Abstract
The human anion exchanger 1 (AE1) is the most abundant integral membrane protein in red cells and is responsible for the exchange of Cl− for HCO3−. However, the detailed role played by the AE1 C-terminal region in the anion translocation and membrane trafficking process remains unclear. In this paper, we created four mutants in the human AE1 C-terminus by deletion of the residues Ala891-Phe895, Asp896-Glu899, Asp902-Glu906 and Val907-Val911, to investigate the role of these sequences in functional expression of AE1. WT AE1 and its deletion mutant constructs were expressed in HEK 293 cells. Western blotting showed that deletions of Ala891-Phe895, Asp896-Glu899, and Val907-Val911 induced high expression of AE1, whereas loss of Asp902-Glu906 results in stable low expression. Pulse chase assays of WT AE1 and its mutants showed that the stability of protein is unaffected by the levels of expression of the AE1 and its mutants. Ala891-Phe895, Asp902-Glu906 and Val907-Val911 mutants exhibited lower levels of trafficking to the plasma membrane compared with WT AE1, while the Asp896-Glu899 mutant was more highly expressed at the plasma membrane. The decreased ability of the mutants to mediate Cl−/HCO3− exchange in transfected cells revealed that the deletion sequences have an important role in transport activity. These results demonstrate that the studied residues in the AE1 C-terminus differently affect the expression, membrane trafficking and functional folding of AE1.
Introduction
AE1, also named Band 3, belongs to a multigene family which consists of three members: AE1, AE2 and AE3. AE1 is found in erythrocytes, and an N-terminal truncated form is present in the kidney; AE2 is found in a variety of tissues; and AE3 is found in the brain, retina, and heart. All AE members mediate Cl−/HCO3− exchange across the plasma membrane (Kopito et al. [Citation1989], Alper [Citation1991]). Among the AE gene family, AE1 is the most extensively studied due to its high expression in erythrocyte membranes, where it constitutes 25% of the total membrane protein (Casey et al. [Citation1989]). AE1 is a 110-kDa protein composed of 911 amino acids and has three separate domains. The 45-kDa N-terminal cytoplasmic domain interacts with glycolytic enzymes and cytoskeletal components. The 55-kDa membrane domain, which is highly conserved with AE2 and AE3, is independent of the N-terminal region, responsible for anion exchange activity, and predicted to span the lipid bilayer 12–14 times. The third domain is the 40 residue acidic cytoplasmic C-terminus (Low [Citation1986], Zhu et al. [Citation2003]).
Unlike the N-terminal cytosolic domain, which functions to interact with ankyrin, protein 4.1, protein 4.2, hemoglobin and aldolase, the function of the C-terminal region still remains elusive (Walder et al. [Citation1984], Gomez & Morgans [Citation1993], Zhu et al. [Citation2003]). Two tryptic sites in the AE1 C-terminal region are accessible only after treatment of erythrocytes with high concentrations of sodium hydroxide (Mori et al. [Citation1995]), suggesting that those regions are related with other protein. Recent studies revealed that the C-terminal 11 amino acid sequence of AE1 is associated with distal renal acidosis (dRTA) which is characterized by defective acid secretion by kidney alpha-intercalated cells (Bruce et al. [Citation1997], Batlle et al. [Citation2001], Toye et al. [Citation2002]). Based on gradual truncations of AE1 C-terminal amino acids, studies revealed that the C-terminal tail of human AE1 is important for efficient AE1 trafficking to the plasma membrane (Cordat et al. [Citation2003]). In addition, carbonic anhydrase II binds to the AE1 C-terminus in the region of Asp887-Asp890 and mutation of this site greatly reduced anion exchange activity of AE1 (Vince & Reithmeier [Citation2000]). Alvarez and co-workers suggested that protein kinase C reduces the carbonic anhydrase II/AE1 interaction, therefore reducing the bicarbonate transport rate (Alvarez et al. [Citation2005]). Our previous research also found that the 19 C-terminal residues directly interact with the glycophorin A C-terminus and cleave glycophorin A at the Leu118-Ser119 bond (Fu et al. [Citation2004]). More recently we reported that the tumor suppressor p16 binds to the AE1 C-terminal region and facilitates the movement of AE1 to the plasma membrane. Furthermore, AE1 up-regulates the expression of p16 (Fu et al. [Citation2005a]). In addition, AE1 is a novel target for the anti-tumor drug As2O3, which acts on the C-terminal 16 residues of AE1 (Fu et al. [Citation2005b]).
