Abstract
Voltage gated potassium channels are tetrameric membrane proteins, which have a central role in cellular excitability. Human Kv1.4 channels open on membrane depolarization and inactivate rapidly by a ‘ball and chain’ mechanism whose molecular determinants have been mapped to the cytoplasmic N terminus of the channel. Here we show that the other terminal end of the channel also plays a role in channel inactivation. Swapping the C-terminal residues of hKv1.4 with those from two non-inactivating channels (hKv1.1 and hKv1.2) affects the rates of inactivation, as well as the recovery of the channel from the inactivated state. Secondary structure predictions of the hKv1.4 sequence reveal a helical structure at its distal C-terminal. Complete removal or partial disruption of this helical region results in channels with remarkably slowed inactivation kinetics. The ionic selectivity and voltage-dependence of channel opening were similar to hKv1.4, indicative of an unperturbed channel pore. These results demonstrate that fast inactivation is modulated by structural elements in the C-terminus, suggesting that the process involves the concerted action of the N- and C-termini.
| Abbreviations | ||
| hKv channel | = | human voltage gated potassium channel |
| TM | = | Transmembrane |
| T1 domain | = | Tetramerization domain |
| HERG | = | human ether-a-go-go related gene |
| KAT | = | K+ transporter of Arabidopsis thaliana |
| EAG | = | ether-a-go-go |
| TREK1 | = | TWIK related K+ channel 1 |
| GIRK1 | = | G-protein coupled inward rectifier |
| AKT1 | = | Arabidopsis K+ transporter 1 |
| HEPES | = | 4-(2-hydroxyethyl)-1-piperazineethane sulphonic acid |
| PHD | = | Profile network prediction HeiDelberg |
| DPM | = | Double Prediction Method |
| DSC | = | Discrimination of protein Secondary structure Class |
| GOR4 | = | Garnier Osguthorpe and Robson |
| HNNC | = | Hierarchical Neural Network C |
| SOPM | = | Self-Optimized Prediction Method |
| Sec. Cons | = | Secondary Consensus |
| CTA | = | C-terminal activation |
| NaV | = | Voltage gated sodium channel |
| SCNA | = | Sodium channel α subunit |
| TEA | = | Tetra Ethyl Ammonium Chloride |
Introduction
Potassium channels are now among the most extensively studied membrane proteins, with a battery of approaches combining to delineate the relationship between form and function in these membrane proteins. The picture that has emerged is of a modular architecture with different portions of the structure contributing to distinct aspects of function. Functional voltage gated potassium (Kv) channels are tetrameric assemblies, each monomer of which can be broadly divided into two structurally distinct regions: the membrane embedded portion containing the ion conducting pathway and the terminal hydrophilic chains. The transmembrane region of each subunit encompasses six transmembrane (TM) segments (S1–S6) and a highly conserved P loop, which constitutes the selectivity filter. The fourth TM segment (S4), with its seven positively charged residues, functions as the principal voltage sensor, and moves in response to changes in the transmembrane potentials Citation[1]. The S5–S6 TM helices along with the P-loop constitute the central pore liner Citation[2] that provides a water-filled passage for the selective passage of K+ ions through the channel. Both the N- and the C-termini of Kv channels are cytoplasmic. The N-terminal contains the inactivation particles and the tetramerization (T1) domain, which is responsible for the subfamily specificity of channel assembly Citation[3], Citation[4].
Rapidly inactivating channels, like Kv1.4, undergo a decrease in ion flux shortly after opening, caused by the physical occlusion of the cytoplasmic vestibule of the channel pore by the inactivating particle Citation[5] (‘ball and chain’ mechanism). N-terminal truncations eliminate this rapid inactivation Citation[5], Citation[6] which can be restored by floating in a peptide with the N-terminal ball sequence Citation[7]. The molecular structures of the Kv1.4 and Kv3.4 ball domains have been determined by NMR spectroscopy Citation[8]. Ball binding to the TM region appears to be influenced by residues in part of the S6 helix Citation[9] and/or the S4–S5 linker Citation[10]. Fast inactivation in sodium channels, however, is not brought about by the N-terminus but involves the III–IV domain linker Citation[11], together with the C-terminus Citation[12].
Apart from being involved in inactivation, cytoplasmic stretches at the N-terminal have been shown to modulate channel activation gating in human ERG channels Citation[13], in plant KAT1 Citation[14] and in Kv channels Citation[15]. The C-terminal has also been shown to be responsible for a variety of channel functions, such as ligand binding Citation[16], gating and membrane targeting of channel proteins Citation[17], and recovery from inactivation Citation[18]. In the case of mammalian ether-a-go-go (EAG) channels, it is known that the C-terminal sequence is important for the production of functional channels and critical for channel assembly Citation[19]. Similarly, C-terminal residues have been shown to be responsible for mechano-gating of the two-pore channel TREK-1 Citation[20]. Structural elements in the C-terminus can also independently control the gating of the G-protein dependent inward rectifier K+ channel, GIRK-1 Citation[21]. Interestingly, a tetramerization domain has also been located in the C-terminus of the plant K+ channel AKT-1 Citation[22]. Predictably, the C-terminus holds clinical significance as evident by the wide array of pathophysiologies associated with either the loss of large stretches Citation[23–25] or point mutations Citation[26] in this region.
