Abstract
CLN6 is a polytopic membrane protein of unknown function resident in the endoplasmic reticulum (ER). Mutant CLN6 causes the lysosomal storage disorder neuronal ceroid lipofuscinosis. Defining the topology of CLN6, and the structural domains and motifs required for interaction with cytosolic and luminal proteins may allow insights into its function. In this study we analysed the topology, ER retention and oligomerization of CLN6. We demonstrated, by differential membrane permeabilization of transfected BHK cells using specific detergents and two distinct antibodies, that CLN6 contains an N-terminal cytoplasmic domain, seven transmembrane domains, and a luminal C terminus. Mutational analyses and confocal immunofluorescence microscopy showed that changes of potential ER localization signals in the N- or C-terminal domain (a triple arginine cluster, and a dileucine motif) did not alter the subcellular localization of CLN6. The deletion of a dilysine motif impaired partially the ER localization of CLN6. Furthermore, expression analyses of fusion and deletion constructs in non-neuronal and neuronal cells suggested that two portions of CLN6 contributed to its retention within the ER. We showed that the N-terminal domain was necessary but not sufficient for ER retention of CLN6 and that deletion of transmembrane domains 6 and 7 was accompanied with the loss of ER localization and, in some instances, trafficking to the cisGolgi. From these data we concluded that CLN6 maintains its ER localization by expressing retention signals present in both the N-terminal cytosolic domain and in the carboxy-proximal transmembrane domains 6 and 7. Additionally, the ability of CLN6 to homodimerize may also prevent exit from the ER via an interaction with membrane-associated factors.
| Acronyms | ||
| NCL | = | neuronal ceroid lipofuscinosis |
| ER | = | endoplasmic reticulum |
| BHK21 cells | = | Baby hamster kidney 21 cells, BS3, bis (sulfosuccinimidyl) suberate, BSA, bovine serum albumin |
| COP | = | cytosolic coat protein |
| DAPI | = | 4’, 6 diamidino-2-phenylindole, DMEM, Dulbecco′s modified Eagle′s medium, ER, endoplasmic reticulum |
| ERGIC | = | ER-Golgi intermediate compartment |
| FCS | = | fetal calf serum |
| FITC | = | fluorescein isothiocyanate |
| IP3 | = | inositol trisphosphate |
| LAMP | = | lysosomal associated membrane protein |
| PDI | = | protein disulfide isomerase |
| SDS-PAGE | = | sodium dodecylsulfate polyacrylamide gel electrophoresis |
| TM | = | transmembrane domain |
Introduction
The neuronal ceroid lipofuscinoses (NCLs), also called Batten disease, are a group of autosomal recessively inherited neurodegenerative diseases with ages of onset ranging from infancy to adulthood. They share common clinical features including visual failure leading to blindness, seizures, myoclonic epilepsies, and progressive mental and motor deterioration (Santavuori et al. [Citation2001]). NCLs are characterized by massive lysosomal storage of autofluorescent ceroid and lipofuscin as well as aggregated proteins such as subunit c of the mitochondrial ATP synthase or sphingolipid activator proteins (Tyynelä [Citation2005]). Currently, seven human NCL genes have been identified. CTSD, CLN1 and CLN2 encode the soluble lysosomal enzymes cathepsin D, palmitoyl protein thioesterase and tripeptidyl peptidase 1, respectively (Vesa et al. [Citation1995], Sleat et al. [Citation1997], Siintola et al. [Citation2006], Steinfeld et al. [Citation2006]), whereas the function of the fourth lysosomal CLN5 protein is unknown (Isosomppi et al. [Citation2002]). CLN3, CLN6 and CLN8 are polytopic membrane proteins with distinct subcellular locations. Overexpressed CLN3 was found in the late endosomal/lysosomal compartment, at the plasma membrane and in neuronal cells in synaptosomes and endosomes (Järvelä et al. [Citation1998], Kyttälä et al. [Citation2004]) and has been reported to play a role in arginine transport (Kim et al. [Citation2003]). CLN6 and CLN8 are localized in the endoplasmic reticulum (ER) and CLN8 traffics to the ER-Golgi intermediate compartment (ERGIC) (Lonka et al. [Citation2000], Heine et al. [Citation2004], Mole et al. [Citation2004]). The function and interacting cytosolic and/or luminal proteins of both CLN6 and CLN8 are unknown.
The CLN6 gene is located on chromosome 15q23 and encodes a highly conserved 311 amino acid protein with six or seven predicted membrane domains (Gao et al. [Citation2002], Wheeler et al. [Citation2002]). The full topology of CLN6 is not known but the accessibility to glycosylation of a neo-N-glycosylation site at residues 151–153 of CLN6 suggests a luminal orientation for the predicted second loop (Heine et al. [Citation2004]). In overexpressing cells CLN6 can form dimers (Heine et al. [Citation2004]). Analysis of fibroblast cells from CLN6 patients or animal models of the disease have documented that the transport, sorting and processing of newly synthesized lysosomal protease cathepsin D is not affected by defective CLN6. In contrast, mutant CLN6 results in an increased mannose 6-phosphate receptor-depending uptake of the recombinant lysosomal enzyme arylsulfatase A and its reduced intracellular degradation (Heine et al. [Citation2004]).