To further investigate the function of the AE1 C-terminal domain, we created four mutants by deleting Ala891-Phe895, Asp896-Glu899, Asp902-Glu906 and Val907-Val911 amino acids using the technique of deletion PCR mutagenesis to study the function of these amino acids in stability, folding, trafficking, and anion exchange activity of the AE1 protein. Our results indicate that deletion of these amino acid residues altered AE1 protein stability, membrane trafficking and anion exchange activity, and that the AE1 C-terminus therefore plays an important role in these processes.
Materials and methods
Materials
Protease inhibitor cocktail, rabbit anti-HA antibody and nigericin were from Sigma-Aldrich. Sulpho-NHS-SS-Biotin and streptavidin-agarose were from Pierce Chemical Company. Molecular Probe 2′,7′-bis (2-carboxyethyl)-5(6)- carboxyfluorescein- acetoxymethyl ester (BCECF-AM) was from Dojindo laboratory Japan. Mouse monoclonal anti-HA antibody was from Santa Cruz Biotechnology. Plasmid midi kit and polyfect transfection reagents were from Qiagen.
Construction of plasmids and mutations
A human AE1 cDNA construct (pcDNA3-AE1) was kindly provided by Dr Joseph R. Casey (University of Alberta, Canada). The hemagglutinin (HA) insertion (position 557) mutant was constructed by inserting the following codons: TAC CCA TAC GAT GTT CCA GAT TAC GCT using a PCR-based megaprimer mutagenesis strategy. Each AE1 deletion mutant was created with the same sense primer and with the antisense primers containing the specific mutation. These antisense primers were: 5′-CAC AGG CAT GGC CAC TTC GTC GTA TTC ATC CCG ACC TTC CTC CTC ATC ATC ATC AGC ATC-3′ (891–895 delete), 5′-TCA CAC AGG CAT GGC CAC TTC GTC GTA TTC ATC CCG ACC AAA GGT TGC CTT GGC ATC ATC-3′ (896–899 delete), 5′-GCG CTC GAG TCA CAC AGG CAT GGC CAC CCG ACC TTC CTC CTC-3′ (902–906 delete), 5′-GCG CTCGAG TCA TTC GTC GTA TTC ATC CCG ACC-3′ (907–911 delete).
Cell culture and western blotting
HEK 293 cells were grown at 37°C in Dulbecco's Modified Eagle Medium supplemented with 10% of calf serum (Gibco Life Technologies) in 5% CO2. Polyfect reagent was used to transiently transfect the cells. Cells were washed with PBS and lysed in sample buffer containing 100 mM Tris-HCl buffer (pH 6.8), 4% SDS, 200 mM DTT and 20% glycerol. The lysates were centrifuged at 20,000 g for 10 min at 4°C. Proteins in the supernatants were quantified and the protein extracts were equally loaded to an acrylamide gel, electrophoresed, and transferred to nitrocellulose membranes (Amersham Bioscience, Buckinghamshire, UK). The membranes were probed with mouse monoclonal anti-AE1 antibody (1:5000, Sigma, CA), followed by horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling). The signals were detected by a chemiluminescence phototope-HRP kit (Cell Signaling) according to manufacturer's instructions.
Pulse-chase assay
Two days after transfection, cells were pulsed with 50 µCi/ml of [35S]-methionine and cysteine (GE Health Care) in methionine and cysteine free DMEM media for 20 min. After labeling, the medium was removed and the cells were chased for 0, 1, 5, and 24 h with DMEM. Each chase time point was a single well of a 12-well plate done in triplicate and cells were lysed with RIPA buffer (1% deoxycholic acid, 1% Triton X-100, 0.1% SDS, 0.15 M NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 7.5) containing protease inhibitor (Cocktail, Sigma). AE1 was immunoprecipitated using anti-HA agarose (Sigma). The immunoprecipitates were analyzed by 8% SDS-PAGE. The gels were dried and exposed to a phosphor screen (Molecular Dynamics) overnight to visualize the radiolabeled AE1.