It has been reported that the N- and the C-termini of Kv2.1 act in concert during channel activation gating Citation[27]. Additionally, the involvement of two arginine residues (R426 and R429 in Kv4.3/R429 and R432 in Kv4.2) at the C-terminus of Kv4 channels have been implicated in the regulation of channel activation, inactivation and its recovery Citation[28]. However a role for the C-terminal residues in inactivation of mammalian Shaker type channels has not been reported so far. Here we report on a series of chimæric channels generated by swapping the C-terminal residues from non-inactivating channels (Kv1.1 and Kv1.2) on to the transmembrane body of the rapidly inactivating channel Kv1.4. Inactivation was specifically affected in one of these chimæras in which the C-terminus of hKv1.4 was replaced with that from hKv1.2. The rate of inactivation was reduced and recovery from inactivation was accelerated in this chimæra while other parameters like ion selectivity remained unchanged. A secondary structure prediction analysis of the C-terminals of the three channels implicated a role of the C-terminal helix in stabilizing the N-type inactivated state of the channel. Deletions of the predicted helix or its replacement with C-terminal stretches that do not possess the helical structure yielded channels where inactivation, as well as recovery from inactivation, was affected. Based on this data and that from other C-terminal deletion constructs of hKv1.4, both retaining and deleting the predicted C-terminal helical stretch, we argue for a role for a helical structure in the C-terminus of hKv1.4 in rapid ‘N-type’ inactivation.
Materials and methods
Molecular biology
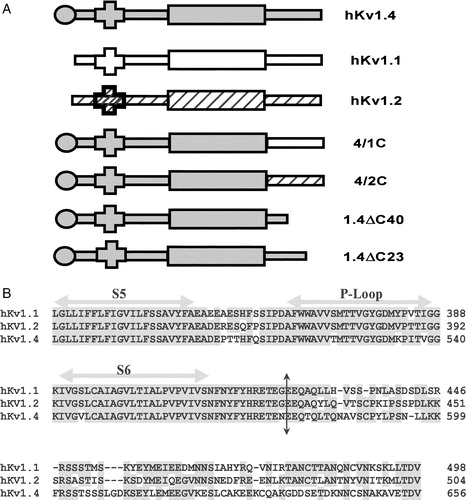
The potassium channel cDNAs used were cloned from human brain stem and subcloned into pGEMA Citation[29]. Silent mutations were introduced to generate an Nhe I restriction site at the proximal end of the C-terminus (10 residues after the S6 TM helix) of hKv1.1, 1.2 and 1.4 using the Quick-Change kit (Stratagene, La Jolla, USA) and used to swap the C-termini. The chimæric constructs we report on, contain the N-terminus and transmembrane body of hKv1.4 and the C-terminus from hKv1.1 (in the case of 4/1C chimæra) or hKv1.2 (in the case of the 4/2C chimæra). The 1.4ΔC23 construct was made by deleting the last 23 residues of hKv1.4 by PCR using a reverse primer which had a stop codon (at position 631), in frame with the gene, thereby leaving 70 amino acids after S6. 1.4ΔC40 was generated using the unique Xba I site of hKv1.4 at nucleotide position 1832. The 5′ overhang of Xba I linearized cDNA was filled by the Klenow fragment of DNA Polymerase I (Promega). This fragment was digested with Sal I to get a 1836 bp fragment of hKv1.4; and was ligated to the Xho I-Stu I DNA fragment of pTM1 plasmid vector with a stop codon (TAA) in frame with rest of the gene. The resulting construct, 1.4ΔC40, contains a deletion of 40 C-terminal amino acid residues of hKv1.4 leaving 53 amino acids after S6. All the reported constructs were confirmed by sequencing on an ABI PRISM-310 automated sequencer. Schematics of these constructs are represented in A.
Figure 1. Schematic representation of K+ channel topologies of the constructs made and sequence comparison of the C-terminus. (A) Bar representations of hKv1.4, hKv1.1, hKv1.2, 4/1C chimæra with the N terminus and TM body of hKv1.4 and the C-terminus from hKv1.1, the 4/2C chimæra with the N-terminus and TM body from hKv1.4 and the C-terminus from hKv1.2, the C-terminal truncation constructs, 1.4ΔC40 and 1.4ΔC23 representing a deletion of 40 and 23 amino acids from the C-terminus of hKv1.4 respectively. Filled circles at the proximal ends of the bar representations symbolize the N-terminal ball present in fast inactivating channels like hKv1.4 and its derivative chimæras. Crosses at the N-terminal region of the bar denote the tetramerization (T1) domain. (B) Sequence comparison of the amino acid residues starting from the S5 TM domain to the C-terminus in hKv1.1, hKv1.2 and hKv1.4. The vertical arrow depicts the splice point of the chimæric constructs. Shading represents the conservation of amino acid residues among the three potassium channels discussed here.
Electrophysiology
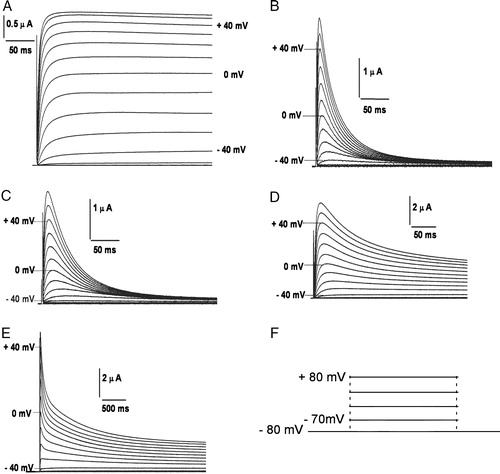
Female Xenopus laevis were anaesthetized using 0.03% Benzocaine and oocytes retrieved by minilaparoctomy. The isolated oocytes were treated with 1 mg/ml Collagenase 1A (Sigma) in a calcium-free solution (mM: 82 NaCl, 2 KCl, 20 MgCl2, 5 HEPES; pH 7.7). The defolliculated oocytes were then stored at 18°C in ND96 solution (mM: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES, with 100 U/ml penicillin and 100 µg/ml streptomycin, pH 7.4) supplemented with 5% heat inactivated horse serum. Capped polyadenylated RNA of all the constructs were generated using T7 RNA polymerase (mMessage mMachine T7 transcription kit, Ambion, USA). Transcribed RNA (46 nL, 150–300 ng/µl) was injected into each oocyte using a Nanoject automatic injector (Drummond, Broomall, PA, USA) and incubated at 18°C for 48 h before recording. Two-electrode voltage clamp of Xenopus oocytes and data analysis were carried out as described earlier Citation[30]. In short, we used an OC-725A oocyte clamp amplifier (Warner Instruments, CT, USA) to maintain the holding potentials and record membrane currents. The currents elicited from the oocytes expressing the constructs studied are shown in . The external recording solution was modified ND96, containing (mM): 96 Sodium Gluconate, 2 Potassium Gluconate, 1.8 Calcium Gluconate, 1 Magnesium Gluconate, 5 HEPES (pH 7.7). All experiments were performed at room temperature. pCLAMP 8.0 (Axon Instruments, CA, USA) software package was used to generate voltage-clamp commands, acquire membrane currents and analyse digitized data.