Several mechanisms have been considered to explain ER retention of ER-resident membrane proteins. The interaction of specific sequence motifs in one or more of the topological domains of an ER-resident protein with effectors has been proposed to inhibit exit of the protein from the ER. Among the few experimentally characterized ER retention motifs are cytoplasmic N-terminal diarginine sequences (Schutze et al. [Citation1994], Zerangue et al. [Citation2001]), C-terminal tyrosine-based signals (Mallabiabarrena et al. [Citation1995]), and transmembrane domains (Sato et al. [Citation1996], Parker et al. [Citation2004]). Recently, a novel retention signal, CVLF, has been reported in a K+ channel subunit splice variant (Zarei et al. [Citation2004]). Additionally, short transmembrane domains with less than 17 amino acid residues and hydrophilic residues promote ER localization (Rayner & Pelham [Citation1997]). Proteins can also be retained indirectly by incorporation into oligomeric structures or large immobile networks (Nikonov et al. [Citation2002]). The ER-resident NCL protein CLN8 contains a functional ER-retrieval signal, KKRP at its C terminus (Lonka et al. [Citation2000]) but it is not known how CLN6 is retained.
The knowledge of the topology, domain structure and the presence of signal motifs is a prerequisite to search for CLN6 interacting proteins which will allow insights into its function. In the present study we investigated the structural properties of CLN6 important for dimerization and retention in the ER of neuronal and non-neuronal cells. Our results show that the N and C terminus of CLN6 are orientated to the cytoplasm and lumen, respectively. Surprisingly, CLN6 contains two ER retention signals, one comprises the N-terminal 49 amino acids and a second dominant motif consists of the distal pair of transmembrane domains.
Materials and methods
Materials
Rainbow™-coloured protein molecular-mass marker was obtained from Amersham Pharmacia Biotech (Freiburg, Germany), and bis (sulfosuccinimidyl) suberate (BS3) from Pierce (Illinois, USA). Opti-MEM™, Lipofectamine™ 2000 and antibiotics (penicillin/streptomycin) were purchased from Gibco Life Technologies (Karlsruhe, Germany). Digitonin and protein A-agarose were obtained from Sigma. Bradford reagent and Trans-Blot nitrocellulose membrane (0.2 µm) were purchased from Bio-Rad (Munich, Germany). Restriction endonucleases, T4 DNA ligase, and other molecular biology reagents were from Invitrogen (Leek, The Netherlands), New England Biolabs (Bad Schwalbach, Germany), Quiagen (Hilden, Germany) and Stratagene (La Jolla, CA, USA).
Antibodies
The polyclonal anti-myc-antibody was obtained from Santa Cruz Biotechnology (California, USA). The monoclonal anti-myc-antibody was a gift from Judith Blanz (ZMNH, Hamburg, Germany), and the monoclonal antibody against protein disulfide isomerase (PDI) and GM130 were purchased from Stressgen Biotechnologies Corp. (Victoria, BC, Canada). The polyclonal anti-CLN6-antibody 1747 was raised in rabbits against a peptide containing amino acids (aa) 1–15 as described recently (Mole et al. [Citation2004]). The polyclonal anti-CLN8 antibody was produced by immunizing rabbits with a peptide corresponding to amino acids 268–286 of CLN8 as described previously (Lonka et al. [Citation2000]). Anti-mouse IgG-Cy3, anti-rabbit-IgG-Cy3, and anti-mouse-IgG-fluorescein isothiocyanate (FITC) were obtained from Sigma. Anti-rabbit IgG-FITC conjugates were from Dianova (Hamburg, Germany) and the peroxidase-conjugated goat-anti-rabbit IgG was from Jackson Immuno Research Laboratories (West Grove, PA, USA).
cDNA constructs
Human full length CLN6-cDNA was cloned without a tag or in frame with a C-terminal myc-His-tag of vector pcDNA3.1/myc-His(−) A (Invitrogen, Leek, The Netherlands) into BamHI/HindIII-restriction sites (Heine et al. [Citation2004]). The substitution of the amino acids Arg5–Arg7 and Leu305–His306 by Ala were introduced by nested PCR using the following primers for the first round of PCR: Arg5–Arg7: R5–7for1 5′gccgccgcccagcacctgggagcgacg 3′and CLN6rev 5′cccaagcttggggtgccgactgctgac 3′; Leu305–His306: CLN6for 5′cgggatccgcgatgg aggcgacgcggagg 3′ and LHrev1 5′ccgactgctgacggcagcggtgtagaaagc 3′. For the second round of PCR the following primers were used for Arg5–Arg7: R5–7for2 5′cgggatccgcgatggaggcgacggccgccg 3′and CLN6rev, and CLN6for and LHrev2 5′cccaagcttgggaacagggtcattccacag 3′ were used for Leu305–His306. Deletion of the amino acid residues 2–10, 2–20, 2–49 and 2–118 was achieved using the primers del10for 5′cgggatccgcgatgggagcgacgggcggccca 3′, del20for 5′cgggatccgcgatgggcgcctccttcctgcag 3′, del49for 5‘gaattcatgatgtacttcacactgcag 3‘ and del118for 5‘ggattcatgatgatcttcatcatgggt 3‘ together with the CLN6rev primer. C-terminal truncation constructs were cloned using the primers del285rev 5‘ggaattcctcagtgccgactgctgac 3‘ and del225rev 5‘tctagactcggtgaccaggtacca 3‘ together with a CLN6for primer. Substitution of the amino acid residues Lys249–Arg252 by Ala was achieved by overlap extension using the primer CLN6for and K249rev 5′tgctgctgctgcctggtgcaggacgagggc 3′, and K249for 5′gcagcagcagcactcttcctggacagcaac 3′and CLN6rev for the first and the primer CLN6for and CLN6 rev for the second round of PCR. All PCR products were cloned into the BamHI/HindIII-sites of the vector pcDNA3.1/myc-His(−) A. The CLN6–CLN8 fusion constructs were made by overlap extensions using the primer CLN6Tfor 5′caccatggaggcgacgcggagg 3′ and CLN68Brev 5′cgtggagcggaaccagaggtcgaggtg 3′to amplify the first 49 amino acid residues of CLN6 and CLN68Cfor 5′ctctggttccgctccacgctgatggtcgct 3′ and CLN68DWTrev 5′ctatggcctcttcttccgcag 3′ or CLN68DWTrev 5′ctatggcctcctcctccgcag 3′ for the amplification of residues 24 to 286 of the wild type or mutant CLN8(K283R/K284R). The CLN6–CLN8 chimeric cDNAs were cloned into pcDNA™3.1D/V5-His-TOPO vector. All constructs were verified by sequencing.