Immunocytochemistry
HEK 293 cells were transiently transfected with pcDNA3-AE1 constructs carrying a HA tag in the third extracellular loop and then grown on glass coverslips. Samples were fixed with 4% formaldehyde, blocked with 3% BSA and incubated for 30 min with mouse monoclonal anti-HA antibody (1/100) for 30 min. Anti-mouse antibody coupled to cy5 was added to the samples for 30 min. Coverslips were finally mounted on slides and observed using a Zeiss laser fluorescence confocal microscope LSM 510.
Cell surface biotinylation
This method has been previously reported (Cordat et al. [Citation2003]). Briefly, the HEK 293 cells were grown for 48 h after transfection. The cells were washed once with borate buffer (10 mM boric acid, 154 mM NaCl, 7.2 mM KCl, 1.8 mM CaCl2, pH 9.0), and treated twice with 0.8 mM EZ-link NHS-SS-biotin (Pierce) in borate buffer for 30 min at room temperature. The unreacted reagent was quenched with buffer containing 192 mM glycine, 25 mM Tris pH 8.3. The cells were lysed in RIPA buffer (containing a protease inhibitor cocktail) and an aliquot of the lysate was saved for measurement of the total AE1 content. The lysate was incubated with immobilized streptavidin (Pierce Biotechnology, Inc., Rockford, IL, USA) for 1 h at 4°C. Streptavidin beads were washed, and the bound proteins were eluted by addition of 2×SDS sample buffer (containing 5% β-mercaptoethanol to cleave disulfide bonds).The protein fractions of total, bound, and unbound were analyzed by western blotting.
Cl−/HCO3− exchange assay
HEK 293 cells, grown on 35 mm glass dishes, were transfected with WT AE1 and its four mutant constructions. Two days post transfection, glass dishes were rinsed in serum-free medium and incubated in 4 ml of serum-free medium containing 2 µM BCECF-AM (37°C, 30 min).The glass dishes were microperfused alternately with Ringer's buffer (5 mM glucose, 5 mM potassium gluconate, 1 mM calcium gluconate, 1 mM MgSO4, 2.5 mM NaH2PO4, 25 mM NaHCO3, 10 mM Hepes pH 7.4) containing either 140 mM NaCl, or 140 mM sodium gluconate. Fluorescence images were acquired and recorded with an image pro plus imaging system equipped with an NIKON TE2000E Microscope, a HAMAMATSU IR CCD, a JVC color video monitor, and a DAD-8VCPP 8 pinch valve superfusion system at excitation wavelengths of 440 and 502.5 nm, and emission wavelength of 528.7 nm. After calibration using the high potassium nigericin technique at four pH values (6.5, 7.0, 7.5 and 8.0), the fluorescence ratios were converted to pHi.
Determination of rate of pHi change
We measured the rate of pHi change from initial dpHi/dt following bath Cl− removal and subsequent restoration in single native and transfected HEK 293 cells by linear regression of the initial or recovery portion of each pHi vs. time record. This method has been previously reported (Fernandez & Malnic [Citation1998], Dantzler et al. [Citation1999]).
Results
Expression and protein stability of the AE1 mutants
In order to accurately evaluate the translocation of AE1 to the cell surface, we inserted a HA tag at position 557 on the third extracellular loop. Immunolocalization of WT AE1 or the deletion mutants by confocal microscopy using the anti-HA antibody was used to detect AE1 expressed at the cell surface. The HA tag insertion was identified and confirmed by PCR analysis and DNA sequencing.
In order to determine the role of the amino acids located in AE1 C-terminus, deletion mutants Ala891-Phe895, Asp896-Glu899, Asp902-Glu906 and Val907-Val911 in the AE1 C-terminus were created. All mutants contained the HA tag (). WT AE1 and the mutant constructs were transiently transfected into HEK 293 cells and the expressed proteins were identified by western blotting of whole cell lysates using the anti-AE1 antibody 24 h post transfection. The results show that Ala891-Phe895, Asp896-Glu899 and Val907-Val911 exhibit higher expression, whereas deleting the Asp902-Glu906 sequence results in lower expression of AE1 protein compared with the WT AE1. A shows a typical experiment, and the quantification of the relative abundance of proteins (includes monomer and dimer) in transfected cells are shown in B (n=3).