Figure 2. Potassium currents through channels expressed in Xenopus laevis oocytes on membrane depolarization. Currents elicited in oocytes expressing (A) non-inactivating hKv1.1; (B) fast inactivating hKv1.4; (C) 4/1C chimæra; (D) slower and incompletely inactivating 4/2C chimæra, upon depolarization from a holding potential of −80 mV and subject to a 1 s hyperpolarizing prepulse of −120 mV prior to depolarizing to potentials indicated against the traces; (E) currents elicited upon depolarization of 4/2C from −80 mV in steps of +10 mV for 3.6 s. The inter-episode time used in this protocol was 10 s. The voltage pulse protocol has been shown in panel F.
Data analysis
Data analysis was performed using Clampfit 8 and exported subsequently to Sigma Plot 5.0 or Origin 6.0 for further analysis, plotting and curve fitting. The entire inactivating currents obtained during a 400 ms depolarizing pulse were fitted to a double exponential function to obtain time constants of activation and inactivation. Voltage-dependent activation and inactivation curves are fit to Boltzmann functions, as explained previously Citation[29]. Voltage-dependent steady-state inactivation curves are fit to the modified Boltzmann function [I = Imax/{1+ exp(V − V1/2)/β}], where I is the peak current during the test pulse following a prepulse at potential V; Imax the maximum peak current observed at the test potential; V1/2 the mid-point of inactivation; and β the slope factor.
In the case of channels not inactivating completely, the inactivating portion of the current was re-normalized and the V1/2 of inactivation estimated. For estimating the fraction of channels capable of opening, channels were inactivated at −10 mV and allowed to recover at −100 mV for varying times before administering a test pulse of −10 mV. The fraction of channels recovered was then fitted to single exponential rise functions.
Secondary structure prediction
The C-terminal sequences of hKv1.1, Kv1.2 and Kv1.4 were subject to secondary structure prediction using the packages PHD, Predator, DPM, DSC, GOR4, HNNC, SIMPA96, SOPM, Sec. Cons and Target 99 (http://npsa-pbil.ibcp.fr).
Experiments to distinguish between N- and C-type inactivation
Effect of varying external K+ concentrations on the rate of inactivation
Currents from oocytes expressing 4/2C channels were recorded in the presence of varying concentrations of external potassium (Potassium replaced the sodium ions in the solution in order to maintain the osmolarity). The normal recording solution contained 2 mM of [K+]out. The time constant of inactivation was calculated for currents recorded from oocytes with 2, 10, 48, 73 and 96 mM of external potassium in the bath solution at 0 mV and was normalized with the τinac values at normal recording conditions (2 mM [K+]out) These values were then plotted against the [K+]out.
Effect of external TEA on the rate of inactivation
The τinac values in the presence of various concentrations of external TEA (a blocker of Kv channels with an ED50 of >50 mM for hKv1.4 Citation[29]), were estimated. The time constant of inactivation was calculated for currents recorded from oocytes with 0, 10, 20 and 40 mM TEA in the bath solution at 0 mV and was normalized with the τinac value estimated in the control oocyte with no TEA in the bath, and this value represented the fold change in τinac upon addition of external TEA. These values were then plotted against the concentration of TEA.
Experiments to modulate PIP2 levels in oocytes
PIP2 levels in oocytes were modulated by (i) Incubating oocytes in 10 µM Wortmannin, or (ii) 20 µM Quercetin. Neomycin sulphate, which complexes PIP2, was injected into oocytes at 5 mM concentration.
Results and discussion
Much of the current knowledge base on ion channels has been built up using a two-pronged approach of molecular biological manipulation and heterologous expression. One of the most frequently used expression systems is the Xenopus laevis oocyte, which does not exhibit any significant endogenous, time or voltage-dependent K+ selective currents Citation[31]. We have previously shown that oocytes microinjected with hKv1.4 message exhibit rapidly inactivating outward currents on membrane depolarization Citation[29]. A schematically represents the transmembrane topologies of the parental potassium channel constructs, hKv1.1 and hKv1.4; the C-terminal chimæric constructs 4/1C and 4/2C; and the C-terminal deletions of hKv1.4: 1.4ΔC23 and 1.4ΔC40, where the last 23 and 40 amino acid residues respectively, have been deleted. An amino acid sequence comparison of hKv1.1, 1.2 and 1.4 demonstrates conservation in the transmembrane core and variations at the N- and the C-termini Citation[29]. B highlights the sequence differences at the C-terminus of these three channels.
We have swapped the C-terminus from hKv1.1 and hKv1.2 onto the transmembrane body of hKv1.4 to generate the 4/1C and 4/2C chimæras (A). Currents elicited upon depolarization in a non-inactivating channel, hKv1.1 and a fast inactivating channel, hKv1.4 are depicted in A and B and that of the chimæric channels, 4/1C and 4/2C in figures 2C and D respectively. The 4/1C chimæra inactivated in a manner similar to the wild type hKv1.4 (C) whereas the 4/2C chimæra surprisingly exhibited a slower and incomplete pattern of inactivation (D). E shows that the outward current from 4/2C channels fail to inactivate completely even upon prolonged depolarization for 3.6 seconds.