Cell cultures and cell transfection
Baby hamster kidney (BHK21) cells were cultured in Dulbecco's modified Eagle's medium (Gibco Life Technologies, Karlsruhe, Germany) containing 10% fetal calf serum (PAA Laboratories, Linz, Austria) and 1% penicillin/streptomycin (DMEM/FCS) in a humidified atmosphere containing 5% CO2/95% O2 at 37°C. BHK21 cells were transiently transfected after seeding on 35 mm plates at a density of 5×105 cells/plate using Lipofectamine™ 2000. For the control, the cells were transfected with pcDNA3.1/myc-His(−) A vector.
Preparation of hippocampal neurons
Hippocampal neurons were prepared from mice brains at embryonic day 17 (Quitsch et al. [Citation2005]) and plated at a density of ∼500 cells/mm2 on glass coverslips coated with poly-L-lysine. Cells were grown in neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 0.5 mM glutamine and 12.5 µM glutamate. Due to a higher transfection efficiency transfection was performed 4 days after plating using the lipofectamine transfection kit. Fifteen hours after transfection cells were fixed with 4% paraformaldehyde/10 mM phosphate buffered saline, pH 7.4 (PBS) and permeabilized with 0.1% Triton X-100/PBS.
Western blotting
Solubilized proteins from transfected and non-transfected cells were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and examined by Western-blot as described previously (Heine et al. [Citation2003]).
Cross-link experiments
BHK21 cells overexpressing CLN6 and/or CLN8 constructs were permeabilized with 0.25% saponin in 50 mM 2-(N-morpholino)ethanesulfonic acid buffer pH 6.5, 150 mM NaCl, 0.5% BSA for 40 min at 4°C. The cross-linker BS3 (0.5 mM) was added in binding buffer (100 mM HEPES buffer pH 7.6 containing 120 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 8 mM glucose, 0.25% saponin) for 30 min at 4°C. The reaction was stopped by the addition of Tris buffer pH 7.4 to a final concentration of 0.2 M. After solubilization the samples were analysed by SDS-PAGE and Western blotting.
Immunofluorescence microscopy
For double immunofluorescence microscopy overexpressing BHK21 cells were plated on poly-L-lysine-coated microscope glass cover slips and transfected either with wild type or mutant CLN6 cDNA constructs or with the vector alone. After transfection (24 h), the cells were fixed with 4% paraformaldehyde and permeabilized with PBS containing 0.1% Triton X-100. To selectively permeabilize the plasma membrane, paraformaldehyde fixed cells were washed with PBS and incubated with PBS containing 1% BSA in the presence of 0, 0.5, 1, 5 or 10 µg/ml digitonin for 15 min at 4°C. The coverslips were then washed with PBS and blocked in PBS containing 3% BSA. After washings the cells were incubated for 2 h at room temperature with the primary antibodies anti-myc (1:50), anti-PDI (1:300), anti-CLN6 (1:1000), anti-CLN8 (1:500), and anti-GM130 (1:100) in PBS containing 3% BSA followed by incubation with the secondary antibodies, conjugated to fluoresecin isothiocyanate (FITC) or Cy3. When indicated the cells were washed three times in phosphate-buffered saline, pH 7.4 and stained with 2 µg/ml 4’, 6 diamidino-2-phenylindole at room temperature for 10 min. The coverslips were mounted in GelMount (Biomeda Corp.) and viewed either with a Zeiss Axiovert S100 microscope and documented with the DP-50 camera (Olympus, Hamburg, Germany) or with Leica DMIRE2 confocal microscope with TCS NT software (Leica Microscope and Scientific Instruments Group).
Results
Opposite orientation of N and C termini in CLN6
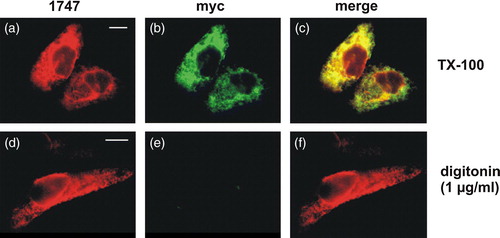
Six or seven transmembrane domains have been predicted for CLN6 (Gao et al. [Citation2002], Wheeler et al. [Citation2002]). To determine the orientation of the N and C termini, the C-terminally myc-tagged CLN6 was expressed in BHK21 cells and analysed by double immunofluorescence microscopy under different membrane permeabilization conditions. The antibody against protein disulfide isomerase (PDI) served as marker for the ER lumen and antibodies 1747 against the N-terminal CLN6 peptide and the C-terminal myc-tag were used to examine the orientation of CLN6 in the ER. As expected, in CLN6myc overexpressing cells fully permeabilized with Triton X-100 both CLN6 antibodies showed a complete overlapping staining (a–1c). Double labelling of CLN6 stained with 1747 or anti-myc antibodies with PDI showed also overlapping staining (not shown). When plasma membranes were selectively permeabilized with increasing concentrations of digitonin, the CLN6myc fusion protein could be stained with the N terminus recognizing antibody 1747 at 1 µg/ml digitonin, whereas no staining was observed with the anti-myc antibody recognizing the C terminus (d–1f). At concentrations >1 µg/ml digitonin CLN6myc was accessible to both antibodies (not shown). These data indicate that the N terminus is directed to the cytoplasm and the C terminus towards the ER lumen. Therefore CLN6 contains seven transmembrane domains.