Figure 1. Deletion mutations created in human AE1. The open circle shows the position of the hemagglutinin (HA) tag inserted at position 557 of the protein. The sequence and position of the deleted residues are shown in the pane.
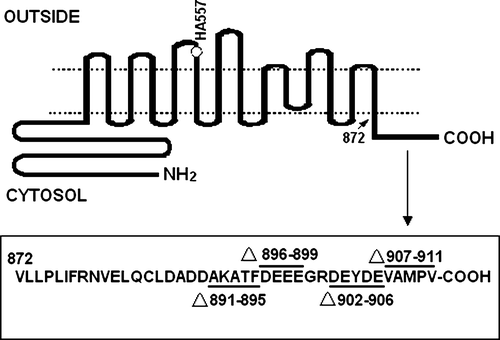
Figure 2. Expression of WT and mutant AE1 in transfected HEK 293 cells. (A) Cells transfected with WT AE1 or deletion mutants were lysed in sample buffer containing 100 mM Tris-HCl buffer (pH 6.8), 4% SDS, 200 mM DTT and 20% glycerol and immunoblotting was performed using an anti-AE1 antibody on the transfected HEK 293 cells lysates. The arrow indicates the position of AE1 at approximately 100 (monomer) and 200 (dimer) kDa. (B) The bar graph shows the relative AE1 monomer and dimer expression over three separate experiments. Densitometric scans of immunoblots were used to quantify the relative amount of AE1 using Quantity one − 4.4.0 software (n=3). The data were normalized to WT AE1. The error bars represent±SD.
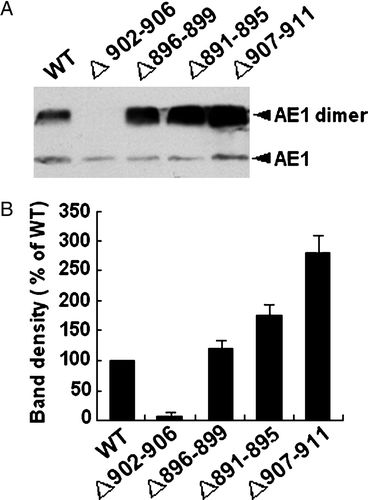
To determine whether the different expression level of AE1mutant in cells is related to the degradation of the protein, the stability of the mutant protein compared to WT was investigated by pulse chase assay (). No significant difference was found in the degradation between WT and Asp896-Glu899 or Ala891-Phe895 mutant. The degradation of Val907-Val911 mutant slightly decreased compared to WT AE1. However, compared to WT AE1 at 0 time point, the amounts of Asp896-Glu899 or Ala891-Phe895 mutant are higher while the amount of Asp902-Glu906 are much lower. These results indicate that the levels of all AE1 mutants are closely related to the regulation of this protein expression, but have no relation to the protein stability. The underlying mechanism of the various levels of expressions of the AE1 mutant warrants further investigated.
Figure 3. Pulse chase assay. Transfected cells were pulsed with 50 µCi/ml of [35S]-methionine and cysteine for 20 min. Cells were collected at 0, 1, 5 and 24 h of the chase and AE1 was immunoprecipitated with anti-HA agarose. The amount of radiolabeled AE1 was determined by 8% SDS-PAGE and autoradiography. The arrows indicate the positions of dimer and monomer AE1.