The entire membrane-spanning portion of the channel, containing the voltage sensing and transduction apparatus derived from hKv1.4, is common to all the constructs studied. In addition, this striking difference in inactivation of the 4/2C chimæra could not be due to a global perturbation of the protein structure since the voltage sensitivity for channel activation is identical for all the constructs as expected (A) as is the ion selectivity (data not shown).
Figure 3. Kinetic analysis of currents. (A) Voltage-dependence of K+ currents: voltage-dependent activation of hKv1.4 (○), 4/1C (•) and 4/2C (▵). The curves through the data were best fit to Boltzmann functions as described in experimental procedures; (B) activation kinetics: time constants for the activation of the currents for hKv1.4 (○), 4/1C (•) and 4/2C (▵), obtained by fitting the currents to a double exponential function and plotting the faster time constant as a function of trans-membrane potential; (C) inactivation kinetics: inactivation time constants for hKv1.4 (○), 4/1C (•) and 4/2C (▵), obtained by fitting the currents to a double exponential function and plotting the slower time constant against the membrane potential. The error bars for all data sets represent SEM. (n≥8).
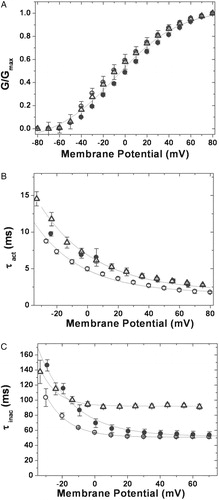
We then sought to analyse the kinetics of channel activation, inactivation and recovery from inactivation of these chimæras and compare them with their parental channels. At a potential of 0 mV, the time constant for channel activation (τact) was 7.0 ms for both chimæras against a value of 5.0 ms for hKv1.4 channels (see B). The τact for hKv1.1 had a value of 6.0 ms and hKv1.2 was 14.0 ms at the same potential (data not shown). The rat Kv2.1 channel activates much faster than its human analog and C-terminal domain swaps of these two channels demonstrate that a region at the C-terminus of Kv2.1 designated as the ‘C-terminal activation’ (CTA) domain accounts for the differences in activation behavior Citation[27]. Our data is consistent with this finding of an effect of a C-terminal influence on activation kinetics of voltage gated potassium channels Citation[27].
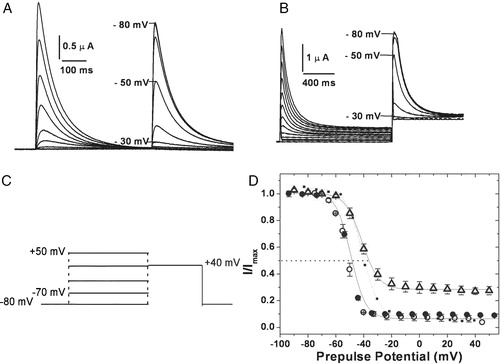
The inactivation kinetics of hKv1.4 and the 4/1C chimæra were similar with time constants of inactivation (τinac) of 58.6±0.43 ms and 68.4±1.8 ms respectively (see C). The rates of inactivation were significantly reduced in the 4/2C chimæra (τinac of 94.7±2.1 ms vs 58.6±0.43 ms for hKv1.4) (C). The voltage dependence of steady state inactivation was estimated by holding the channels at a range of potentials for 500 ms (or for 1.5 seconds in case of slower inactivating constructs such that a steady state of inactivation could be attained) and then stepping to a common test potential of +40 mV (see A and 4B). The current at the test potential represents channels still available for opening. These normalized currents are presented in D as a function of pre pulse potentials. Boltzmann fits of these data yield mid points of inactivation of −50.0±0.1 mV, −42.0±0.3 mV (p ≪ 0.01) and −52.0±0.3 mV for 4/1C, 4/2C and hKv1.4 respectively (D). The shift in the V1/2 of inactivation of 4/2C by 10 mV towards the positive direction is indicative of a change in the relative stabilities of the open and inactive states compared to hKv1.4. The C-terminus from hKv1.2 may be responsible for this effect since such a shift is not observed in the case of the 4/1C chimæra with residues from the C-terminus of hKv1.1.
Figure 4. Voltage dependence of inactivation. Steady state inactivation kinetics: steady state inactivated channels obtained by delivering a series of depolarizing prepulses ranging from −80 mV to +50 mV until a steady state of inactivation is achieved, were stepped to a test potential of +40 mV (C). The currents elicited upon depolarization are shown for hKv1.4 (A) and 4/2C chimæra (B). The normalized peak currents at the test potential of +40 mV are plotted as a function of the prepulse potentials and fitted to Boltzmann functions (D). The V1/2 for 4/2C was estimated by renormalizing to the minimum value, thereby analyzing only the inactivating portion of the currents (represented as the dotted line). The calculated slope factors were 5.32±0.24 for hKv1.4 and 4/1C and 5.96±0.62 for the 4/2C construct. The renormalized curve for 4/2C had a slope factor of 6.74±0.32. The error bars for all data sets represent SEM. (n≥8).
The dramatic decline in inactivation rates does not however, quite reach typical C-type inactivation rates with τ in the range 89–95 ms. Further, C-type inactivation has been shown to be sensitive to external K+ and TEA Citation[32]. A shows that inactivation kinetics are insensitive to external K+, while B shows that the rates are not influenced by external TEA up to 40 mM. The inactivation seen in the chimæric channels is thus of the N-type.
