Abstract
Some BK channels are activated in response to membrane stretch. However, it remains largely unknown which membrane component transmits forces to the channel and which part of the channel senses the force. Recently, we have shown that a BK channel cloned from chick heart (named SAKCa channel) is a stretch activated channel, while deletion of a 59 amino acids splice insert (STREX) located in the cytoplasmic side, abolishes its stretch-sensitivity. This finding raised a question whether stress in the bilayer is crucial for the mechanical activation of the channel. To address this question we examined the effects of membrane perturbing amphipaths on the stretch activation of the SAKCa channel and its STREX-deletion mutant. We found that both anionic amphipath trinitrophenol (TNP) and cationic amphipath chlorpromazine (CPZ) could dose-dependently activate the channel by leftward shifting the voltage activation curve when applied alone. In contrast, TNP and CPZ compensated each other's effect when applied sequentially. These results can be understood in the framework of the bilayer couple hypothesis, suggesting that stress in the plasma membrane can activate the SAKCa channel. Interestingly, the STREX-deletion mutant channel has much less sensitivity to the amphipaths, suggesting that STREX acts as an intermediate structure that can indirectly convey stress in the membrane to the gate of the SAKCa channel via an unidentified membrane associated protein(s) that can detect or transmit stress in the membrane.
Introduction
Mechanosensitive (MS) ion channels have been identified in a variety of organisms from bacteria to humans, and are believed to play an important role in the process of transducing mechanical stimuli into intracellular signals, such as Ca2 + and electrical signals. Actually MS channels have been known to be involved in several specialized cell functions, such as sound detection (Corey et al. [Citation2004]), cell volume regulation (Christensen [Citation1987]), touch sensation (García-Añoveros et al. [Citation1998]), hypo-osmotic shock protection (Levina et al. [Citation1999]), and cell locomotion (Lee et al. [Citation1999]), and in pathology including muscular dystrophy (Obregón-Franco & Lansman [Citation2002]) and cardiac arrhythmias (Bode et al. [Citation2001]). Notwithstanding little is known on the molecular and biophysical mechanisms of mechano-gating in eukaryotic MS channels.
To date, two different but not mutually exclusive mechanisms of MS channel gating have been proposed. One mechanism proposes that gating tension is exerted through their interaction with ancillary structures/proteins (e.g., membrane–cytoskeleton interactions); while the other supposes that the tension is exerted within the lipid bilayer. It is well known that the bacterial MS channels, MscL and MscS, are activated by membrane tension alone (Martinac et al. [Citation1990]; Perozo et al. [Citation2002]; Sukharev [Citation2002]). On the other hand, it has been generally supposed that the high mechanosensitivity of eukaryotic cells are derived from their interactions with ancillary structures/proteins (such as extracellular matrix, cytoskeleton) that focus and transmit mechanical forces to the channel (Hamill & Martinac [Citation2001]). For example, it has been shown that a stomatin–related protein (MEC-2) contributes to the activation of a MS channel in caenorhabditis elegans (Goodman et al. [Citation2002]). However, recent results show that the stretch-activated TRPC1 cation channel retains its mechanosensitivity after detergent solubilization and reconstitution in liposomes (Maroto et al. [Citation2005]; Barritt & Rychkov [Citation2005]). Although it is not known if their result truly reproduced the mechanical activation in situ, it clearly demonstrated that some vertebrate MS channels can be gated solely by tension in the lipid bilayer as in various prokaryotic MS channels.