![Figure 3. Pulse chase assay. Transfected cells were pulsed with 50 µCi/ml of [35S]-methionine and cysteine for 20 min. Cells were collected at 0, 1, 5 and 24 h of the chase and AE1 was immunoprecipitated with anti-HA agarose. The amount of radiolabeled AE1 was determined by 8% SDS-PAGE and autoradiography. The arrows indicate the positions of dimer and monomer AE1.](/cms/asset/f9a032ae-6df4-464e-9d71-d30c4bf8e1c1/imbc_a_194439_f0003_b.gif)
Deletion of Asp896-Glu899 in AE1 C-terminus results in facilitated movement to plasma membrane
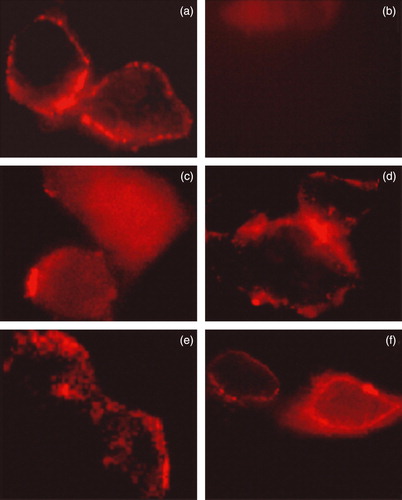
The plasma membrane trafficking of WT AE1 or the mutants was assessed using immunofluorescence and a surface biotinylation assay. First, WT AE1 or the deletion constructs carrying a HA tag were transfected into HEK 293 cells. At 24 h post transfection, AE1 expressed on the cell surface was detected using mouse anti-HA antibody. The rabbit polyclonal anti-HA antibody was incubated with nonpermeabilized cells. This method allowed us to differentiate by immunofluorescence the protein present at the plasma membrane of the cells from the total protein. The antibody was detected using an anti-rabbit antibody coupled to cy5 (red staining) (). The result shows that deletion of Asp896-Glu899 significantly increases the plasma membrane level of the protein, whereas very low surface signals were observed for the deletion mutants Ala891-Phe895 and Asp902-Glu906 compared to WT AE1. Although the Val907-Val911 deletion mutant had a lower level of membrane trafficking compare to the WT AE1 (also refer to cell surface biotinylation experiment), it is higher than mutants of Asp902-Glu906 and Ala891-Phe895. These results indicate that deletion of Ala896-Phe899 did not impair trafficking of AE1 to the plasma membrane.
Figure 4. Immunolocalization of WT AE1 and its mutants by confocal microscopy. Transiently transfected HEK 293 cells with WT and four mutant constructs carrying the HA tag were fixed, blocked and incubated with mouse monoclonal anti-HA antibody. After three washes, goat anti-mouse antibody coupled to cy5 were added to samples for 30 min prior to mounting the coverslips on slides and observation using a Zeiss laser fluorescence confocal microscope LSM 510. (a) WT AE1; (b) mock transfection; (c) Δ902–906; (d) Δ896–899; (e) Δ891–895; (f) Δ907–911. This Figure is reproduced in colour in Molecular Membrane Biology online.
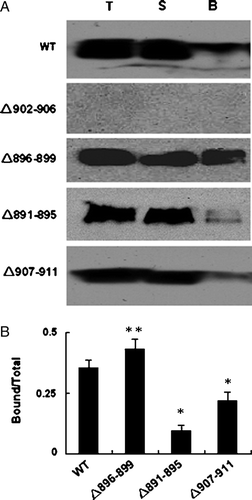
These results were further confirmed by the cell surface biotinylation experiments. Cell surface biotinylation has emerged as an important tool for studying the expression of membrane proteins. Once the cell surface proteins have been biotinylated, the labeled proteins can be immobilized on a streptavidin resin. Intact HEK 293 cells were treated with biotinylation reagent and labelled protein was captured by adding streptavidin beads. The total, unbound and bound fractions were loaded on 9% SDS-PAGE gel and AE1 protein was analyzed by western blotting. The cell surface biotinylation experiments with WT AE1 and deletion mutants are consistent with the immunofluorescence observations. A shows that deleting Asp896-Glu899 results in high expression and membrane trafficking of AE1. Deletions of Ala891-Phe895 and Val907-Val911 visibly affected the targeting of AE1 to the plasma membrane. The quantification of the protein in fractions of total, supernatant and bound are shown in B (n=3).
Figure 5. Biotinylation of WT AE1 or deletion mutants. (A) HEK 293 cells were transfected with constructs encoding WT and mutated AE1.Transfected cells were harvested and incubated twice with biotinylation reagent at room temperature before quenching. The fraction of bound (line B) was obtained by eluting the beads with 2×sample loading buffer. Fractions from total cell lysate (lane T), supernatant (lane S) or bound fraction (10 times overloaded) were detected by western blot using anti-AE1 antibody (1/5000), and anti-rabbit antibody coupled to horseradish peroxidase enzyme (1/2000). (B) The quantification of the bands (AE1 dimer) was done using Quantity one − 4.4.0 software. The error bars represent±SD (n=3) (*compare with WT, p < 0.01; **compare with WT, p < 0.05).
