Abstract
Neurosecretion is catalyzed by assembly of a soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE)-complex composed of SNAP-25, synaptobrevin and syntaxin. Munc 18-1 is known to bind to syntaxin in vitro. This interaction prevents assembly of the SNARE-complex, but might also affect intracellular targeting of the proteins. We have fused syntaxin and Munc 18 to the yellow- (YFP) or cyan-fluorescence-protein (CFP) and expressed the constructs in CHO- and MDCK-cells. We have studied their localization with confocal microscopy and a possible protein-protein interaction with fluorescence-resonance energy transfer (FRET). YFP-syntaxin localizes to intracellular membranes. CFP-Munc 18 is present in the cytoplasm as expected for a protein lacking membrane targeting domains. However, Munc 18 is redirected to internal membranes when syntaxin is coexpressed, but only limited transport of the proteins to the plasma membrane was observed. An interaction between Munc 18 and syntaxin could be demonstrated by FRET using two methods, sensitized acceptor fluorescence and acceptor photobleaching. A mutation in syntaxin (L165A, E166A), which is known to inhibit binding to Munc 18 in vitro, prevents colocalization of the proteins and also the FRET signal. Thus, a protein-protein interaction between Munc 18 and syntaxin occurs on intracellular membranes, which is required but not sufficient for quantitative transport of both proteins to the plasma membrane.
Introduction
Neuronal soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE)-proteins have been identified as key players in neurosecretion. They are located on synaptic vesicles (synaptobrevin 2, also called VAMP 2) or on the presynaptic plasma membrane (syntaxin 1 and SNAP-25). Formation of a SNARE-complex triggers fusion of vesicles with the plasma membrane. The neuronal SNARE-complex consists of a four-helix bundle, with one helix from synaptobrevin, one from syntaxin and two from SNAP-25 Citation[1], Citation[2].
The regulation and fine-tuning of SNARE-complex formation are now beginning to be elucidated. One important regulator of SNARE-complex formation is Munc 18-1 Citation[1]. Munc 18-1 (also called nSec1) was identified by its high affinity interaction with syntaxin in pull down experiments with brain extracts Citation[3–5]. Subsequent experiments with recombinant proteins showed that binding of syntaxin to the SNARE-complex and to Munc 18-1 are mutually exclusive Citation[6]. The structural basis for this observation was elucidated by X-ray crystallography of the Munc 18-1-syntaxin complex. Besides the domain present in the SNARE-complex, syntaxin contains an additional helical domain. This Habc domain can bind to the SNARE-domain thereby blocking its accessibility for SNARE-complex formation. Munc 18-1 binds to this closed conformation of syntaxin, and stabilizes it Citation[7], Citation[8]. Thus, Munc 18-1 reduces the amount of free, fusion-competent syntaxin and should inhibit neurosecretion. It therefore came as a surprise that mice with a deletion of the Munc 18-1 gene had a complete block in neurosecretion. Formation of synapses and docking of synaptic vesicles was not impaired in the knock-out mice Citation[9]. Thus, Munc 18-1 might also play an active role during neurosecretion, probably after SNARE-mediated docking of synaptic vesicles.
The interaction between syntaxin 1 and Munc 18-1 might also be required for intracellular trafficking and membrane binding of the proteins. In neurons Munc 18-1 is found in a soluble and a membrane-associated form. Munc 18-1 does not contain a transmembrane region and is not modified by lipids which might anchor the protein to membranes. It is therefore believed that the interaction with syntaxin is required for membrane binding of Munc 18-1. Indeed, syntaxin 1 and Munc 18-1 colocalize in neurons along the axonal membrane Citation[10]. Likewise, coexpression of syntaxin in non-neuronal cells redirects Munc 18-1 from the cytoplasm to the plasma membrane Citation[11]. However, a direct interaction between both proteins was not demonstrated in these studies.
On the other hand, Munc 18-1 also influences intracellular trafficking of syntaxin 1. Syntaxin, like other SNARE-proteins, probably travels via ER and Golgi apparatus to the plasma membrane. Syntaxin does not contain a classical N-terminal signal sequence for cotranslational insertion into the ER. Instead, the C-terminal hydrophobic transmembrane region is essential for its association with the membrane. When syntaxin is transfected into non-neuronal cells it does not travel to the plasma membrane, but accumulates in the Golgi apparatus. In certain cell types this has a profound influence on vesicular trafficking. General transport along the exocytotic pathway is blocked and the whole Golgi complex disassembles. Coexpression of Munc 18-1 rescues the transport block and also allows syntaxin 1 to reach the plasma membrane Citation[12].