BK channels have been found in a wide range of cell types being implicated to contribute to the regulation of a variety of cellular functions such as firing in neurons, secretion in endocrine or exocrine cells, and myogenic tone in arterial smooth muscle (Gribkoff et al. [Citation2001]). Recently, activation of BK channels in response to membrane stretch without an increase in intracellular [Ca2 + ] has been observed in osteoblast-like cells (Davidson et al. [Citation1990]; Allard et al. [Citation2000]), renal cells (Pacha et al. [Citation1991]), smooth cells (Kirber et al. [Citation1992]; Dopico et al. [Citation1994]), skeletal muscle cells (Mallouk & Allard [Citation2000]), and neuroepithelium (Mienville et al. [Citation1996]). However, the mechanism of mechano-gating of these mechanosensitive BK channels is not well understood. Previously, we found a stretch-activated BK channel in cultured chick embryo ventricular myocytes (Kawakubo et al. [Citation1999]). A unique property of this channel is that it is activated not only by transmembrane voltage and an increase in intracellular [Ca2 + ] like other BK channels, but also by membrane stretch. Because of this property, we named the channel as SAKCa channel. Recently, this channel has been cloned from chick embryo ventricular myocytes and its properties were compared with the native one (Tang et al. [Citation2003]). These studies have shown that the SAKCa channel can be activated by membrane stretch, while deletion of a 59 amino acids insert, called Stress-axis Regulated Exon (STREX), in the c-terminus abolishes stretch-sensitivity of the channel. The latter finding suggests that STREX constitutes a part of mechanosensing apparatus of the SAKCa channel. However, as STREX is supposed to face cytoplasm and not to interact directly with the plasma membrane, it may not be able to sense directly the stress in the membrane. To resolve this difficulty, at least two mechanisms can be speculated: First, sub-membranous cytoskeletal structures that could generate tension during membrane stretch directly connect with STREX to transmit the tension. Second, an interfacing protein that links STREX and the plasma membrane can detect tension in the plasma membrane and transmit it to STREX. In the first model tension in the membrane would not necessarily be required for channel activation, while in the second model, membrane tension would be crucial to activate the SAKCa channel.
The present study was designed to examine if membrane stress contributes to the activation of the SAKCa channel by using ionic amphipaths that can generate stress in the plasma membrane possibly without generating tension in cytoskeletal structures. The experimental results converged to support the idea that stress generated in the plasma membrane can contribute to the activation of the SAKCa channel. Some preliminary results of this work have been presented in abstract form (Qi et al. [Citation2003]).
Materials and methods
Cell culture and gene transfection
CHO-K1 cells were grown in Ham's F-12 nutrient mixture (Invitrogen, Co. Grand Island, N.Y.) supplemented with 10% fetal bovine serum. Cells were grown in a 37 °C incubator with 5% CO2 humidified environment and passaged twice weekly through exposure to 0.05% trypsin, 0.5 mM EDTA in PBS(−) solution. For gene transfection, CHO-K1 cells were transferred to fibronectin coated glass coverslips. After cell density reaches 50–70% confluence, pEGFP (Clontech, Palo Alto, CA) was transiently coexpressed with the SAKCa channel gene that is in mammalian expression vectors (pTarget®-, Promega, Madison, WI) at a ratio of 5:1 (weight /weight) using LipofectAMINE Plus(TM) reagent (Invitrogen, Co. Grand Island, N.Y.). Cells were used for electrophysiological studies 1–3 days after transfection.
Electrophysiology
Membrane currents were measured using a conventional tight seal whole cell recording technique at room temperature. Only GFP-positive cells that were identified by Olympus IX750 inverted fluorescence microscope were selected for the patch-clamp experiments. Virtually no detectable endogenous BK channel activity was observed in non-transfected CHO cells. The pipette solution contained (in mM): 145 KCl, 10 EGTA, 10 HEPES (pH 7.3 with Tris), 1 CaCl2, while the extracellular solution was Hanks’ balanced salts solution (HBSS, Sigma): 1.3 CaCl2, 0.8 MgSO4, 5.4 KCl, 0.4 KH2PO4, 136.9 NaCl, 0.3 Na2PO4, 10 D-glucose, 4.2 NaHCO3. Patch pipettes (Drummond Scientific Co.) were fire-polished on a microforge (MG-83, Narishige) to give a resistance in a range of 3–7 MΩ in recording solution. Negative pressure was applied to the patch pipette through the suction port of the pipette holder using a pneumatic transducer tester (DPM-IB, BIO-TEK Instruments). Compensation for cell capacitance and series resistance was made automatically by the EPC-9 amplifier, only recordings with stable series resistances ≤25 megohms were included in the study. Currents were amplified using an EPC-9 patch-clamp amplifier (HEKA), sampled at 2∼5 kHz and filtered at 1.5∼2.9 kHz via a 4-pole low-pass Bessel filter. The program package PULSE + PULSEFIT was used for data acquisition and analysis. Data were obtained from at least three experiments and presented as mean±standard error (SE), unless otherwise noted. We have observed that TNP transiently increased intracellular Ca2 + concentration in a few CHO cells (data not shown). Besides, it has been reported that TNP can produce influx of Ca2 + in the red cell plasma membrane (Kaila & Juusela [Citation1982]). To avoid concentration change of intracellular Ca2 + that will affect the gating of SAKCa channel, intracellular [Ca2 + ] was buffered to be at nanomolar level with 10 mM EGTA (free [Ca2 + ]i ∼30 nM) on the basis of calculations using a free software CALCON3 (Takeshi Tojo & Go Nakayama 1995).