Figure 1. Opposite orientation of N and C termini in CLN6. BHK21 cells transiently expressing the C-terminally myc-tagged CLN6 fusion protein were fixed and membranes either permeabilized with Triton X-100 (a–c) or selectively with 1 µg/ml digitonin (d–f). The N terminus was detected using purified primary CLN6 antibody 1747, and secondary rhodamine-conjugated antibody (a and d). The C terminus was visualized with the anti-myc and a secondary FITC-conjugated antibody (b and e). The images were merged (c and f). Yellow indicates the simultaneous accessibility of primary antibodies to the cytosolic and luminal epitopes. Bars, 5 µm. This Figure is reproduced in colour in Molecular Membrane Biology online.
CLN6 contains two ER retention signals
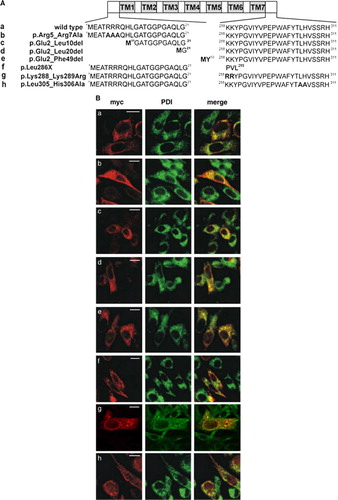
The CLN6 protein contains four potential ER retention or retrieval signals in the N and C terminus (Cosson & Letourneur [Citation1994], Hardt et al. [Citation2003], Michelsen et al. [Citation2005]) comprising 5RRR7 (residues 5–7), 249KRKR252 (residues 249–252), 288KK289 (residues 288–289), and 305LH306 (residues 305–306). To study the importance of single motifs or the proposed N-terminal cytosolic domain for ER retention, various substitutions of amino acid residues or N-terminal truncations were introduced into CLN6myc constructs, as illustrated in A. Furthermore, amino acid motifs in the C-terminal domain were substituted either by alanine or arginine residues or the C-terminal domain (residues 287–311) was deleted and the localization of CLN6 constructs tested by double immunofluorescence microscopy. The majority of all CLN6 mutants expressed in BHK21 cells were found to be entirely colocalized with the ER marker PDI (B). For distinct CLN6myc constructs, however, such as p.Arg5_Arg7Ala, p.Leu286X, or p.Lys288_Lys289Arg the extent of colocalization with PDI varied resulting in a heterogenous granular staining of CLN6 that was dispersed around the nucleus throughout the cytosol.
Figure 2. ER retention of CLN6 is not mediated by dibasic or dileucine-motifs. (A) Wild type (a) and various CLN6myc constructs (b–h) are represented schematically. The triple arginine motif in the N terminus was substituted by alanine residues (p.Arg5_Arg7Ala = b) or sequential N-terminal truncations were introduced (p.Glu2_Leu10del; p.Glu2_Leu20del; p.Glu2_Phe49del = c–e). Potential retention motifs in the C terminus of CLN6 were substituted by alanine residues (p.Lys288_Lys289Arg; p.Leu305_His306Ala g, h). (B) The wild type (a) and mutant CLN6myc constructs (b–h) were transiently expressed in BHK21 cells, stained for the myc-tag in red, and double-labeled with an antibody against PDI (green). Merged images (yellow) indicate overlapping localization of CLN6myc constructs with the ER marker. Bars, 5 µm. Figure 2B is reproduced in colour in Molecular Membrane Biology online.
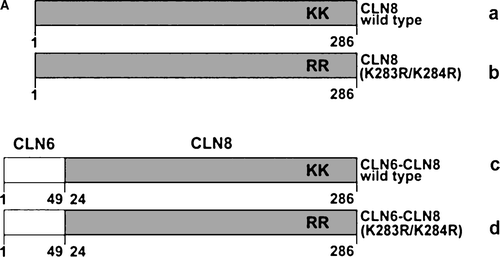
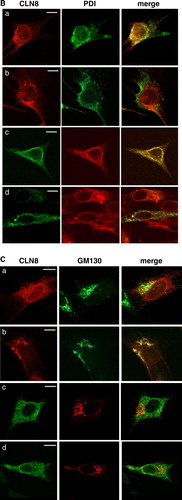
ER-retention of various membrane proteins examined so far is mediated by signal structures localized either in cytosolic or transmembrane domains or require both cytosolic and transmembrane domains (Yang et al. [Citation1997], Barre et al. [Citation2005], Michelsen et al. [Citation2005]). To examine whether the N-terminal cytoplasmic domain of CLN6 contains information for ER retention independent of the full-length CLN6 protein, the N-terminal cytosolic domain of wild type CLN8 and CLN8 (K283R/K284R) were replaced by the N-terminal CLN6 domain (A). Substitution of lysine residues by arginines in the ER retrieval signal 283KKRP286 of the CLN8 protein prevents its return to the ER and results in localization of CLN8 in the Golgi complex (Lonka et al. [Citation2000]). The constructs were expressed in BHK21 cells and the subcellular localization was determined by double immunofluorescence microscopy. Both wild type CLN8 and CLN6–CLN8 wild type were colocalized with the ER marker PDI (B). The mutant CLN8 (K283R/K284R) demonstrated an immunostaining pattern different to that of PDI (B; Lonka et al. [Citation2000]) but the CLN6–CLN8 (K283R/K284R) was detected in the ER. The data were confirmed by the colocalization of CLN6–CLN8 (K283R/K284R) with the Golgi marker protein GM130. The CLN8 (K283R/K284R) protein was detected in GM130-positive structures whereas the N terminus of CLN6 altered the intracellular localization of the CLN8 (K283R/K284R) fusion protein (C). The data indicate that the N-terminal 49 amino acids of CLN6 are sufficient to retain the CLN8 (K283R/K284R) protein in the ER.