Figure 5. External potassium and TEA independence of inactivation kinetics. (A) Fold change in τinac upon varying the external K+ concentrations: the time constant of inactivation at 0 mV estimated from oocytes expressing 4/2C channels in the presence of 10, 48, 73 and 96 mM [K+]out was normalized using the τinac value at 2 mM [K+]out. These values were then plotted against the [K+]out values and fit to a straight line function. (B) Fold change in τinac in the presence of different concentrations of externally applied TEA: the time constant of inactivation was calculated for currents recorded from oocytes expressing 4/2C channels with 0, 10, 20 and 40 mM TEA in the bath solution at 0 mV. The τinac values estimated for these currents recorded in the presence of 10, 20 and 40 mM TEA were normalized using the τinac value estimated in the control oocyte with no TEA in the bath. These values were then plotted against the concentration of TEA and fit to a straight line function.
![Figure 5. External potassium and TEA independence of inactivation kinetics. (A) Fold change in τinac upon varying the external K+ concentrations: the time constant of inactivation at 0 mV estimated from oocytes expressing 4/2C channels in the presence of 10, 48, 73 and 96 mM [K+]out was normalized using the τinac value at 2 mM [K+]out. These values were then plotted against the [K+]out values and fit to a straight line function. (B) Fold change in τinac in the presence of different concentrations of externally applied TEA: the time constant of inactivation was calculated for currents recorded from oocytes expressing 4/2C channels with 0, 10, 20 and 40 mM TEA in the bath solution at 0 mV. The τinac values estimated for these currents recorded in the presence of 10, 20 and 40 mM TEA were normalized using the τinac value estimated in the control oocyte with no TEA in the bath. These values were then plotted against the concentration of TEA and fit to a straight line function.](/cms/asset/5812f069-8ddd-423f-859e-97cb3d18660d/imbc_a_119049_f0005_b.jpg)
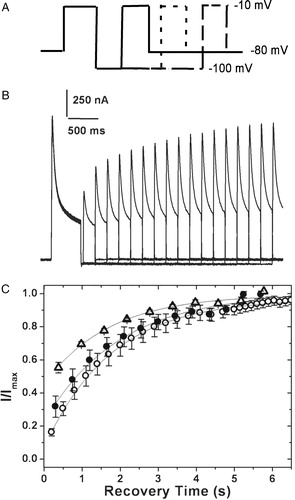
All of the chimæric constructs examined have the same inactivating particle and presumably the same binding site, which is expected to lie within the transmembrane domain Citation[9]. Variation in the rate and extent of inactivation may then reflect perturbations in either the accessibility of the ‘ball’ binding site or stabilization of the ball-bound inactivated state. Recovery from inactivation, on the other hand, is expected to be dominated by the rate of ball ‘unbinding’ from its receptor or the off rate. Release of the ball from its binding site is also voltage-dependent and this recovery is favored at hyperpolarizing potentials. presents data on channels which were allowed to recover at −100 mV following steady state inactivation at −10 mV. The relative current amplitudes obtained on stepping back to the test potential of −10 mV have been plotted as a function of recovery time (see C). The observed time constants of recovery from inactivation are 1678 ms for the 4/2C chimæra, as against values of around 1950 ms for both hKv1.4 and the 4/1C chimæra. This observation suggests that the altered inactivation kinetics seen in the case of the 4/2C chimæra may be due to perturbations affecting the stability of the inactivated state.
Figure 6. Recovery from inactivation. (A) Pulse protocol used: the membrane potential was stepped from a holding potential of −80 mV to −10 mV for 750 ms. The cell was then hyperpolarized to −100 mV for varying periods of time before stepping back to the test potential of −10 mV. (B) The currents obtained in oocytes expressing 4/2C when subjected to the pulse protocol described above. (C) The fraction of channels recovered was plotted as a function of recovery time and fitted to a single exponential function. The 4/2C (▵) chimæra data fit with a single time constant of 1678 ms as against 1953 ms for channels encoded by hKv1.4 (○), and 4/1C (•) channels. Only one in every three data points recorded are represented in order to retain clarity of the figures. The error bars for all data sets represent SEM. (n≥8).
To investigate this in detail, the divergent C-terminal sequences of hKv1.1, 1.2 and 1.4 were analyzed using several secondary structure predicting algorithms (PHD, Predator, DPM, DSC, GOR4, HNNC, SIMPA96, SOPM, Sec. Cons, Target 99 (http://npsa-pbil.ibcp.fr)). Structural elements predicted by more than seven out of ten programs have been indicated in A. The structures predicted for the three C-terminal segments are not identical. A secondary structure element that is common to hKv1.1 and hKv1.4, but absent in hKv1.2 is the distal-most helix predicted in the former pair. It is conceivable that the presence of this helical segment correlates with rapid inactivation in the family of chimæric proteins reported here. Cormier and coworkers have earlier reported the involvement of a C-terminal helical segment in rapid inactivation of the cardiac voltage gated sodium channel, Nav1.5 (SCNA5) Citation[33]. In order to extend these correlates to Kv channels, we generated two C-terminal truncation mutants of hKv1.4: the 1.4ΔC40 construct where deletion of the last 40 C-terminal residues results in disruption of the predicted helical structure; and the 1.4ΔC23 construct where truncation of the last 23 residues retains the helical stretch (A). Outward currents elicited from 1.4ΔC40 and 1.4ΔC23 constructs are shown in B and 7D respectively. C presents outward currents from 1.4ΔC40 on prolonged depolarization of 7 seconds. Note the residual steady state current at the end of the pulse amounting to 25% of the peak current. Such inactivation as is seen in this construct is significantly slower than in hKv1.4 (τinac=88.9±1.2 ms against 58.6±0.4 ms for hKv1.4; p≪0.01) (see A). The V1/2 of inactivation for this construct was estimated to be −44.01±1.1 mV as against −52.0±0.3 mV for hKv1.4 (p=0.015) (B). The recovery from inactivation was faster than for hKv1.4 (τrec of 1096 ms, C). The physiology of this construct resembles that of the 4/2C chimæra in that inactivation is slow and incomplete and that the recovery of inactivation is faster than that observed for hKv1.4.