From these studies it appears that membrane targeting and trafficking of syntaxin and Munc 18-1 are intimately connected. However, it is not known whether both proteins actually interact in a compartment along the exocytotic pathway. The upcoming technology of fluorescence-resonance energy transfer (FRET) allows now to analyze protein-protein interactions inside cells. Proteins of an interacting pair are fused to the cyan fluorescence protein (CFP) or to the yellow fluorescence protein (YFP), spectral variants of the green fluorescence protein (GFP). If both proteins are in close proximity (usually below 50Å) the energy emitted by the donor CFP is transferred to the acceptor YFP. As a result the fluorescent emission of the acceptor YFP is enhanced by excitation of the donor CFP Citation[13], Citation[14]. Using this technology, Liu and colleagues demonstrated FRET between YFP-synaptobrevin and CFP-SNAP 25 in neurons. The FRET signal increases during stimulation of the neurons suggesting that SNARE-complexes are already present in resting cells, but more complexes are formed during neurosecretion Citation[15]. The same group showed FRET between YFP-SNAP 25 and CFP-syntaxin on the plasma membrane of PC-12 cells Citation[16]. Thus, the CFP/YFP pair of fluorescent proteins is suitable for FRET-analysis. Here we have analyzed whether the described protein-interaction between Munc 18-1 and syntaxin can be demonstrated by FRET and whether it occurs on the plasma membrane or in an intracellular compartment.
Materials and methods
Cloning of syntaxin and Munc 18-1
Plasmids containing syntaxin-1b and Munc 18-1, both cloned from rat, were obtained from James E. Rothman (MSKCC, New York). The plasmids pECFP-N1, pECFP-C1, and pEYFP-C1 were from CLONTECH.
The syntaxin gene was amplified using pGEX-KG syntaxin plasmid as template. The sense primer 5′GTTCCGCGTGGATCCCCGG GAATT 3′ contained a BamHI restriction site and the anti sense primer 5′GGTCCTGGGGAAGAGAAGGGTACCGGCCTACAAGCC 3′ contained a KpnI site. The amplified product was cloned into the MCS region between BglII and KpnI of pEYFP-C1. The resultant clone (YFP-syntaxin) expressed a fusion protein with YFP at the N-terminus of the syntaxin 1b insert. The sequence between the colour-tag and syntaxin 1b is: eYFP-SGLRSPGISGGGGGIPDRTQ- syntaxin 1B. RTQ are the first amino acids from the syntaxin 1b gene.
The Munc 18-1 gene was tagged with CFP either at the N-terminus (CFP-Munc 18-1) or at the C-terminus (Munc 18-1-CFP). For the construction of CFP-Munc 18-1 the plasmid pEGFP-C1 Munc 18-1 was cut with the restriction enzymes EcoRI and XbaI and the Munc 18-1 gene was ligated into the respective cloning sites of pECFP-C1. The sequence between the colour-tag and Munc 18-1 is: eCFP-SGLRSRAQASNSAVDGTAGPM-Munc 18-1. M is the authentic start codon of Munc 18-1.
For the construction of Munc 18-1-CFP, PCR was performed using GFP-Munc 18-1 gene as a template contained in pEGFP-C1 plasmid. The sense primer 5′ GAC GGT AGA TCT GGC CCC ATG GCC CCC ATT GGC 3′ contained a BglII restriction site and the anti-sense primer 5′ AGG GCT GCA GAG GTA CCC TGC TTA TTT CTT CGT CTG TTT T 3′ contained a KpnI restriction site. The amplified Munc 18-1 gene was cloned into the MCS region between Bgl II and Kpn I of pECFP-N1. The sequence between the colour-tag and Munc 18-1 is Munc 18-1: K T Y E E I S R V P R A R D P P V A T M- eCFP. S and M (in bold) are the end of the Munc 18-1 gene and start of eCFP, respectively.
A mutant of syntaxin (L165A E166A) was constructed with a substitution of both the Leu-165 and Glu-166 to alanine. Mutagenesis was carried out using the ‘QuikChange’ Site-directed Mutagenesis kit (Stratagene, USA). The sense primer was 5′ATCCAGAGGCAGGC- GGCGATCACCGGTAGGACTACTACC 3′ and the anti-sense primer was 5′GGTAGTAGTCC- TACCG GTGATCGCCGCTGCCTCTGGAT 3′. All of the plasmid DNAs were prepared using Qiagen plasmid purification kit and the constructs were confirmed by gene sequencing.