All the deleted amino-acids are important for AE1 anion exchange activity
Deletion of the residues important in functional folding could impair AE1 exchange activity. To examine the role of the deleted sequences in anion exchange activity, HEK 293 cells were transfected with WT and mutant AE1 cDNA. Exchange activity was measured in cells loaded with the pH-sensitive dye BCECF-AM. Cells were alternately perfused with Ringer's buffer containing Cl− or gluconate. All solutions were bubbled continuously with 5% CO2 in air. AE1-mediated efflux of HCO3− acidifies the cell, while influx alkalinizes the cell. Removal of the Cl− from the perfusion buffer would initialize the efflux of Cl− and influx of HCO3− that results in intracellular alkalinization. shows that all AE1 mutants exhibited lower initial or recovery rate of the exchange activity (c,d,e,f) compared with WT AE1 (a) and empty vector (b) transfections. Surprisingly, even the Asp896-Glu899 mutant, which is highly expressed and trafficked to the plasma membrane in HEK 293 cells, exhibited impaired exchange activity. These findings demonstrate the important role of these amino acids for AE1 activity. In order to compare the difference of the anion exchange activity in WT AE1 and its mutants, we calculated the initial and recovery rate of pHi (dpHi/dt, pH units/min) by measuring these values for the first 36 sec after perfusion with Cl−-free Ringer's buffer, and an additional 36 sec after reintroduction of Cl−-containing Ringer's buffer, respectively. The results show that the activity of mutants Asp896-Glu899 and Val907-Val911 are higher than that of the other mutants ().
Figure 6. Anion exchange assay. HEK 293 cells transiently transfected with WT AE1 or its mutants were loaded with BCECF-AM. Cells were perfused alternately with Cl−-containing (starting from solid bar) and Cl−-free (starting from open bar) Ringer's buffer, and fluorescence in single cells was monitored using excitation wavelengths of 440 and 502 nm and an emission wavelength of 528.7 nm. (a) WT AE1; (b) mock transfection; (c) Δ902–906; (d) Δ896–899; (e) Δ891–895; (f) Δ907–911.
Figure 7. Anion exchange activity was calculated as the dpHi/dt. (A) The initial rate during the first 36 sec after perfusion with Cl_-free Ringer's buffer. (B) The recovery rate during the 36 sec after reintroduction of Cl−-containing Ringer's buffer. Values from AE1 and mutants transfection experiments were replicated in >10 separate transfections respectively (*compare with WT, p < 0.01).
Discussion
The C-terminal region of membrane proteins can provide specific binding sites for other proteins and play a vital role via protein-protein interaction (Quilty et al. [Citation2002]). Truncation of the C-terminus of other membrane proteins led to improper trafficking of the proteins (Kasir et al. [Citation1999]). The C-terminal tail of some proteins contains a signal that sorts the protein to the apical membrane in polarized cells (Chuang & Sung [Citation1998]).
Determination of AE1 topology reveals an exposed intracellular area of residues from Val972 to Val911 in the AE1 C-terminal tail (Zhu et al. [Citation2003]). This region of AE1 protein has a negatively charged cluster. Two peptides within the C-terminal domain, Asn880-Lys892 and Ala893-Val911 can be liberated as fragments from ghost membranes with trypsin treatment. However, the release of the two peptides requires pretreatment with 100 mM NaOH, suggesting that the C-terminal region might be hidden by interaction with other proteins. In this paper, we designed four deletion sites in the AE1 C-terminal region based on their location and function. Ala891-Phe895 is nearest the sequence Asp887-Asp890 which is responsible for the interaction of AE1 with carbonic anhydrase II. Asp896-Glu899 is a negatively-charged cluster and Asp902-Glu906 and Val907-Val911 carry the putative AP-1 binding site. In addition, the motif Thr-Phe-Asp-Glu, which is found in other membrane proteins and is commonly implicated in the promotion of normal membrane insertion of polytopic membrane proteins (Mori et al. [Citation1995]), is present in the sequence of AE1 chosen for mutation in this study.