Results
Activation of the SAKCa channel by TNP and CPZ
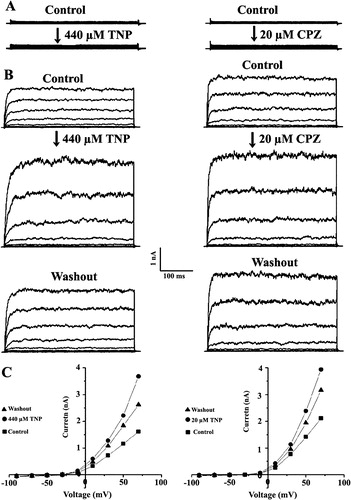
Current-voltage relationships obtained from CHO cells transfected with GFP alone showed that the whole-cell current was less than 300 pA at + 80 mV (A, upper traces) and the reversal potential was close to zero (data not shown). In contrast, the SAKCa/GFP co-expressed cells showed large and strong voltage-dependent current at nA level (2.5±0.9 nA at +20 mV, n=69). The reversal potential was −44±4 mV (n=3), close to the K+ equilibrium potential in our experimental conditions (C). Thus, SAKCa forms a functional ion channel in the plasma membrane of CHO cells. To judge if TNP and CPZ have effects on SAKCa currents, we compared the effects of these drugs on the cells transfected with GFP alone and co-transfected with SAKCa/GFP. After the whole cell current became stable (∼10 min), 440 µM TNP or 20 µM CPZ was added to the external bath solution. Upon addition of the chemicals for about 1 min, the whole cell current was significantly enhanced with time and reached a stable value in about 5–10 min. Both TNP (n=57) and CPZ (n=43) strongly enhanced the whole-cell SAKCa current (B and C) whereas almost no current increase was observed in the presence of 440 µM TNP (n=18) and 20 µM CPZ (n=13) in the cells transfected with GFP alone (A). These results suggest that both TNP and CPZ specifically activated the SAKCa channel.
Figure 1. Activation of the SAKCa channel by TNP and CPZ. Effect of TNP (left) and CPZ (right) on whole-cell currents from a cell transfected with GFP alone (A) or with SAKCa/GFP (B). Cell membrane potential was held at −40 mV and stepped to various potentials for 1 sec duration from −80 to +80 mV in 10 mV increments. Each pair of recordings was from the same cell. (C) Current–voltage relationships corresponding to the above recordings.
TNP and CPZ increase whole cell currents of the SAKCa channel in a dose-dependent manner
A clearly shows graded increase in the whole-cell current of the SAKCa channel in response to increases in the concentration of TNP (27.5 to 440 µM) and CPZ (10 to 20 µM). The whole-cell current at +80 mV increased from a control level of approximately 2.5 nA to about 5.5 nA at 440 µM TNP. Similarly, when the concentration of CPZ increased from 10 to 20 µM, the whole cell current increased about two times compared to the control (B). The dose-dependent activation of the SAKCa channel by TNP and CPZ can also be shown in conductance–voltage (G–V) curves (). It is clear that both TNP and CPZ activated the channel as indicated by a parallel leftward shift of G–V relations on the voltage axis without affecting the slope of the curve, suggesting that the voltage-sensitivity of the channel was not altered by the drugs.
Figure 2. Dose-dependent activation of the SAKCa channel by TNP and CPZ. Representative whole-cell current traces from the same cell corresponding to the addition of TNP from 27 µM to 440 µM (A) or CPZ from 10 µM to 20 µM (B). The I-V curves before and after addition of TNP and CPZ were obtained with a voltage ramp of 1 s in duration starting from −80 to +80 mV.