Figure 3. The N terminus of CLN6 mediates ER retention. (A) Schematic representation of CLN constructs used in the experimental approach: the ER-resident wild type (a) and the mutant CLN8 (K283R/K284R = b) protein localized in the Golgi apparatus, and the respective CLN6-CLN8 fusion proteins in which the N terminally 24 amino acids of CLN8 were substituted by the 49 amino acid N terminus of CLN6 (c, d). The position of the dibasic motif (KK) in the C-terminal domain of CLN8 is indicated. (B and C) the CLN8 (a, b) and CLN6-CLN8 constructs (c, d) were expressed in BHK21 cells and visualized using a polyclonal antibody directed against the C terminus (amino acid residues 268–286) of CLN8 (red). The cells were double-labeled with monoclonal antibodies against PDI (B) or GM130 (C) shown in green. Yellow indicates in the merged images overlapping of CLN constructs with the ER or Golgi markers. Bars, 5 µm. B and C are reproduced in colour in Molecular Membrane Biology online.
Because loss of the N- or C-terminal domains did not appear to abolish the ER localization of CLN6 (B), we assessed the contribution to location of more internal domains. Truncated CLN6 proteins lacking either N-terminal, transmembrane domains TM1 and TM2, and the first cytosolic loop (CLN6 p.Glu2_Ile118del) or the luminal C terminus, TM6 and TM7 and the third cytoplasmic loop (CLN6 p.Glu225X) (A) were expressed in BHK21 cells. Western blot analysis showed that deletions at the N terminus (p.Glu2_Phe49del and p.Glu2_Ile118del) affected the stability of CLN6 (B) but deletions at the C terminus (p.Leu286X and p.Glu225X) did not. Importantly, whilst the N-terminal truncated CLN6 p.Glu2_Ile118del was found to colocalize with the ER marker PDI (), the loss of the C-terminal distal pair of transmembrane domains (p.Glu225X) altered the subcellular localization of the mutant CLN6. Immunostaining of CLN6 p.Glu225X was found in compact juxtanuclear structures which were positive for the cis Golgi marker GM130 (). The data indicate that retention of CLN6 in the ER is mediated by the most distal pair of transmembrane domains TM6 and TM7.
Figure 4. Expression of N- and C-terminal truncated CLN6 mutants. (A) Schematic representation of CLN6myc truncation mutants affecting the N terminus (p.Glu2_Leu20del = 2; p.Glu2_Phe49del = 3; p.Glu2_Ile118del = 4) or the C terminus (p.Leu286X = 5; p.Glu225X = 6). (B) The expression of the wild type and CLN6myc mutants were analysed by Western blotting using anti-myc antibodies 24 h after transfection of BHK21 cells.
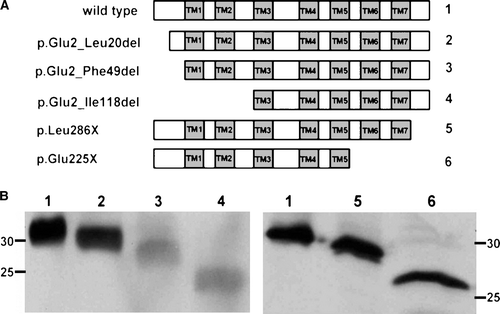
Figure 5. The distal pair of transmembrane domains of CLN6 represent a dominant ER retention structure. The localization of N- and C-terminally truncated CLN6myc mutants (p.Glu2_Ile118del and p.Glu225X corresponding to construct number 4 and 6, respectively, described in A) were analysed by double immunofluorescence microscopy 24 h after transfection using either the anti-myc antibody or antibodies against PDI, or GM130. Blue staining represents nuclear 4’, 6 diamidino-2-phenylindole (DAPI) staining used to show location of the cells. Bars, 10 µm. This Figure is reproduced in colour in Molecular Membrane Biology online.