Figure 7. C-terminal truncation constructs. (A) Secondary structure prediction: a secondary structure prediction of the C-terminal residues of hKv1.1, hKv1.2 and hKv1.4, done as described in experimental procedures. Shaded regions represent predicted helical stretches. Arrows indicate residues where stop codons were introduced while generating the 1.4ΔC23 and 1.4ΔC40 constructs. The absence of the predicted helical structure in hKv1.2 is to be noted. Currents elicited in oocytes expressing the 1.4ΔC40 construct upon depolarization to (B) 320 ms and (C) 7 seconds. (D) 1.4ΔC23 construct in response to depolarizing pulses from a holding potential of −80 mV to the potentials indicated against the traces, with an interval of 10 s between two successive sweeps.
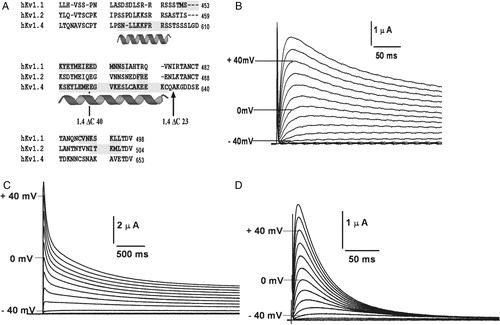
Figure 8. Inactivation kinetics and recovery from inactivation of the C-terminal truncation constructs. (A) Inactivation kinetics: inactivation time constants for hKv1.4 (○), 1.4ΔC40 (▴) and 1.4ΔC 23 (□), obtained by fitting the currents to a double exponential function and plotting the slower time constant against the membrane potential. The error bars for all data sets represent SEM. (n≥8). (B) Voltage dependence of inactivation: steady state inactivation kinetics: steady state inactivated channels obtained by delivering a series of depolarizing prepulses ranging from −80 mV to +50 mV until a steady state of inactivation is achieved, were stepped to a test potential of +40 mV. The normalized peak currents at this potential are plotted as a function of the prepulse potentials and fitted to Boltzmann functions. The mid points of inactivation obtained thus were −43.01 mV and −52.0±0.1 mV for 1.4ΔC40 and hKv1.4 respectively. The V1/2 for 1.4ΔC40 was estimated by renormalizing to the minimum value, thereby analyzing only the inactivating portion of the currents. The calculated slope factors were 5.32±0.24 for hKv1.4, and 1.4ΔC23 and 5.96±0.62 for the 1.4ΔC40 construct. The renormalized curve for 1.4ΔC40 had a slope factor of 6.74±0.32. The error bars for all data sets represent SEM. (n≥8). (C) Recovery from inactivation: the membrane potential was stepped from a holding potential of −80 mV to −10 mV for 750 ms. The cell was then hyperpolarized to −100 mV for varying periods of time before stepping back to −10 mV. The fraction of channels recovered was plotted as a function of recovery time and fitted to a single exponential function. The 1.4ΔC40 (▴) construct data fit with a single time constant of 1096 ms as against 1953 ms for channels encoded by hKv1.4 (○) and 1.4ΔC 23 (□) constructs. Only one in every three data points recorded are represented in order to retain clarity of the figures. The error bars for all data sets represent SEM. (n≥8).
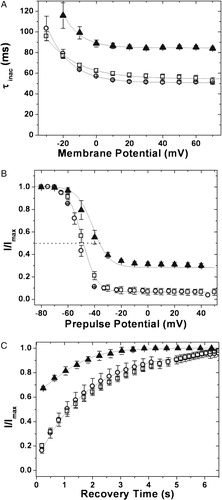
On the other hand, oocytes expressing the 1.4ΔC23 construct with an intact helix at the C-terminal, exhibited currents that inactivated rapidly and completely (D) with a time constant of inactivation of 62.0±1.1 ms at 0 mV (A). The V1/2 of inactivation and the rate of recovery from inactivation for this construct are comparable to hKv1.4 (B and 8C).
A cumulative analysis of the data, in particular the time constants of recovery from inactivation, obtained from the chimæric channels as well as truncation mutants, suggests that the C-terminal helix plays a role in stabilizing the inactivated state. Loss of this helix does not, however, perturb the structure of the channel much, as evident from similar voltage dependence and kinetics of activation (see A) as well as ion selectivity (data not shown) of all the constructs investigated here.
The significant non-inactivating component in the 4/2C chimæra and the 1.4ΔC40 construct could arise from a fraction of the channels undergoing modifications that render it incapable of rapid N type inactivation. Alternately, the observed physiology could arise from rapid unbinding of the ball even at depolarizing potentials as reported for a Shaker construct Citation[18]. Full-length hKv1.4, 1.4ΔC23 and the 4/1C chimæra, all of which have an intact predicted C-terminal helix, recover with time constants of recovery of around 1950 ms at −100 mV. The 4/2C chimæra on the other hand, recovers more rapidly with a time constant of 1678 ms at −100 mV (C). The 1.4ΔC40 construct, which also lacks the predicted C-terminal helix, recovers even more rapidly from inactivation with a time constant of 1096 ms at −100 mV (C). A critical glutamate in the S4–S5 linker has been implicated to be important in the binding of the inactivation particle in Shaker channels Citation[10]. However, the corresponding residue, E 477 in hKv1.4, is present in all the constructs studied and so cannot be responsible for the variations in ball unbinding seen.