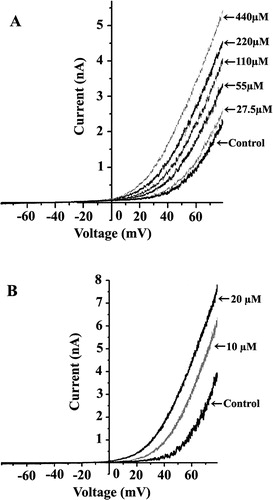
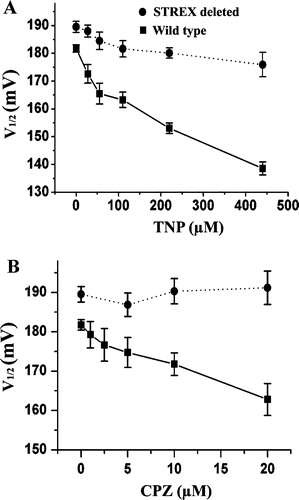
Figure 3. Conductance–voltage (G–V) curves for the TNP (A) and CPZ (B) activation of the channel. Each point represents mean±S.E. from at least five experiments. The G–V curves were fitted with the Boltzmann distribution G/Gmax=1/[1 + exp((V−V1/2)/k)], where k is a factor affecting the steepness of the relationship, and V1/2 is the voltage at which the conductance (G) is half the maximum conductance (Gmax). The error bars for each data point between the control and the maximal concentration of TNP and CPZ are omitted for clarity.
![Figure 3. Conductance–voltage (G–V) curves for the TNP (A) and CPZ (B) activation of the channel. Each point represents mean±S.E. from at least five experiments. The G–V curves were fitted with the Boltzmann distribution G/Gmax=1/[1 + exp((V−V1/2)/k)], where k is a factor affecting the steepness of the relationship, and V1/2 is the voltage at which the conductance (G) is half the maximum conductance (Gmax). The error bars for each data point between the control and the maximal concentration of TNP and CPZ are omitted for clarity.](/cms/asset/f9e39621-6f2a-4b8b-88b2-307ecd81b989/imbc_a_137053_f0003_b.jpg)
Compensatory effect between TNP and CPZ
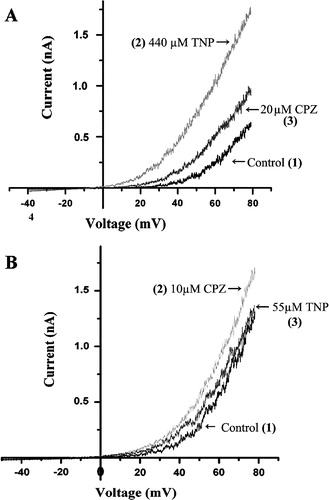
Above results indicated that the activation of the channel was not dependent on the charge of the drug, because both negatively charged TNP and positively charged CPZ could activate the channel in a dose dependent manner when applied alone. It is unlikely that TNP and CPZ exert their effects by acting at the same site in the channel protein considering their differences in the charge and chemical structures. These results may be interpreted qualitatively on the basis of the bilayer couple hypothesis (Sheetz & Singer [Citation1974]). The anionic amphipath TNP would preferentially insert into the outer leaflet, while cationic CPZ in the inner leaflet, because of the asymmetry in lipid composition of the inner and outer monolayers of biological membranes. Such insertion will increase area of the membrane and consequently generate tension in the membrane irrespective of the sign of the effective charges of the drug (Markin & Martinac [Citation1991]). An inference from the bilayer hypothesis is that the effects on channel activation by cationic and anionic amphipaths will be cancelled out when cationic and anionic amphipaths are present simultaneously. This prospect is clearly shown in A, where 440 µM TNP increases the whole cell current from the control of 0.6 nA to 1.8 nA, which is gradually decreased after an addition of 20 µM CPZ. Similarly, the increased currents by 10 µM CPZ was reversed by 55 µM TNP (B). Such a phenomenon was first shown in bacterial MS channels (Martinac et al. [Citation1990]), strongly suggesting that the SAKCa channel can be activated by stress in the membrane.
Figure 4. Compensatory effect between TNP and CPZ on the whole cell SAKCa current. (A) The TNP activated current was compensated by the CPZ. (B) The CPZ activated current was compensated by the TNP. Numbers in the parentheses indicate sequence of treating the same cell with the drugs: (1) for the SAKCa current without addition of any drug, (2) for addition of TNP (A) or CPZ (B) that was followed by (3) for addition of CPZ (A) or TNP (B).