Localization of CLN6 in neuronal cells
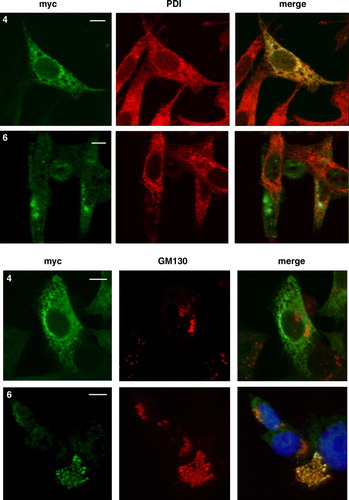
Because the symptoms of the variant form of late infantile NCL manifest only in the central nervous system, the distribution of CLN6 transiently expressed in primary hippocampal neurons four days after plating was investigated by double immunofluorescence microscopy. The wild type CLN6 was localized in cell soma and along neural extensions (). In neuronal soma the CLN6 staining colocalized completely with the ER marker PDI (A). A higher magnification of the neuronal extensions showed partial colocalization but also a punctate immunostaining different to PDI which was found in tubular-like structures. Whereas the staining of the N-terminally truncated CLN6 mutant p.Glu2_Phe49del resembled that of the wild type CLN6, no colocalization of CLN6 p.Glu225X with the PDI marker was observed. CLN6 p.Glu225X appeared to be present in small vesicular-like structures which were not positive for GM130 (B).
Figure 6. Localization of wild type and mutant CLN6 forms in hippocampal neurons. Mouse hippocampal primary neurons were transfected with CLN6 cDNAs encoding wild type (1) or p.Glu2_Phe49del (3) and p.Glu225X (6) mutants according the numbering of constructs described in A. Neurons were double immunostained for CLN6 (green) and the ER marker PDI or the Golgi marker GM130 (red). Yellow indicates overlap of CLN6 protein and subcellular markers. The square at the right is a magnified view of the indicated region. Insets represent 8-fold higher magnification images of the regions marked by the white rectangles. Bars, 5 µm. This Figure is reproduced in colour in Molecular Membrane Biology online.
CLN6 forms homodimers
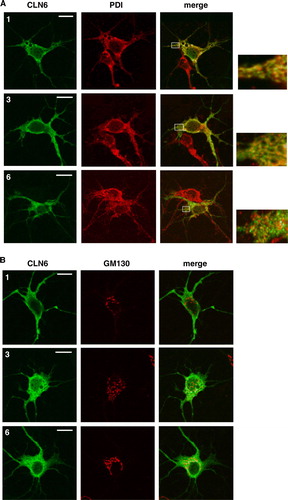
Cross-linkage experiments have already shown that CLN6 forms dimers (Heine et al. [Citation2004]). Two experimental approaches were carried out to examine whether CLN6 can form homodimers or heterodimeric complexes with the ER-localized NCL protein CLN8. In the first approach, full length CLN6 (27 kDa) and the C-terminal CLN6myc fusion protein (30 kDa) were co-expressed in BHK21 cells and cross-linked with the non-cleavable cross-linker BS3. Upon cross-linkage dimeric forms of 60 and 54 kDa were observed in CLN6myc and CLN6 expressing cells, respectively (A). In cells cotransfected with CLN6myc and CLN6, three immunoreactive dimeric forms of 60, 57, and 54 kDa were detectable representing CLN6myc/CLN6myc, CLN6myc/CLN6, and CLN6/CLN6 complexes, respectively. These data demonstrate the capability of CLN6 to form homodimers.
Figure 7. CLN6 forms homodimers. (A) BHK21 cells were transfected either with the empty vector, or with cDNAs coding for full-length CLN6, CLN6myc, or both CLN6 and CLN6myc. After 24 h the cells were permeabilized and treated (+) or not (−) with 0.5 mM BS3. The cell extracts were separated by SDS-PAGE and analysed by Western blotting using anti-CLN6-specific 1747 antibody. The positions of the molecular mass marker proteins and the different dimeric CLN6 forms are indicated. (B) BHK21 cells overexpressing either wild type CLN8, CLN6myc or both CLN8 and CLN6myc were permeabilized and treated with (+) or without (−) BS3. The cell extracts were separated by SDS-PAGE and analysed first by Western blotting using anti-CLN8 antibody. Thereafter, the blot was stripped and reprobed with anti-myc antibody. Vector-transfected cells were used as control. The blots were densitometrically scanned and the percentage of dimeric forms formed by cross-linkage are given.
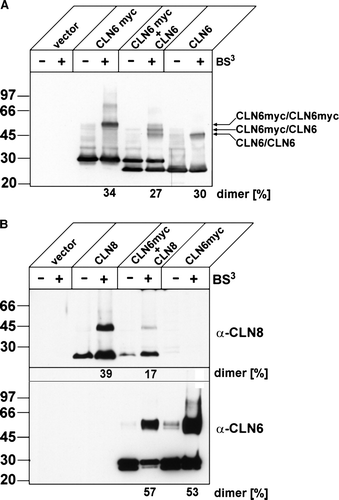
In the second approach, the capability of CLN6 to form heterodimers with CLN8 was tested. BHK21 cells were transiently transfected with the cDNAs of CLN8, CLN6myc or both CLNB and CLN6myc and after BS3 cross-linkage the polypeptides were detected by Western blotting for CLN8 or CLN6. B (upper panel) shows that about 40% of expressed CLN8 can also form dimers of 46 kDa. The absence of CLN8 immunoreactive bands of approximately 53 kDa in cells coexpressing CLN8 (23 kDa) and CLN6myc (30 kDa) indicated that CLN6 cannot form heterodimers with CLN8 when BS3 was used as cross-linker. This conclusion was confirmed after reprobing the blot for CLN6 immunoreactive forms (B, lower panel). After cross-linkage only homodimeric CLN6 forms of 60 kDa were observed but no heterodimeric forms of 53 kDa with CLN8. The identity of the second band with increased electrophoretic mobility reacting occasionally with the anti-myc antibody in BHK21 cells is unknown.