Transfection of CHO- and MDCK-cells
Chinese hamster ovary (CHO)-cells, Madin-Darby canine kidney (MDCK)-cells, PC-12 cells and BON-cells (a human pancreatic carcinoid cell line) were cultured in Dulbecco's mimimal essential-medium (DMEM) supplemented with 10% fetal calf serum. For transfections cells were seeded at different densities onto 3.5 cm diameter culture plates containing sterile 22×22 mm glass coverslips (no. 1 thickness, Fisher Scientific, Pittsburgh, PA) and incubated overnight. Transfections for confocal microscopy and for FRET-analysis were done on approximately 50% confluent cells. Typically, each transfection was done using 2 µg plasmid DNA and 2 µl of lipofectamine (Invitrogen). Both the plasmid DNA and lipofectamine were diluted respectively in 100 µl each serum free DMEM medium for 15 min. The plasmid DNA and lipofectamine were pooled and incubated for 30 min. The DNA-lipofectamine complex was diluted with an additional 800 µl of serum free medium. The cells grown in small petri-dishes were washed once with serum free medium and the DNA-lipofectamine mixture was overlaid on to the cells and further incubated at 37°C and 5% CO2. The medium containing DNA-lipofectamine was substituted with growth medium after 3–12 h after visualizing the morphology of the cells hourly for cytotoxicity. The cells were usually assayed for FRET or confocal microscopy at the end of 36 h.
Establishment of CHO-cell lines for permanent expression of Munc 18-1-CFP
The CHO cells were transfected with the Munc 18-1-CFP plasmid as described above and were incubated for 48 h on normal growth medium at 37°C, 5% CO2. The normal growth medium was substituted with a selective medium containing Geneticin (G418 sulphate) to a final concentration of 500 µg/ml. The cells were grown on selective medium for 10–14 days with a daily change of the medium. The positive clones were selected after screening for fluorescence. The Geneticin resistant colonies were picked and seeded into 6 well plates for further growth and maintained on the antibiotic at a concentration of 250 µg/ml. The selective medium was substituted with normal growth medium during the expression of the Munc 18-1-CFP fusion protein.
Confocal microscopy
After transfection, cells were cultured for 36 h. To identify intracellular compartments cells were then incubated with TRITC-wheat germ agglutinin (200 µg/ml, EY laboratories, California) or Bodipy-TR C5-ceramide complexed to BSA (5 µM, Molecular Probes, Oregon) for 30 min. Cells were then rinsed with medium and incubated in fresh medium for a further 90 (TRITC-WGA) or 30 (Bodipy-Cer) min at 37°C. Cells were washed in PBS, fixed with paraformaldehyde (3.7% in PBS) for 10 min at room temperature and again washed with PBS. Cover slips were mounted in ImmuMount (Shandon, Pittburgh, PA, USA) with the use of nail polish as a sealant. Expression and localization of proteins were observed under an inverted confocal microscope (Leica TCS-SP II system; Leica, Germany) controlled by Leica-confocal software (Version 2.5). Images of cells transfected with CFP-constructs were obtained with 458 nm excitation and an emission wave length of 470 to 600 nm. Images of cells transfected with the syntaxin-YFP-construct were obtained with 514 nm excitation and an emission wavelength of 530 to 600 nm. For experiments with double transfected cells emission wave length for CFP was 465 to 485 nm, for YFP 544 to 580 nm. To localize TRITC-WGA or Bodipy-ceramide cells were excited at 543 nm and emission was recorded at 555 to 620 nm. In these experiments YFP-syntaxin was excited at 488 nm and emission was recorded at 500 to 540 nm. To minimize cross-talk between CFP (or TRITC and Bodipy) and YFP-channels the PMT gain settings were established on each day when an experiment was performed on cells transfected with a single plasmid. The CFP cross-talk channel was defined as 514 nm laser line, but with CFP detection. The YFP cross-talk channel was defined as the 458 nm laser line, but with YFP detection. In each cross-talk channel, the appropriate PMT gain was lowered until there was no detectable signal in the channel. Double transfected cells were then observed with sequential image recording (mode: between frames, Leica confocal application letter, No 15, Nov. 2002) and a Leica×63 oil immersion objective. Final image processing was done with Adobe Photoshop 7.01 software.
FRET-analysis
FRET microscopy was performed using a Zeiss Axiovert 100 fluorescence microscope (Zeiss, Germany) equipped with separate filter wheels for excitation and emission operated by a Ludl MAC5000 controller (Ludl Electronic Products Ltd., Hawthorne, NY, USA) using a Chroma 86002v2 cfp/yfp filter set with a multi wavelength dichroic beam splitter. The following filter combinations were used: CFP – Excitation (Ex.) 430/25 nm, Emission (Em.) 470/30 nm, YFP – Ex. 500/20 nm, Em. 545/30 nm and FRET – Ex. 430/25 nm, Em. 545/30 nm. Images were acquired with a cooled CCD camera Coolsnap fx (Photometrics, AZ, USA) and analysed using the Metamorph 6.1 (Universal Imaging Corporation, PA, USA.) image analysis software.
In order to correct for instrument and sample specific backround fluorescence, Net FRET (nF) was calculated as follows Citation[16]:where I FRET, I YFP, and I CFP are intensities in each region of interest (ROI) under FRET, YFP, and CFP filter sets, respectively. A is a norm of the percentage of CFP bleed-through, and B is a norm of the percentage of YFP bleed-through under the FRET filter set. There were no bleed-through signals from CFP under YFP filter sets and vice versa. The norms A and B for the system used in the present study were 0.06 (YFP-syntaxin wild-type), 0.11 (YFP-syntaxin mutant) and 1.12 (CFP-Munc 18-1), 1.432 (nSec-1-CFP), respectively, which were determined by analysing images of cells expressing only CFP or YFP and quantifying the relative intensity ratio under the FRET/CFP or FRET/YFP filter sets.