STREX is critical for the channel activation by TNP and CPZ
It has been reported that deletion of STREX abolished mechanosensitivity of the SAKCa channel (Naruse et al. [Citation2003]). Therefore, we tested if the STREX is also critical for the TNP and CPZ activation of the channel by comparing the effect of TNP and CPZ on the currents of wild-type SAKCa with that of a STREX-deletion mutant of the SAKCa. The whole cell current was increased two folds for the wild type channel at +80 mV by TNP (440 µM) and CPZ (20 µM) as shown in , while only a slight increase in the currents from the STREX deletion mutant was observed (). Essentially the same conclusion could be drawn from . The value of the half activation voltage (V1/2) for the wild type SAKCa channel decreased with increases in the concentration of TNP and CPZ. In contrast, this value scarcely changed in the STREX-deletion mutant (), suggesting that STREX is critical for TNP and CPZ activation of the SAKCa channel.
Figure 5. Effect of TNP and CPZ on whole cell current of the STREX deleted SAKCa channel. Effect of only 55 and 440 µM TNP (A) as well as 10 and 20 µM CPZ (B) on the whole cell current is shown for clarity. Each curve is an average from three traces when the currents were stable. n=3 or more.
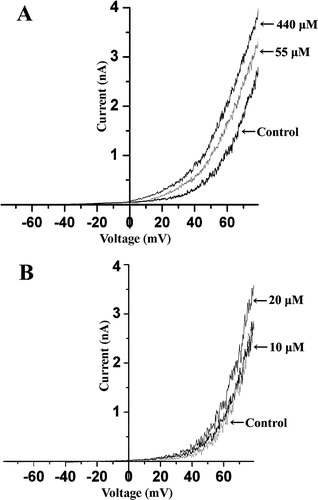
Figure 6. Effect of TNP (A) and CPZ (B) on the half activation voltage (V1/2) shift of the SAKCa and its STREX deleted channel. Estimates of the V1/2 value for a given condition represent the mean value for a set of at least three individual patches.
Discussion
The inner and outer leaflets of the cell membrane are asymmetrically charged. In HEK cells, the cytoplasmic side of the cell membrane is estimated 4 times more negative than the outside (Zhang et al. [Citation2001]). Because of this property of the cell membrane, anionic amphipaths preferentially insert into the outer half of the bilayer, while cationic ones into the negatively charged inner half of the bilayer. Apparently, the preferential insertion of amphipaths into one of the monolayers will cause it to expand. According to the bilayer couple hypothesis (Sheetz & Singer [Citation1974]), the two monolayers are viscously coupled. Therefore, the expansion of one monolayer would stretch another monolayer and thereby activate the mechanosensitive channel irrespective of the charge sign of applied chemicals. On the other hand, when both monolayers contained equal amounts of amphipath, the membrane area increased equally in both halves with no increase in tension in either monolayer and the channel could not be activated (Sachs & Morris [Citation1998]). This property has given a basis for the idea that the mechano-gating force can come from the surrounding lipids to activate the mechanosensitive channels, such as MscS from E. coli (Martinac et al. [Citation1990]) and some of the two pore domain K+ channels (Patel et al. [Citation1998]; Maingret et al. [Citation1999]; Bang et al. [Citation2000]; Maingret et al. [Citation2000]; Patel et al. [Citation2001]).
In this study, we have obtained the following three major results on the amphipaths’ actions on the SAKCa channel: (1) Both CPZ and TNP activated the SAKCa channel when applied alone ( and ), despite their opposite charges, (2) CPZ and TNP compensated for each other's effects on the SAKCa channel when applied sequentially (), (3) Both TNP and CPZ shifted the G-V curve of the SAKCa channel toward lower voltage but did not alter the slope of the curve (). These results fit well the bilayer coupling theory (Sheetz & Singer [Citation1974]), suggesting that the SAKCa channel can be activated by stress generated by TNP or CPZ in the bilayer membrane. In terms of the bilayer couple hypothesis, Markin and Martinac ([Citation1991]) have estimated that adsorption of 20 µM CPZ in the inner leaflet of the membrane may generate membrane tension of 2.4 mN/m, which corresponds to a suction of ∼35 mm Hg in the patch pipette. This amplitude of suction can increase substantially the open probability of the SAKCa channel (Kawakubo et al. [Citation1999]; Tang et al. [Citation2003]), consistent with our results here ( and ). Collectively, these results suggest that chemically induced membrane “tension” can contribute to the activation of the SAKCa channel. A decade ago, BK channels in lipid bilayers were shown to respond to cholesterol as if bilayer stress modulated their gating (Chang et al. [Citation1995]), a conclusion which is in agreement with ours here. More recently, Hamill's group (Maroto et al. [Citation2005]) has drawn a similar conclusion for the TRPC1 channel, which can be activated solely by tension developed in the lipid bilayer membrane.