Structural requirements for CLN6 dimerization
To examine the structural requirements for CLN6 dimerization N- and C-terminal truncated CLN6 mutants were tested for their ability to form dimers upon BS3 cross-linkage. CLN6 mutants lacking half or the complete N-terminal domain (p.Glu2_Leu20del; p.Glu2_Phe49del) were still able to form dimeric complexes (A, 8B; ). Cross-linkage of the CLN6 mutant p.Glu2_Leu20del resulted even in a higher percentage of dimers than for wilde type CLN6 (74 vs. 38% of total, respectively). Upon cross-linkage of the CLN6 mutants p.Glu2_Ile118del, p.Leu286X, and p.Glu225X, no dimeric forms were detectable. Reduced amounts of monomeric CLN6 forms, particularly for pGlu225X (A), suggest either that cross-linkage occurred with other components in higher molecular mass complexes which did not enter the separation gel or alternatively that crosslinking prevented accessibility by the antibody. This data implicate both N and C termini of CLN6 in interaction with other proteins.
Figure 8. Capability of truncated CLN6 to dimerize. In BHK21 cells overexpressing wild type (1) or the indicated CLN6myc truncation mutants (p.Glu2_Leu20del = 2; p.Glu2_Phe49del = 3; p.Glu2_Ile118del = 4; p.Leu286X = 5; p.Glu225X = 6) membranes were permeabilized and proteins chemically cross-linked with (+) or without (−) 0.2 mM BS3. The cell extracts were separated by SDS-PAGE followed by Western blot analysis using anti-myc antibodies. The blots were densitometrically scanned and the percentage of dimeric forms formed by cross-linkage are given
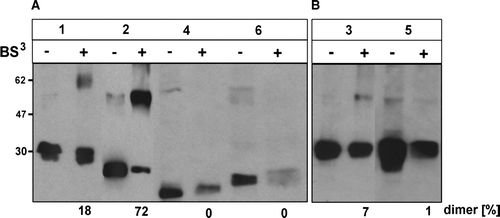
Table I. Relative expression of dimeric CLN6 forms.
Discussion
Our structural studies on CLN6 have provided evidence for (i) the location of the N and C termini, (ii) a signal-dependent retention in the ER, and (iii) the capability of CLN6 to form homodimers.
Analysis of the deduced amino acid sequence of CLN6 by different computer programs resulted in different predictions on the topology of CLN6. One study suggested six transmembrane domains and the N and C terminus at the same site of the membrane (Gao et al. [Citation2002]). A second model proposed seven transmembrane domains with the N terminus in the cytoplasm and a luminal C terminus (Wheeler et al. [Citation2002]). Using a differential digitonin permeabilization protocol and a specific antibody against the N-terminal 15 amino acid residues, the latter prediction has been supported showing the orientation of the N and C termini towards the cytosol and lumen of the ER, respectively, separated by seven transmembrane domains (). Consistent with this model, the introduction of a neo N-glycosylation site at Asn151 and its glycosylation after expression in BHK21 cells (Heine et al. [Citation2004]) confirmed that the hydrophilic residues 137–179 form a luminal loop.
Exit of membrane proteins from the ER is a selective process mediated by positive sorting signals such as diacidic and dihydrophobic motifs (Sevier et al. [Citation2000], Nufer et al. [Citation2002], Wang et al. [Citation2004]). The sorting signals by interaction with coat proteins or vesicle membrane proteins, mediate concentration of the membrane proteins in selected regions of the ER, where they are packed into COP II (cytosolic coat protein II)-transport vesicles. ER-resident membrane proteins are partly maintained by the well characterized linear signal K(X)KXX (Teasdale & Jackson [Citation1996]). Another class of sorting motifs is represented by arginine-based ER localization signals and can be active at various positions of diverse polytopic membrane proteins (Michelsen et al. [Citation2005]). Recently, a new ER retention motif, CVLF has been described in a splice variant of a K+ channel subunit (Zarei et al. [Citation2004]). Interestingly, primary determinants mediating localization of type I and II membrane proteins to the ER have been mapped to their transmembrane (TM) domains (Yang et al. [Citation1997], Letourneur & Cosson [Citation1998]). A relatively short length of the hydrophobic domain (≤17 residues) is a common feature of these proteins. In the polytopic IP3 (inositol trisphosphate) receptor, however, ER retention signals have been reported in pairs of TM domains linked by a luminal loop (Parker et al. [Citation2004]). Finally, integral membrane proteins can also be retained indirectly by incorporation into oligomeric structures or large immobile networks (Nikonov et al. [Citation2002]). CLN6 contains four potential active ER retention/retrieval motifs such as 5RRR7, 249KRKR252, 288KK289, and 305LH306. Neither alanine substitution of the KRKR-motif (C. Heine unpublished results) or changes of the LH residues nor the complete deletion of the C-terminal domain altered the localization of CLN6 in the ER (B). The staining pattern of the p.Lys288_Lys289Arg mutant, however, was not completely identical with the ER marker localization. Whether this changed distribution of the CLN6 mutant is simply due to overexpression or to reduced retention in the ER is unclear. Further studies are required to define the importance of the KK residues in more detail and to examine their potential cooperative effects with other ER retention signals. On the other hand, the KRKR-motif, the KK and LH residues in CLN6 are located either in the predicted TM domain 6 or in the C-terminal domain exposed on the luminal side of the ER membrane (Wheeler et al. [Citation2002]) and it is perhaps not surprising that these signals do not act as an ER retention motif.
