koisti MJ, Sandgren O, Li W, Bergen AAB, Andreasson S, Rosenberg T, Petrukhin K, Wadelius C. The mutation spectrum of the bestrophin protein—functional implications. Hum Genet 1999; 104: 383–89
- Boese SH, Aziz O, Simmons NL, Gray MA. Kinetics and regulation of a Ca2 + -activated Cl− conductance in mouse renal inner medullary collecting duct cells. Am J Physiol Renal Physiol 2004; 286: F682–F692
- Booth IR, Louis P. Managing hypo-osmotic stress: Aquaporins and mechanosensitive channels in Escherichia coli. Curr Opin Microbiol 1999; 2: 116–169
- Caldwell GM, Kakuk LE, Griesinger IB, Simpson SA, Nowak NJ, Small KW, Maumenee IH, Rosenfeld PJ, Sieving PA, Shows TB, Ayyagari R. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics 1999; 58: 98–101
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res 1984; 12: 387–395
- Felsenstein J. Phylogeny Inference Package (version 3.2). Cladistics 1989; 5: 164–166
- Feng D-F, Doolittle RF. Progressive alignment and phylogenetic tree construction of protein sequences. Methods Enzymol 1990; 183: 375–387
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci 1998; 23: 403–405
- Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol 2004; 338: 1027–1036
- Krogh A, Larsson B, von Heijne G, Sonnhammer E. Predicting transmembrane protein topology with a hidden Markov model. Application to complete genomes. J Mol Biol 2001; 305: 567–580
- Kuo MM-C, Saimi Y, Kung C. Gain-of-function mutations indicate that Escherichia coli Kch forms a functional K+ conduit in vivo. EMBO J 2003; 22: 4049–4058
- Marchant D, Gogat K, Boutboul S, Pequignot M, Sternberg C, Dureau P, Roche O, Uteza Y, Hache JC, Puech B, et al. Identification of novel VMD2 gene mutations in patients with Best vitelliform macular dystrophy. Hum Mutat 2001; 17: 235
- Marmorstein LY, McLaughlin PJ, Stanton JB, Yan L, Crabb JW, Marmorstein AD. Bestrophin interacts physically and functionally with protein phosphatase 2A. J Biol Chem 2002; 277: 30591–30597
- Page RD. TreeView: An application to display phylogenetic trees on personal computers. Comput Appl Biosci 1996; 12: 357–358
- Persson B, Argos P. Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J Mol Biol 1994; 237: 182–192
- Persson B, Argos P. Topology prediction of membrane proteins. Prot Sci 1996; 5: 363–371
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998; 19: 241–247
- Pivetti CD, Yen M-R, Miller S, Busch W, Tseng Y-H, Booth IR, Saier MH, Jr. Two families of prokaryotic mechanosensitive channel proteins. Microbiol Mol Biol Rev 2003; 67: 66–85
- Qu Z, Fischmeister R, Hartzell C. Mouse bestrophin-2 is a bona fide Cl− channel: Identification of a residue important in anion binding and conduction. J Gen Physiol 2004; 123: 327–340
- Qu Z, Wei RW, Mann W, Hartzell HC. Two bestrophins cloned from Xenopus laevis oocytes express Ca2 + -activated Cl− currents. J Biol Chem 2003; 278: 49563–49572
- Saier MH, Jr. Computer-aided analyses of transport proteins sequences: Gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol Rev 1994; 58: 71–93
- Saier MH, Jr. Tracing pathways of transport protein evolution. Mol Microbiol 2003a; 48: 1145–1156
- Saier MH, Jr. Answering fundamental questions in biology with bioinformatics. ASM News 2003b; 69: 175–181
- Stohr H, Marquardt A, Nanda I, Schmid M, Weber BH. Three novel human VMD2-like genes are members of the evolutionary highly conserved RFP-TM family. Eur J Hum Genet 2002; 10: 281–284
- Sun H, Tsunenari T, Yau K-W, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA 2002; 99: 4008–4013
- Swofford, DL. 2003. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, MA: Sinauer Associates
- Tavsanli BC, Pappu KS, Mehta SQ, Mardon G. Dbest1, a Drosophila homolog of human Bestrophin, is not required for viability or photoreceptor integrity. Genesis 2001; 31: 130–136
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 25: 4876–4882
- Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau K-W, Nathans J. Structure-function analysis of the Bestrophin family of anion channels. J Biol Chem 2003; 278: 41114–41125
- Tusnady GE, Simon I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J Mol Biol 1998; 283: 489–506
- von Heijne G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J 1986; 5: 3021–3027
- von Heijne G. Proline kinks in transmembrane alpha-helices. J Mol Biol 1991; 218: 499–503
- Zhai Y, Saier MH, Jr. A web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins. J Mol Microbiol Biotechnol 2001a; 3: 285–286
- Zhai Y, Saier MH, Jr. A web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence. J Mol Microbiol Biotechnol 2001b; 3: 501–502
- Zhai Y, Saier MH, Jr. A simple sensitive program for detecting internal repeats in sets of multiply aligned homologous proteins. J Mol Microbiol Biotechnol 2002; 4: 29–31
- Zhai Y, Tchieu J, Saier MH, Jr. A web-based Tree View (TV) program for the visualization of phylogenetic trees. J Mol Microbiol Biotechnol 2002; 4: 69–70
- Zhou X, Yang NM, Tran CV, Hvorup RN, Saier MH, Jr. Web-based programs for the display and analysis of transmembrane α-helices in aligned protein sequences. J Mol Microbiol Biotechnol 2003; 5: 1–6