Acceptor photobleaching
In order to verify the FRET-signal, the method of selective photobleaching of the acceptor was used. CFP, YFP and FRET images were taken as described above. Then the acceptor fluorophor was photobleached by YFP-excitation (500/20 nm) for 3 min and CFP, YFP and FRET images were taken again. This procedure abolishes FRET by destroying the ability of the acceptor to take up energy from the donor, in regions where FRET occurred this will result in an increase of the donor emission. To analyse the data, CFP images before and after photobleaching were compared and all images were processed to calculate net-FRET (nF).
Results and discussion
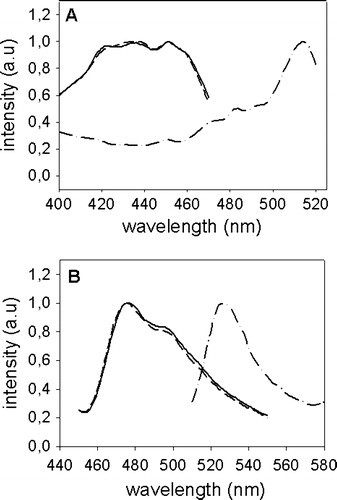
Syntaxin 1 and Munc 18-1 were fused at their N-termini to YFP and CFP by cloning the genes into the plasmids pEYFP-C1 and pECFP-C1, respectively. The resulting constructs were designated CFP-Munc 18-1 and YFP-syntaxin. Munc 18-1 was also cloned into pECFP-N1 (construct Munc 18-1-CFP) thereby attaching the C-terminus to the fluorescent protein. The plasmids were transfected into non-neuronal CHO-cells, which do not express endogenous syntaxin 1 and Munc 18-1. One day later cells were permeabilized and the excitation and emission spectra of the expressed proteins were measured (). Only the excitation spectra of CFP-Munc-18 and Munc-18-CFP differ slightly from published spectra of purified CFP and YFP alone, probably due to binding to Munc 18, but our constructs are nevertheless suitable for colocalization studies and FRET-analysis.
Figure 1. Excitation- and emission-spectra of CFP-Munc 18-1, Munc 18-1-CFP and YFP-syntaxin. CHO-cells were transfected with CFP-Munc 18-1 (——), Munc 18-1-CFP (-------) or YFP-syntaxin (—•—•—). Thirty-six hours later cells were lysed and excitation (A) and emission (B) spectra were measured using an Aminco Bowman Series II fluorescence spectrophotometer (Polytec, Waldbronn, Germany). Intensity in arbitrary units (a.u.) is plotted against the wavelength (nm).
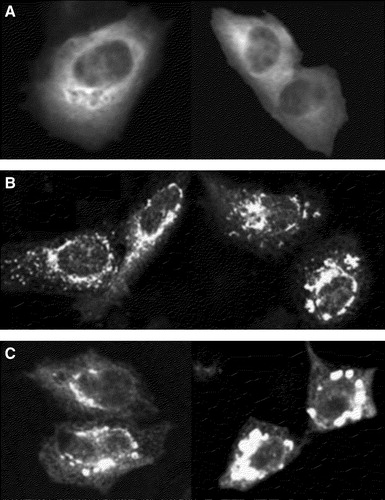
Next we tested the subcellular distribution of the proteins in transfected cells with confocal microscopy. CHO-cells expressing CFP-Munc 18-1 showed a diffuse cytosolic staining throughout the cell (A). A similar immunofluorescence pattern was observed by Perez-Branguli et al. for Munc 18-1 in transfected 29.3T human kidney cells Citation[11]. The cytosolic distribution of Munc 18-1 in non-neuronal cells suggests that the protein does not contain membrane targeting information.
Figure 2. Localization of Munc 18-1 and syntaxin in CHO-cells. CHO-cells were transfected with CFP-Munc 18-1 (A), YFP-syntaxin wild-type (B) or YFP syntaxin mutant L165A, E166A (C). Thirty-six hours later intracellular localization of proteins was visualized with confocal microscopy.