The aggregation of mutant membrane proteins is often caused by their incomplete folding or misfolding (Kopito [Citation1997], Aridor & Hannan [Citation2002]). In addition, hetero-oligomer formation of mutants also results in intracellular retention of the protein (Quilty et al. [Citation2002], Li et al. [Citation2000]). Literature indicated that the only way to dissociate AE1 in its native dimeric state was to denature the protein with SDS (Boodhoo & Reithmeier [Citation1984]). That, combined with other data (Casey & Reithmeier [Citation1991]), suggests that AE1 is dimeric. AE1 on SDS PAGE gels represents an SDS-denatured form of AE1 that was almost certainly full dimeric. Migration at the dimeric position reflects a failure to dissociate the dimers to monomers. Three of the AE1 deletion mutants exhibiting the increased expression with dimer may reflect enhanced protein dimerization. Cell surface biotinylation and immunochemistry experiments shows that the deletion of Asp896-Glu899 induced increased expression and membrane trafficking, however, Ala891-Phe895 and Val907-Val911 mutants, despite increased expression, do not show increased membrane trafficking, indicating that it was the intracellular reservation of the dimers which impaired the trafficking ability of AE1 to the plasma membrane.
The sequence of Asp902-Glu906 (Asp-Glu-Tyr-Asp-Glu) is similar to the apoptosis-associated sequence of Asp-Glu-Val-Asp, which is an inhibitor of caspase frequently used in apoptotic experiments (D'Mello et al. [Citation1998]). In addition, the potential AP-1 (Adaptor Protein) binding site is also in this region (Tyr-X-X-Val). This information suggests that the Asp902-Glu906 sequence may actively interact with other proteins and participate in cellular signal transduction, especially in the apoptotic pathway. Deletion of Asp902-Glu906 in the AE1 C-terminus results in very low expression which is not related to low stability of the protein compared with other mutants, indicating that the sequence is very important for AE1 expression. Deletion of Asp902-Glu906 in the AE1 C-terminus would be expected to disrupt the structure that may be essential for AE1 interaction with other proteins, which may play an important role in AE1 expression.
Transport assays suggest that all the four deleted sequences are important for AE1 functional expression because the anion exchange activity of the four mutants was reduced by 50% or more compared with WT AE1. In the case of Asp902-Glu906, Ala891-Phe895 and Val907-Val911, the decrease in transport could be a result of decreased protein expression at the plasma membrane. Finally, since Asp896-Glu899 is expressed and trafficked at a comparable level to WT AE1, the defect in transport activity likely indicates a vital role for these residues in the transport event. Our results indicate that the activity of the deletion mutants are not completely consistent with the protein level expressed at the cell surface, suggesting that the membrane targeting and transport activity are governed by different amino acids.
This paper was first published online on prEview on 29 September 2006.
We thank Haley J. Shandro (University of Alberta, Canada) and Stephen Buscemi (University of Arizona, USA) for editing this manuscript, and Dr Joseph R. Casey (University of Alberta, Canada) for kindly providing pcDNA3-AE1 construct. This work was supported in part by the Key (NO30230160) and general (NO30570697) Project of the National Natural Science Foundation of China, the National Key Program (973) for Basic Research of China (NO2002CB512805), Key Project for International Collaboration of the Ministry of Science and Technology of China (2003DF000038), and 100-Talent Program of the Chinese Academy of Sciences.