A classical voltage-sensitive Shaker channel has recently been shown to be tension sensitive and the activation-dependent left-shifts with stretch, without affecting voltage sensitivity implies that the voltage-dependent activation of the Shaker channel is inherently stretch sensitive (Tabarean & Morris [Citation2002]; Laitko & Morris [Citation2004]). In this study, we showed a similar phenomenon, say, a leftward parallel shift of G–V curves for the SAKCa channel caused by amphipath-induced “tension” in the bilayer. Another noteworthy phenomenon is that TNP activates the 2P domain K+ channel hTREK-1 (KCNK2), while CPZ caused inhibition of the channel (Patel et al. [Citation1998]; Miller et al. [Citation2003]). Then an interesting question would be why the hTREK-1 is activated by TNP but inhibited by CPZ, whereas the SAKCa channel is activated by both TNP and CPZ. It has been indicated that a mechanosensitive channel is tension sensitive if it can be activated by both positive and negative pressure applied to the membrane, or it is pressure sensitive if it can only be activated by positive or negative pressure (Sokabe et al. [Citation1991]). For the 2P domain K+ channel, such as TREK-1 and TRAAK channels, only negative, but not positive pressure induces channel opening in inside-out patches, while it is only opened by positive pressure in the outside-out patches, suggesting that the opening of these channels is purely pressure but not tension dependent (Maingret et al. [Citation1999]). On the other hand, we observed that the SAKCa channel can be activated by both positive and negative pressure (data not shown), suggesting that the SAKCa channel is tension sensitive. Therefore, the SAKCa channel can be activated by both TNP and CPZ, while the hTREK-1 channel can only be activated by TNP but not by CPZ.
Both TNP and CPZ had much less effect on activating the STREX deleted SAKCa channel than its wild-type, indicating that the STREX is critical for CPZ and TNP activation of the channel. STREX was previously found to be important for the mechanosensing in the stretch activation of the SAKCa channel (Naruse et al. [Citation2003]), though it is situated in the cytoplasmic side of the SAKCa channel protein. In contrast, CPZ almost could not increase the currents while TNP only slightly increased the current from the STREX deletion mutant ( and ). One explanation for the different effects of TNP and CPZ may be that the channel is more sensitive to tension in one monolayer compared to the other and therefore responds to a different extent to TNP compared to CPZ. Then a question arises how the STREX senses and transmits the force in the membrane to the channel. A simple explanation may be that the STREX interacts, directly or indirectly via a membrane associated protein, which could sense or transmit the force in the membrane. Consequently, the STREX acts as an intermediate structure that can indirectly convey the forces in the membrane to the gating apparatus of the SAKCa channel. Crucial role of a periplasmic loop for the mechanosensitivity of the bacterial mechanosensitive ion channel MscL has already been shown based on a functional reconstitution of N- and C-halves of the channel in artificial liposomes (Park et al. [Citation2004]). In addition, the cytoplasmic C-terminus containing a charged region has been shown to be critical for chemical and mechanical activation of the 2P domain K+ channel (Patel et al. [Citation1998]; Maingret et al. [Citation1999]; Chemin et al. [Citation2005]).