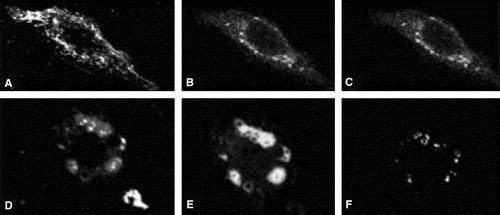
YFP-syntaxin localizes mainly to the perinuclear region (B). In order to identify this compartment, CHO-cells expressing YFP-syntaxin were costained with TRITC-wheat-germ-agglutinin (WGA), a marker for compartments along the endocytotic route (e.g., endosomes and the trans-Golgi-network (TGN)) or with Bodipy-ceramide, which is enriched in the Golgi-complex. YFP-syntaxin partially colocalizes with WGA, especially in the perinuclear region probably representing the TGN ( A–C). Perinuclear structures accumulating YFP-syntaxin are also stained with Bodipy-ceramide (, D–E). However, both stains do not precisely overlap indicating that ceramide-lipids and YFP-syntaxin segregate in this compartment. In summary, our results show that YFP-syntaxin does not travel to the plasma membrane, but is stuck in a perinuclear compartment, probably the Golgi, when expressed in non-neuronal cells Citation[12].
Figure 3. Colocalization of YFP-syntaxin with Golgi-markers in CHO-cells. CHO-cells were transfected with YFP-syntaxin wild-type. Thirty-six hours later cells were costained with TRITC-WGA (A–C) or Bodipy-ceramide (D–E) and intracellular localization of proteins and marker-stains was visualized with confocal microscopy. A and D: marker stains TRITC-WGA (A) and Bodipy-ceramide (D). B and E: YFP-syntaxin. C and F = pixels which are labelled both with YFP-syntaxin and the marker-stain.
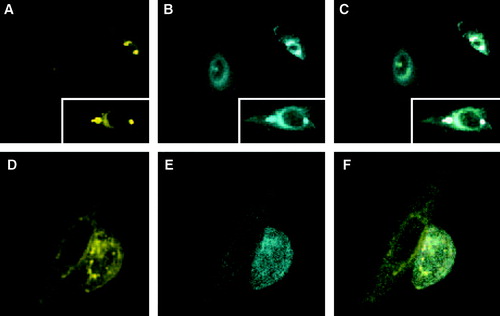
Does the subcellular distribution of syntaxin and Munc 18-1 change when both proteins are expressed in the same cell? In contrast to published studies with other cell types Citation[12], Citation[17] we did not observe transport of syntaxin to the plasma membrane after cotransfection of CHO-cells with CFP-Munc 18-1. One day (A) and two days (not shown) after transfection syntaxin still accumulates in the perinuclear region. However, a dramatic rearrangement of the distribution of CFP-Munc 18-1 was observed (B). Although some diffuse cytosolic fluorescence is still detectable, the majority of the CFP-Munc 18-1 signal now overlaps with the staining of YFP-syntaxin in the perinuclear region (C). This is in line with a recent publication describing colocalization of Munc 18-1 and syntaxin 1 in the Golgi-apparatus of epithelial cells Citation[17]. Thus, expression of syntaxin apparently provides a signal for membrane-binding of Munc 18-1.
Figure 4. Localization of Munc 18-1 and syntaxin in double transfected CHO-cells. (A–C) Munc 18-1 does colocalize with syntaxin wild-type. CHO-cells were double transfected with CFP-Munc 18-1 and YFP-syntaxin wild-type. Thirty-six hours later intracellular localization of proteins was visualized with confocal microscopy. (A) YFP channel, (B) CFP channel, (C) merged images. The insets are from a different optical field. (D–F) Munc 18-1 does not colocalize with the syntaxin mutant L165A, E166A. CHO-cells were double transfected with CFP-Munc 18-1 and YFP-syntaxin mutant L165A, E166A. Thirty-six hours later intracellular localization of proteins was visualized with confocal microscopy. (A) YFP channel, (B) CFP channel, (C) merged images.
Next we analyzed whether the colocalization of Munc 18-1 and syntaxin is due to a direct protein-protein interaction. CHO-cells were either single or double transfected with the plasmids and cells were observed with a digital camera under three sets of filters, a CFP-set, a YFP-set, and a FRET-set (). With cells expressing only CFP-Munc 18-1 or only YFP-syntaxin we did observe a bleed-through in the FRET-channel. This background was subtracted from the observed signal in the FRET-channel of double transfected cells according to a published procedure Citation[16]. The resulting image was pseudo-colored such that yellow and especially red signifies the highest FRET-signal. The resulting image (C) shows FRET inside the cell in the perinuclear region.
Figure 5. FRET with CFP-Munc 18-1 and YFP-syntaxin wild-type in CHO-cells. CHO-cells were double transfected with CFP-Munc 18-1 and YFP-syntaxin wild-type. Thirty-six hours later localization of proteins was visualized with a CCD camera. (A) CFP-channel, (B) YFP-channel, (C) FRET-channel. To correct for the bleed-through Net-FRET was calculated as described in ‘Methods’. Images were then pseudo-colored. The color bar represents relative degree of Net-FRET within the cells.