References
- Alper SL. The band 3-related anion exchanger (AE) gene family. Annu Rev Physiol 1991; 53: 549–564
- Alvarez BV, Vilas GL, Casey JR. Metabolon disruption: a mechanism that regulates bicarbonate transport. EMBO J 2005; 24: 2499–2511
- Aridor M, Hannan LA. Traffic jams II: an update of diseases of intracellular transport. Traffic 2002; 3: 781–790
- Batlle D, Ghanekar H, Jain S, Mitra A. Hereditary distal renal tubular acidosis: new understandings. Annu Rev Med 2001; 52: 471–484
- Boodhoo A, Reithmeier RA. Characterization of matrix-bound Band 3, the anion transport protein from human erythrocyte membranes. J Biol Chem 1984; 259: 785–790
- Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest 1997; 100: 1693–1707
- Casey JR, Lieberman DM, Reithmeier RA. Purification and characterization of band 3 protein. Methods Enzymol 1989; 173: 494–512
- Casey JR, Reithmeier RA. Analysis of the oligomeric state of band 3, the anion transport protein of the human erythrocyte membrane, by size exclusion high performance liquid chromatography. Oligomeric stability and origin of heterogeneity. J Biol Chem 1991; 266: 15726–15737
- Chuang JZ, Sung CH. The cytoplasmic tail of rhodopsin acts as a novel apical sorting signal in polarized MDCK cells. J Cell Biol 1998; 142: 1245–1256
- Cordat E, Li J, Reithmeier RA. Carboxyl-terminal truncations of human anion exchanger impair its trafficking to the plasma membrane. Traffic 2003; 4: 642–651
- Dantzler WH, Serrano OK, Abbott DE, Kim YK, Brokl OH. Basolateral regulation of pHi in isolated snake renal proximal tubules in presence and absence of bicarbonate. Am J Physiol 1999; 276: R1673–1681
- D'Mello SR, Aglieco F, Roberts MR, Borodezt K, Haycock JWA. DEVD- inhibited caspase other than CPP32 is involved in the commitment of cerebellar granule neurons to apoptosis induced by K+ deprivation. J Neurochem 1998; 70: 1809–1818
- Fernandez R, Malnic G. H+ ATPase and Cl− interaction in regulation of MDCK cell pH. J Membr Biol 1998; 163: 137–145
- Fu G, Wang T, Yang B, Lv F, Shi C, Jiang X, Tian L, Yu W, Hamasaki N. Purification and characterization of the human erythrocyte band 3 protein C-terminal domain. Biochemistry 2004; 43: 1633–1638
- Fu GH, Wang Y, Xi YH, Shen WW, Pan XY, Shen WZ, Jiang XS, Chen GQ. Direct interaction and cooperative role of tumor suppressor p16 with band 3 (AE1). FEBS Lett 2005a; 579: 2105–2110
- Fu GH, Wang Y, Xi YH, Guo ZW, Liu XB, Bai SZ, Yang BF, Chen GQ. As2O3 enhances the anion transport activity of band 3 and the action is related with the C-terminal 16 residues of the protein. J Drug Target 2005b; 13: 235–243
- Gomez S, Morgans C. Interaction between band 3 and ankyrin begins in early compartments of the secretory pathway and is essential for band 3 processing. J Biol Chem 1993; 268: 19593–19597
- Kasir J, Ren X, Furman I, Rahamimoff H. Truncation of the C terminus of the rat brain Na + –Ca2+ exchanger RBE-1 (NCX1.4) impairs surface expression of the protein. J Biol Chem 1999; 274: 24873–24880
- Kopito RR, Lee BS, Simmons DM, Lindsey AE, Morgans CW, Schneider K. Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell 1989; 59: 927–937
- Kopito RR. ER quality control: the cytoplasmic connection. Cell 1997; 88: 427–430
- Li J, Quilty J, Popov M, Reithmeier RA. Processing of N-linked oligosaccharide depends on its location in the anion exchanger, AE1, membrane glycoprotein. Biochem J 2000; 349: 51–57
- Low PS. Structure and function of the cytoplasmic domain of band 3: center of erythrocyte membrane-peripheral protein interactions. Biochim Biophys Acta 1986; 864: 145–167
- Mori A, Okubo K, Kang D, Hamasaki N. A structural study of the carboxyl terminal region of the human erythrocyte band 3 protein. J Biochem 1995; 118: 1192–1198
- Quilty JA, Cordat E, Reithmeier RA. Impaired trafficking of human kidney anion exchanger (kAE1) caused by hetero-oligomer formation with a truncated mutant associated with distal renal tubular acidosis. Biochem J 2002; 368: 895–903
- Toye AM, Bruce LJ, Unwin RJ, Wrong O, Tanner M.J. Band 3 Walton, a C-terminal deletion associated with distal renal tubular acidosis, is expressed in the red cell membrane but retained internally in kidney cells. Blood 2002; 99: 342–347
- Vince JW, Reithmeier RA. Identification of the carbonic anhydrase II binding site in the Cl−/HCO3− anion exchanger AE1. Biochemistry 2000; 39: 5527–5533
- Walder JA, Chatterjee R, Steck TL, Low PS, Musso GF, Kaiser ET, Rogers PH, Arnone A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. J Biol Chem 1984; 259: 10238–10246
- Zhu Q, Lee DWK, Casey JR. Novel topology in C-terminal region of the human plasma membrane anion exchanger, AE1. J Biol Chem 2003; 278: 3312–3120