Phosphorylation of certain key residues has been shown to affect inactivation in Kv1.4 Citation[34] and in Kv3.4 Citation[35]. However, the residues reported on are conserved among all the constructs studied. Further, a motif search for a potential PKA phosphorylation site, as reported for Shaker channels Citation[36], was performed on hKv1.4 using the NetPhos 2 server (http://tw.expasy.org/cgi-bin/niceprot.pl?KCNA4_HUMAN). The results revealed a potential Serine at position 599 which may be phosphorylated by PKA. However this site as well as additional putative phosphorylation sites, was conserved among the three channels, hKv1.1, 1.2 and 1.4. The issue of accessibility of these sites remains an open question.
N-type inactivation is also modulated by interaction with lipids such as PIP2. The residues involved are present on the N-terminus Citation[37] and hence conserved across all the constructs studied. The issue of accessibility was addressed by modulating PIP2 levels. Wortmannin, which depletes cellular PIP2 by inhibiting PI 4 Kinase Citation[38], has no effect on inactivation kinetics (data not shown). Treating oocytes with another PI 4 Kinase inhibitor, Quercetin Citation[39] also was without effect, as was complexing PIP2 with Neomycin sulphate Citation[40] (data not shown).
The difference seen in inactivation kinetics hence could be in the variant C-termini and its interactions with the inactivating particle. The importance of a C-terminal helical structure in fast inactivation of a cardiac sodium channel Nav1.5 (SCNA5) Citation[33] has been reported. A recent report from the same group demonstrates an interaction between the sodium channel inactivating domain (III-IV linker) and the C-terminal domain Citation[41].
A growing body of evidence suggests that the C-terminal end of the K+ channel may interact with the T1 domain. Cryo-electron microscopic images of Shaker and Kv1.1 channels are mushroom-shaped with the T1 domain suspended like a ‘hanging gondola’ below the transmembrane domain. Excess density in the ‘gondola’ has been interpreted in terms of the presence of C-terminal residues Citation[42]. 3-D reconstruction of single particle electron microscopy images suggests that the C-terminal tail together with the T1–S1 linker constitute the cable suspending the T1 gondola Citation[43]. The distal end of the tail is proposed to lie on the membrane-facing surface of the gondola. Schulteis and co workers have shown that residues C96 (in the T1 domain) and C505 (in the C-terminal tail of an adjacent subunit) are within disulfide bonding distance Citation[44] and the C-terminus has been proposed to proceed back along the T1 surface by wrapping around it and forming a compact density on the surface facing the membrane Citation[43]. The T1 surface has exposed hydrophobic residues which might interact with the C-terminal helix. Disruption of the helix or its complete absence may release the tail into the fluid volume of the basket and interfere with N type inactivation. Conversely, the shift in the V1/2 of inactivation from −52.0±0.3 mV (for hKv1.4) to approximately −43 mV (for 4/2C and 1.4ΔC40 channels), along with the faster recovery from inactivation seen in the 4/2C chimæra and 1.4ΔC40 suggest the involvement of the C-terminal helix in interactions that stabilize the inactivated state.
Early evidence for a possible N- and C-terminal interaction was seen in electrophysiological studies on a series of N- and C-terminal deletions in drk 1 channels, where C-terminal deletions were found to profoundly affect both activation and inactivation kinetics Citation[45]. Later investigations of C-terminal deletions in Kv channels have revealed a role in inactivation. In the case of mouse Shal K+ channel, mKv4.1, an NH2 terminal truncation abolished the rapid phase of inactivation and altered voltage-dependent gating as expected but surprisingly, these effects could be mimicked by deletions in the hydrophilic C-terminus Citation[46]. The deletion of 149 C-terminal amino acids in Kv4.3a significantly slowed its rate of recovery from inactivation. However, neither the rate of inactivation nor voltage dependence of activation or inactivation were affected Citation[47]; suggesting a specific perturbation of unbinding of the ‘ball’. Domain swap experiments with Shaker had implicated the C-terminus in recovery from inactivation Citation[18]. A report on HERG double-deletion constructs showed additive effects of the N- and C-termini on the voltage dependence of activation, the kinetics of inactivation and recovery from inactivation Citation[48]. The additivity of the effects would suggest that deletions in the two regions bring about changes in these parameters by independent mechanisms.
Presence of the helical structure at the C-terminus appears to stabilize the inactivated state of hKv1.4 channel. Mutations affecting N-type inactivation are of clinical significance as evidenced by the number of channelopathies associated with mutations affecting inactivation. Hence a study to investigate the elements of the molecular apparatus involved in fast inactivation would be of clinical interest. Our study establishes that the C-terminus is implicated in the process of fast inactivation in symphony with the N-terminus. We have localized the possible area of interaction to a helical structure at the distal end of C-terminus.
This paper was first published online on prEview on 19 July 2005.
K.S. and A.V. acknowledge support from the Kanwal Rekhi Scholarship of the TIFR Endowment Fund. This work was supported by core funds from NCBS-TIFR and a research grant (# P81102) from CSIR, India.