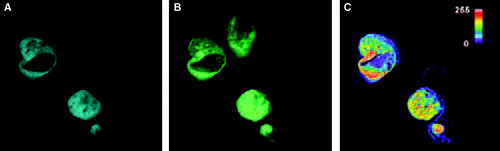
It is well documented that coexpression of syntaxin and Munc 18-1 in non-neuronal cells causes almost quantitative transport of both proteins to the plasma membrane Citation[11]. In MDCK-cells, which are known to allow plasma membrane transport of native (untagged) Munc 18-1 and syntaxin 1 Citation[17], our CFP-Munc 18-1 and YFP-syntaxin constructs colocalize mainly to intracellular compartments, but staining of the plasma membrane was also observed (, A and B). Colocalization of CFP-Munc 18-1 and YFP-syntaxin mainly at internal structures was also observed when the cells were analyzed at different times after transfection and when Munc 18-1-CFP instead of CFP-Munc 18-1 was used for cotransfection (data not shown). Thus, attachment of the colour-tag apparently interferes with quantitative transport of the Munc 18-syntaxin-complex to the plasma membrane.
Figure 6. Localization of CFP-Munc 18-1 and YFP-syntaxin in MDCK-cells and FRET-analysis with acceptor photobleaching. MDCK-cells were double transfected with CFP-Munc 18-1 and YFP-syntaxin wild-type. Thirty-six hours later localization of proteins was visualized with a CCD camera as decribed in . The acceptor fluorophor was then photobleached with YFP-excitation for 3 min and additional images were taken at all wavelengths. Upper panel: before bleaching. Lower panel: after bleaching. (A, D) CFP-channel, (B, E) YFP-channel, (C, F) FRET-channel corrected for bleed-through (Net-FRET calculated as described in ‘Methods’).
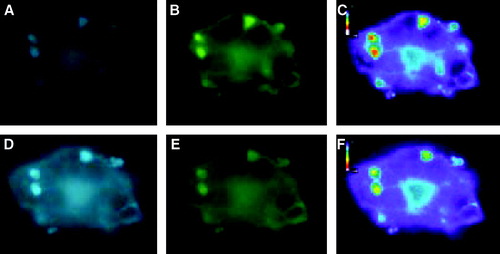
In the same experiment we observed a FRET-signal, mainly from internal structures (, C). The FRET-signal could also be verified with the acceptor photobleach method. The emission of light by the CFP-Munc 18-1 donor became more intense following photobleach of the YFP-syntaxin acceptor (, D and E) and the FRET-signal disappeared (, F). The intensity of the donor- and acceptor-fluorescence located on internal membranes and on the plasma membrane was also calculated before and after photobleaching. The fluorescence in the YFP-channel was reduced to 25% (internal) and 50% (membrane), whereas the intensity in the CFP-channel increased to 190% (internal) and 150% (membrane). In contrast, both the acceptor (to 10%) and the donor signal (to 50%) decreased after photobleaching MDCK-cells coexpressing the YFP-syntaxin mutant L165A E166A together with CFP-Munc 18-1 (data not shown). The acceptor photobleaching experiments are additional proof for the validity of our FRET-signal and show that the interaction between YFP-syntaxin and CFP-Munc 18-1 occurs also in MDCK-cells.
To verify the FRET-signal further we created a mutation in syntaxin (L165A, E166A) which is known to completely block binding to Munc 18-1 in pull-down experiments with recombinant proteins Citation[7]. First we analyzed the subcellular distribution of the YFP-syntaxin mutant in transfected CHO-cells with confocal microscopy. Like the wild-type protein YFP-syntaxin L165A E166A is also not transported to the plasma membrane, but remains stuck in the perinuclear region (C). Apparently, the mutation does not affect intracellular targeting of syntaxin. However, one major difference between wild-type and mutant YFP-syntaxin is obvious in CHO-cells also expressing Munc 18-1. Munc 18-1 does not colocalize with the syntaxin mutant, but remains in the cytosol (, D–F). Thus, intracellular accumulation of the mutant form of syntaxin does not attract Munc 18-1 to a perinuclear location.
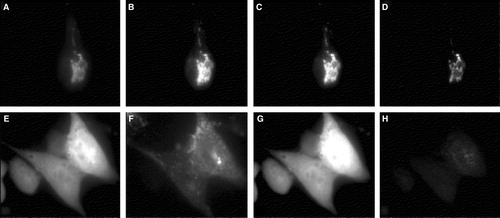
Based on this result we hypothesized that no FRET signal should be observed when the mutant form of syntaxin is coexpressed with Munc 18-1 providing also a convincing control of our approach. By simple cotransfection of CHO-cells, only few cells express both proteins. This limitation made the evaluation of the data tedious as in the previous FRET-experiment. We therefore established a CHO-cell line stably expressing Munc 18-1-CFP. These cells were then transfected with either wild-type or mutant YFP-syntaxin and observed with the cooled CCD camera under the CFP-, YFP-, and FRET-filter set. Without correction of the images for background and bleed-through, fluorescence is visible in the FRET-channel of cells expressing the wild-type as well as the mutant form of syntaxin (, ‘raw FRET’). The data were then mathematically corrected pixel by pixel as described in the methods section. The resulting images in the FRET-channel revealed a signal only for wild-type syntaxin (, ‘net-FRET’). The absence of the FRET-signal for the mutant form of syntaxin is convincing proof for the reliability of our FRET-data for wild-type syntaxin. Furthermore, we could demonstrate FRET between YFP-syntaxin and both CFP-Munc 18-1 () and Munc 18-1-CFP (). Thus, the FRET-signal is unlikely to be caused by oligomerization of CFP or YFP.