Insertion of charged amphipaths may change the surface charge distribution of the membrane, which in turn may influence voltage-dependent gating of the channel (Cukierman et al. [Citation1988]). However, we think that the surface charge effect is not the main factor in our observed effects of TNP and CPZ because of the following two reasons: (1) If surface charge of the amphipaths takes their effect, insertion of positively charged CPZ and/or negatively charged TNP in the intracellular and extracellular leaflet of the plasma membrane would depolarize the membrane, which in turn would activate the channel. Therefore, the result that CPZ and TNP compensated for each other's effects on the SAKCa channel suggests that the surface charge of TNP and CPZ does not take much effect on the channel activation. (2) The STREX deletion mutant was much less sensitive to TNP and CPZ even though there is no charged amino acid in the STREX sequence. Besides, the compensatory effects between TNP and CPZ as well as the result that the STREX-deletion mutant was much less sensitive to both TNP and CPZ may imply that the channel activation by the amphipaths was not due to an increase in intracellular Ca2 + induced by TNP and CPZ. This result is consistent with the reported results that activation of BK channels in response to membrane stretch is not mediated by an increase in Ca2 + entry in osteoblast-like cells (Davidson et al. [Citation1990]), renal cells (Pacha et al. [Citation1991]), smooth cells (Kirber et al. [Citation1992]; Dopico et al. [Citation1994]), skeletal muscle cells (Mallouk & Allard [Citation2000]), and neuroepithelium (Mienville et al. [Citation1996]).
In summary, our findings strongly suggest that stress in the plasma membrane can contribute to the mechanical activation of the SAKCa channel, though the possibility for the involvement of ancillary structures/proteins in the activation cannot be ruled out. In addition, STREX acts as an intermediate structure that can directly or indirectly convey and/or amplify the forces in the membrane to the gate of the SAKCa channel.
We thank Dr K. Furuya for his great help and Ms MY Huang for technical assistance. This work was partly supported by research grants from NSFC 30340041 and 30470447 (to ZQ) and a grant for Scientific Research on Priority Areas (15086270), and Creative Scientific Research (16GS0308) from MEXT, and a grant from Japan Space Forum to MS.
References
- Allard B, Couble ML, Magloire H, Bleicher F. Characterization and gene expression of high conductance calcium-activated potassium channels displaying mechanosensitivity in human odontoblasts. J Biol Chem 2000; 275: 25556–15561
- Bang H, Kim Y, Kim D. TREK-2, a New Member of the Mechanosensitive Tandem-pore K+ Channel Family. J Biol Chem 2000; 275: 17412–17419
- Barritt G, Rychkov G. TRPs as mechanosensitive channels Nature Cell Biol 2005; 7: 105–107
- Bode F, Sachs F, Franz MR. Tarantula peptide inhibits atrial fibrillation. Nature 2001; 409: 35–36
- Chang HM, Reitstetter R, Mason RP, Gruener R. Attenuation of channel kinetics and conductance by cholesterol: an interpretation using structural stress as a unifying concept. J Membr Biol 1995; 143: 51–63
- Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honore E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J 2005; 24: 44–53
- Christensen O. Mediation of cell volume regulation by Ca2 + influx through stretch-activated channels. Nature 1987; 330: 66–68
- Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature 2004; 432: 723–730
- Cukierman S, Zinkand WC, French RJ, Krueger BK. Effects of membrane surface charge and calcium on the gating of rat brain sodium channels in planar bilayers. J Gen Physiol 1988; 92: 431–447
- Davidson RM, Tatakis DW, Auerbach AL. Multiple forms of mechanosensitive ion channels in osteoblast-like cells. Pflugers Arch 1990; 416: 646–651
- Dopico AM, Kirber MT, Singer JJ, Walsh JV, Jr. Membrane stretch directly activates large conductance Ca(2 + )-activated K+ channels in mesenteric artery smooth muscle cells. Am J Hypertens 1994; 7: 82–89
- García-Añoveros J, García JA, Liu JD, Corey DP. The nematode degenerin UNC-105 forms ion channels that are activated by degeneration- or hypercontraction-causing mutations. Neuron 1998; 20: 1231–1241
- Goodman MB, Ernstrom GG, Chelur DS, O'Hagan R, Yao CA, Chalfie M. MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature 2002; 415: 1039–1042