References
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000; 80: 555–592
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998; 280: 69–77
- Kreusch A, Pfaffinger PJ, Stevens CF, Choe S. Crystal structure of the tetramerization domain of the Shaker potassium channel. Nature 1998; 392: 945–948
- Shen NV, Chen X, Boyer MM, Pfaffinger PJ. Deletion analysis of K+ channel assembly. Neuron 1993; 11: 67–76
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990; 250: 533–538
- Varshney A, Mathew MK. Cytoplasmic residues influence the voltage-dependence of the gating of human K+ channels. Neuroreport 2000; 11: 2913–2917
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 1990; 250: 568–571
- Antz C, Geyer M, Fakler B, Schott MK, Guy HR, Frank R, Ruppersberg JP, Kalbitzer HR. NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature 1997; 385: 272–275
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001; 411: 657–661
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature 1991; 353: 86–90
- Moorman JR, Kirsch GE, Brown AM, Joho RH. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science 1990; 250: 688–691
- Deschenes I, Trottier E, Chahine M. Implication of the C-terminal region of the alpha-subunit of voltage-gated sodium channels in fast inactivation. J Membr Biol 2001; 183: 103–114
- Wang J, Myers CD, Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K(+) channels. J Gen Physiol 2000; 115: 749–758
- Marten I, Hoshi T. Voltage-dependent gating characteristics of the K+ channel KAT1 depend on the N and C termini. Proc Natl Acad Sci USA 1997; 94: 3448–3453
- Varshney A, Mathew MK. Inward and outward potassium currents through the same chimeric human Kv channel. Eur Biophys J 2003; 32: 113–121
- Vanoye CG, MacGregor GG, Dong K, Tang L, Buschmann AS, Hall AE, Lu M, Giebisch G, Hebert SC. The carboxyl termini of K(ATP) channels bind nucleotides. J Biol Chem 2002; 277: 23260–23270
- Bentley GN, Brooks MA, O'Neill CA, Findlay JB. Determinants of potassium channel assembly localised within the cytoplasmic C-terminal domain of Kv2.1. Biochim Biophys Acta 1999; 1418: 176–184
- Iverson LE, Rudy B. The role of the divergent amino and carboxyl domains on the inactivation properties of potassium channels derived from the Shaker gene of Drosophila. J Neurosci 1990; 10: 2903–2916
- Ludwig J, Owen D, Pongs O. Carboxy-terminal domain mediates assembly of the voltage-gated rat ether-a-go-go potassium channel. Embo J 1997; 16: 6337–6345
- Maingret F, Honore E, Lazdunski M, Patel AJ. Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K(+) channel. Biochem Biophys Res Commun 2002; 292: 339–346
- Pessia M, Bond CT, Kavanaugh MP, Adelman JP. Contributions of the C-terminal domain to gating properties of inward rectifier potassium channels. Neuron 1995; 14: 1039–1045
- Daram P, Urbach S, Gaymard F, Sentenac H, Cherel I. Tetramerization of the AKT1 plant potassium channel involves its C-terminal cytoplasmic domain. Embo J 1997; 16: 3455–3463
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science 1998; 279: 403–406
- Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. Embo J 1997; 16: 5472–5479
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995; 80: 795–803
- Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, Richard P, Funke H, Schwartz K, Coumel P, Hainque B, Guicheney P. C-terminal HERG mutations: The role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation 1999; 99: 1464–1470
- Ju M, Stevens L, Leadbitter E, Wray D. The Roles of N- and C-terminal determinants in the activation of the Kv2.1 potassium channel. J Biol Chem 2003; 278: 12769–12778
- Hatano N, Ohya S, Muraki K, Clark RB, Giles WR, Imaizumi Y. Two arginines in the cytoplasmic C-terminal domain are essential for voltage-dependent regulation of A-type K+ current in the Kv4 channel subfamily. J Biol Chem 2004; 279: 5450–5459
- Ramaswami M, Gautam M, Kamb A, Rudy B, Tanouye MA, Mathew MK. Human potassium channel genes: Molecular cloning and functional expression. Mol Cell Neurosci 1990; 1: 214–223
- Varshney ASK, Mathew MK. Modulation of voltage sensitivity by N-terminal cytoplasmic residues in human Kv1.2 channels. Eur Biophys J 2002; 31: 365–372
- Dascal N. The use of Xenopus oocytes for the study of ion channels. CRC Crit Rev Biochem 1987; 22: 317–387
- Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res 1998; 82: 739–750
- Cormier JW, Rivolta I, Tateyama M, Yang AS, Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: Evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem 2002; 277: 9233–9241
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2 + /calmodulin-dependent protein kinase. J Neurosci 1997; 17: 3379–3391
- Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron 1994; 13: 1403–1412
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+ channel inactivation gating by the cAMP-dependent protein kinase. Neuron 1994; 12: 1097–1109
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 2004; 304: 265–270
- Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol 2002; 4: 329–336
- Yagyu K, Kitagawa K, Irie T, Wu B, Zeng XT, Hattori N, Inagaki C. Amyloid beta proteins inhibit Cl(-)-ATPase activity in cultured rat hippocampal neurons. J Neurochem 2001; 78: 569–576
- Arbuzova A, Martushova K, Hangyas-Mihalyne G, Morris AJ, Ozaki S, Prestwich GD, McLaughlin S. Fluorescently labeled neomycin as a probe of phosphatidylinositol-4, 5-bisphosphate in membranes. Biochim Biophys Acta 2000; 1464: 35–48
- Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol 2004; 123: 155–165
- Sokolova O, Kolmakova-Partensky L, Grigorieff N. Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure (Camb) 2001; 9: 215–220
- Sokolova O, Accardi A, Gutierrez D, Lau A, Rigney M, Grigorieff N. Conformational changes in the C terminus of Shaker K+ channel bound to the rat Kvbeta2-subunit. Proc Natl Acad Sci USA 2003; 100: 12607–12612
- Schulteis CT, Nagaya N, Papazian DM. Intersubunit interaction between amino- and carboxyl-terminal cysteine residues in tetrameric shaker K+ channels. Biochemistry 1996; 35: 12133–12140
- VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM. Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron 1990; 5: 433–443
- Jerng HH, Covarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J 1997; 72: 163–174
- Ohya S, Tanaka M, Oku T, Furuyama T, Mori N, Giles WR, Watanabe M, Imaizumi Y. Regional expression of the splice variants of Kv4.3 in rat brain and effects of C-terminus deletion on expressed K+ currents. Life Sci 2001; 68: 1703–1716
- Aydar E, Palmer C. Functional characterization of the C-terminus of the human ether-a-go-go-related gene K(+) channel (HERG). J Physiol 2001; 534: 1–14