Figure 7. Comparison of FRET with syntaxin mutant L165A, E166A. CHO-cells permanently expressing Munc 18-1-CFP were transfected with either wild-type YFP-syntaxin (upper panel) or the YFP-syntaxin mutant L165A, E166A (lower panel). (A, E) CFP-channel, (B, F) YFP-channel (C, G) Raw-Fret as observed in the FRET-channel, (D, H) Net-FRET signals were calculated as described in ‘Methods’. More than 50 images were analyzed in this way and in each case Munc 18-1-CFP showed FRET with syntaxin wild-type, but not with syntaxin L165A, E166A.
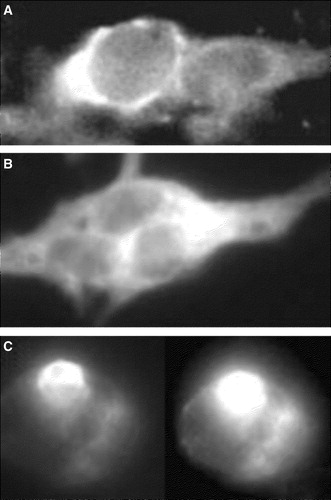
We finally asked if the transport block of YFP-syntaxin and CFP-Munc 18-1 could be rescued by expression of the colour-tagged proteins in cells containing native, endogenous partner proteins. However, neither PC-12 cells nor BON-cells support quantitative transport of singly expressed YFP-syntaxin or CFP-Munc 18-1 to the plasma membrane ().
Figure 8. Localization of CFP-Munc 18-1 and YFP-syntaxin in secretory cells containing endogenous Munc 18-1 and Syntaxin 1. Bon-cells (A, B) or PC-12 cells (C) were transfected with YFP-syntaxin (A, C) or CFP-Munc 18-1 (B) and results were recorded 36 h later with a CCD-camera.
In summary, we have shown that the FRET-technology is suitable to demonstrate an interaction between syntaxin 1 and Munc 18-1 inside cells. In principle, this is in accordance with another study published very recently Citation[18]. However, Liu et al. did observe FRET between CFP-Munc 18-1 and YFP-syntaxin 1 at the plasma membrane, whereas our constructs interact mainly in a compartment along the exocytotic pathway. The intracellular interaction between YFP-syntaxin 1 and CFP-Munc 18-1 we did observe is likely to be of physiological relevance since colocalization of native Munc 18-1 and syntaxin 1 in the Golgi-apparatus of epithelial cells has been reported Citation[17]. Furthermore, Liu et al. Citation[18] also mention FRET between YFP-syntaxin 1 and CFP-Munc 18-1 in an intracellular compartment, but only at early times after transfection.
What might be the function of the intracellular Munc 18-1-syntaxin interaction? Assembly of SNARE-complexes not only triggers neurosecretion, but probably every membrane fusion process in a cell. According to the SNARE-hypothesis, each vesicular trafficking step is catalyzed by a unique set of SNARE-proteins and this specificity helps to preserve the membrane boundaries of the cell Citation[2], Citation[19]. However, SNARE-complex formation is promiscuous, at least with recombinant proteins in vitro. Syntaxin 1 (in combination with SNAP-25) has been shown to form SNARE-complexes not only with synaptobrevin 2, but also with synaptobrevin types 4, 7 and 8, with endobrevin and with sec22b Citation[20], Citation[21]. These SNARE-proteins travel along the exocytotic pathway or even exert their function there. Thus, syntaxin 1 likely encounters other SNAREs during its intracellular transport and the cell must ensure that syntaxin 1 does not engage in complex formation with other SNAREs in the exocytotic pathway. This might be achieved by binding to Munc 18-1, which locks the protein in a fusion-incompetent state not accessible for SNARE-complex formation Citation[6], Citation[8]. Furthermore, syntaxin functions as an anchor for membrane binding of Munc 18-1, which does not possess intrinsic membrane targeting information.