- Gribkoff VK, Starrett JE, Jr, Dworetzky SI. Maxi-K potassium channels: Form, function, and modulation of a class of endogenous regulators of intracellular calcium. Neuroscientist 2001; 7: 166–177
- Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev 2001; 81: 685–740
- Kaila K, Juusela A. Calcium-dependent increase in the potassium permeability of human red blood cells by pentachlorophenol and 2,4,6-trinitrophenol. Med Biol 1982; 60: 260–266
- Kawakubo T, Naruse K, Matsubara T, Hotta N, Sokabe M. Characterization of a newly found stretch-activated KCa,ATP channel in cultured chick ventricular myocytes. Am J Physiol 1999; 276: H1827–1838
- Kirber MT, Ordway RW, Clapp LH, Walsh JV, Jr, Singer JJ. Both membrane stretch and fatty acids directly activate large conductance Ca(2 + )-activated K+ channels in vascular smooth muscle cells. FEBS Lett 1992; 297: 24–28
- Laitko U, Morris CE. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J Gen Physiol 2004; 123: 135–154
- Lee J, Ishihara A, Oxford G, Johnson B, Jacobson K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999; 400: 382–386
- Levina N, Totemeyer S, Stokes NR, Louis P, Jones MA, Booth IR. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO J 1999; 18: 1730–1737
- Maingret F, Fosset M, Lesage F, Lazdunski M, Honore E. TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem 1999; 274: 1381–1387
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J Biol Chem 2000; 275: 10128–10133
- Mallouk N, Allard B. Stretch-induced activation of Ca(2 + )-activated K(+) channels (BK) in mouse skeletal muscle fibers. Am J Physiol 2000; 278: C473–479
- Markin VS, Martinac B. Mechanosensitive ion channels as reporters of bilayer expansion: A theoretical model. Biophys J 1991; 60: 1120–1127
- Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biol 2005; 7: 179–185
- Martinac B, Adler J, Kung C. Mechanosensitive ion channels of E. coli activated by amphipaths. Nature 1990; 348: 261–263
- Mienville J, Barker L, Lange GD. Mechanosensitive properties of BK channels from embryonic rat neuroepithelium. J Membr Biol 1996; 153: 211–216
- Miller P, Kemp PJ, Lewis A, Chapman CG, Meadows HJ, Peers C. Acute hypoxia occludes hTREK-1 modulation: Re-evaluation of the potential role of tandem P domain K+ channels in central neuroprotection. J Physiol 2003; 548: 31–37
- Naruse K, Tang Q, Kishio M, Sokabe M. 2003. Strex turns large-conductance “BK” calcium-gated channels into stretch-activated channels. Jpn J Physiol 53:S209 (Abstr.).
- Obregón-Franco A, Lansman JB. Changes in mechanosensitive channel gating following mechanical stimulation in skeletal muscle myotubes from the mdx mouse. J Physiol 2002; 539: 391–407
- Pacha J, Frindt G, Sackin H, Palmer LG. Apical maxi K channels in intercalated cells of CCT. Am J Physiol 1991; 261: F696–705
- Park KH, Berrier C, Martinac B, Ghazi A. Purification and functional reconstitution of N- and C-halves of the MscL channel. Biophys J 2004; 86: 2129–2136
- Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 1998; 17: 4283–4290
- Patel AJ, Lazdunski M, Honore E. Lipid and mechano-gated 2P domain K(+) channels. Curr Opin Cell Biol 2001; 13: 422–428
- Perozo E, Kloda A, Cortes DM, Martinac B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat Struct Biol 2002; 9: 696–703
- Qi Z, Naruse K, Sokabe M. 2003. Ionic amphipaths affect the gating of a stretch activated BK channel (SAKCa) cloned from chick heart. Biophys J 84:A234 (Abstr.).
- Sachs F, Morris CE. Mechanosensitive ion channels in nonspecialized cells. Rev Physiol Biochem Pharmacol 1998; 132: 1–77
- Sheetz MP, Singer SJ. Biological membranes as bilayer couples. A molecular mechanism of drug–erythrocyte interactions. Proc Natl Acad Sci USA 1974; 71: 4457–4461
- Sokabe M, Sachs F, Jing ZQ. Quantitative video microscopy of patch clamped membranes stress, strain, capacitance, and stretch channel activation. Biophys J 1991; 59: 722–728
- Sukharev S. Purification of the small mechanosensitive channel of Escherichia coli (MscS): The subunit structure, conduction, and gating characteristics in liposomes. Biophys J 2002; 83: 290–298
- Tabarean IV, Morris CE. Membrane stretch accelerates activation and slow inactivation in Shaker channels with S3-S4 linker deletions. Biophys J 2002; 82: 2982–2894
- Tang QY, Qi Z, Naruse K, Sokabe M. Characterization of a functionally expressed stretch-activated BKca channel cloned from chick ventricular myocytes. J Membr Biol 2003; 196: 185–200
- Zhang PC, Keleshian AM, Sachs F. Voltage-induced membrane movement. Nature 2001; 413: 428–432