What might be the reason that our constructs are not quantitatively transported to the plasma membrane? We have used syntaxin 1b whereas Liu et al. utilized the 1a variant of syntaxin. However, the amino acid sequences of rat syntaxin 1a and 1b are almost identical (77% identity and 85% similarity). Furthermore, Perez-Branguli et al. Citation[11] have shown that Munc 18-1 is able to facilitate transport of syntaxin 1b to the plasma membrane. Thus, it is more likely that inherent properties of our constructs, i.e., the precise amino acid sequence at the transition between the colour-protein and Munc 18-1 or syntaxin 1 (as specified in the methods section) affect transport of the fusion proteins.
Our study shows that the interaction between syntaxin and Munc 18-1 in a compartment along the exocytotic pathway is not sufficient to cause quantitative transport of both proteins to the plasma membrane. We assume that a further reaction, for example inclusion of the proteins into transport vesicles, is inhibited in our constructs. It has been shown that export of SNARE-proteins from the endoplasmic reticulum is dependent on an interaction with the Sec24 subunit of the COP II coat complex Citation[22], Citation[23]. If similar interactions with coat-proteins are required for export of SNAREs from the Golgi-complex, they might be inhibited by unfavourable attachment of the GFP-proteins.
Liu et al. also analysed FRET between Munc 18-1 and the syntaxin mutant L165A E166A. In contrast to our study, where essentially no FRET was present (), they report a reduced (to 70%), albeit detectable FRET-signal. Besides differences in the computation of the FRET-data (see ‘materials and methods’ for details), it is also conceivable that Munc 18-1 occurs in two conformations with different affinities for syntaxin. Munc 18-1 localized to internal membranes does not bind to syntaxin L165A E166A, whereas the protein present at the plasma membrane interacts in a different mode with syntaxin such that the L165A E166A mutations do not completely eliminate binding.
This paper was first published online on prEview on 14 October 2005.
This work was conducted by A. R. in partial fulfilment of the requirements for a PhD from the Humboldt University Berlin. M.V. is grateful to Michael F. G. Schmidt for encouragement. We also thank Jim Rothman and Thomas Söllner for providing the clones used in this study. This project was supported by the Deutsche Forschungsgemeinschaft through grant Ve 141/5-1 to M. V., G. A-H. and A. H.
References
- Jahn R. Sec1/Munc18 proteins: Mediators of membrane fusion moving to center stage. Neuron 2000; 27: 201–204
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science 1996; 272: 227–234
- Garcia EP, Gatti E, Butler M, Burton J, De Camilli P. A rat brain Sec1 homologue related to Rop and UNC18 interacts with syntaxin. Proc Natl Acad Sci USA 1994; 91: 2003–2007
- Hata Y, Slaughter CA, Sudhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature 1993; 366: 347–351
- Pevsner J, Hsu SC, Scheller RH. n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci USA 1994; 91: 1445–1449
- Yang B, Steegmaier M, Gonzalez LC, Jr, Scheller RH. nSec1 binds a closed conformation of syntaxin1A. J Cell Biol 2000; 148: 247–252
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: Role of munc18. Embo J 1999; 18: 4372–4382
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 2000; 404: 355–362
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000; 287: 864–869
- Garcia EP, McPherson PS, Chilcote TJ, Takei K, De Camilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol 1995; 129: 105–120
- Perez-Branguli F, Muhaisen A, Blasi J. Munc 18a binding to syntaxin 1A and 1B isoforms defines its localization at the plasma membrane and blocks SNARE assembly in a three-hybrid system assay. Mol Cell Neurosci 2002; 20: 169–180
- Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P. Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci 1999; 112: 1865–1877
- Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol 2003; 160: 629–633
- Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol 2003; 3: 906–918
- Xia Z, Zhou Q, Lin J, Liu Y. Stable SNARE complex prior to evoked synaptic vesicle fusion revealed by fluorescence resonance energy transfer. J Biol Chem 2001; 276: 1766–1771
- Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J 2001; 81: 2395–2402
- Rowe J, Calegari F, Taverna E, Longhi R, Rosa P. Syntaxin 1A is delivered to the apical and basolateral domains of epithelial cells: The role of munc-18 proteins. J Cell Sci 2001; 114: 3323–3332
- Liu J, Ernst SA, Gladycheva SE, Lee YY, Lentz SI, Ho CS, Li Q, Stuenkel E. Fluorescence resonance energy transfer reports properties of syntaxin1a interaction with Munc18-1 in vivo. J Biol Chem 2004; 279: 55924–55936
- McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, Paumet F, Sollner TH, Rothman JE. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature 2000; 407: 153–159
- Fasshauer D, Antonin W, Margittai M, Pabst S, Jahn R. Mixed and non-cognate SNARE complexes. Characterization of assembly and biophysical properties. J Biol Chem 1999; 274: 15440–15446
- Yang B, Gonzalez L, Jr, Prekeris R, Steegmaier M, Advani RJ, Scheller RH. SNARE interactions are not selective. Implications for membrane fusion specificity. J Biol Chem 1999; 274: 5649–5653
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 2003; 114: 497–509
- Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell 2003; 114: 483–495