The sequential truncation of the N-terminal cytosolic 49 amino acids or the replacement of the triple arginine motif by alanines also did not result in alterations of the subcellular localization of mutant CLN6 (B). However, the substitution of the N-terminal domain of the polytopic mutant CLN8 (K283R/K284R) localized in the Golgi (Lonka et al. [Citation2000]) by the N-terminal 49 amino acids of CLN6 retained the chimera in the ER (). From these data we conclude that the N-terminal CLN6 domain contributes to retention of CLN6 within the ER. However, the failure of a CLN6 mutant lacking the first 49 amino acids to exit the ER indicates the presence of an ER retention signal elsewhere in the CLN6 molecule. Because pairs of TM domains in the polytopic IP3 receptor 1 target the protein to the ER (Parker et al. [Citation2004]), we expressed CLN6 mutants either lacking the N-terminal and the proposed TM1 and TM2 domains (p.Glu2_Ile118del) or the C-terminal domain and TM6 and TM7 (p.Glu225X). The data showed clearly that loss of the distal pair of transmembrane domains altered the subcellular localization in non-neuronal cells towards structures positive for the cis Golgi marker GM130 (). However, when the CLN6 mutant lacking TM6 and TM7 was expressed in primary mouse neurons, the retention in the ER was lost but no colocalization with the Golgi marker GM130 was detectable. The data suggest that the C-terminally truncated CLN6 protein can exit the ER and may become sorted into a PDI-negative ER subcompartment that is unable to fuse with cis Golgi membranes, or, alternatively that the membrane region containing the mutant CLN6 protein fails to be recognized by the vesicular transport machinery in neuronal mouse cells required for proper targeting. Together, CLN6 appeared to contain two ER retention signals, one in the N-terminal cytosolic domain and a second dominant determinant comprising the distal pair of transmembrane domains. Additionally, the 288KK289 motif may function in an additive manner to ER retention. Two signal structures, a dilysine motif and the transmembrane domain, have also been reported to be required for ER residency of the type I membrane protein glucuronosyltransferase 1A (Barre et al. [Citation2005]).
Both the N and C termini of CLN6 were implicated in homodimerization or interaction with other proteins, perhaps explaining the ability of the N terminus to prevent the mutant CLN8 (K283R/K284R) from leaving the ER. It remains to be studied which residues in the N-terminal domain and in the transmembrane domains of CLN6 are important for intermolecular interactions and which proteins may be involved in the recognition and binding of the signals. Short transmembrane domains with less than 18 amino acid residues have been reported to promote ER localization of single membrane spanning proteins in yeast (Rayner & Pelham, [Citation1997]). Because the seven TM domains of CLN6 comprise between 19 and 29 amino acid residues, it is rather unlikely that the length of TM domains determines ER localization of CLN6. However, the capability to form homodimeric CLN6 complexes is drastically reduced in N- and C-terminal truncated CLN6 () suggesting that dimerization may contribute to retention of CLN6 in the ER. There are several examples of polytopic proteins where dimeric or multimeric assembly result in steric masking of signal structures such as arginine-based motifs preventing the exit from the ER (Michelsen et al. [Citation2005]). Additionally, of interest, the cross-linkage experiments demonstrated that CLN6 cannot dimerize with the other ER-resident NCL protein CLN8 whereas both CLN6 and CLN8 form homodimers.
Two mutations were found in CLN6 patients predicting the loss of transmembrane domain 6 and 7 (p.Tyr221X) or the C-terminal domain (p.Val277ProfsX5; http://ucl.ac.uk/ncl/cln6.shtml). At present it is not known whether truncated CLN6 polypeptides are expressed in tissues of these patients capable to form dimers. Therefore, expression analyses of mutant CLN6 will be required to determine the effect of the mutation on dimerization and subcellular localization which might also allow correlations to the clinical phenotype of the patients.
This work was supported by the Commission of the European Communities grant (503051), and the Batten Disease Support and Research Association, USA (YM).
References
- Barre L, Magdalou J, Netter P, Fournel-Gigleux S, Ouzzine M. The stop transfer sequence of the human UDP-glucuronosyltransferase 1A determines localization to the endoplasmic reticulum by both static retention and retrieval mechanisms. FEBS J 2005; 272: 1063–1071
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science 1994; 263: 1629–1631
- Gao H, Boustany RM, Espinola JA, Cotman S L, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet 2002; 70: 324–335
- Hardt B, Kalz-Füller B, Aparicio R, Völker C, Bause E. (Arg)3 within the N-terminal domain of glucosidase I contains ER targeting information but is not required absolutely for ER localization. Glycobiology 2003; 13: 159–168
- Heine C, Koch B, Storch S, Kohlschütter A, Palmer DN, Braulke T. Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J Biol Chem 2004; 279: 22347–22352
- Heine C, Tyynelä J, Cooper JD, Palmer DN, Elleder M, Kohlschütter A, Braulke T. Enhanced expression of manganese-dependent superoxide dismutase in human and sheep CLN6 tissues. Biochem J 2003; 376: 369–376
- Isosomppi J, Vesa J, Jalanko A, Peltonen L. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum Mol Genet 2002; 11: 885–891
- Järvelä I, Sainio M, Rantamaki T, Olkkonen VM, Carpen O, Peltonen L, Jalanko A. Biosynthesis and intracellular targeting of the CLN3 protein defective in Batten disease. Hum Mol Genet 1998; 7: 85–90
- Kim Y, Ramirez-Montealegre D, Pearce DA. A role in vacuolar arginine transport for yeast Btn1p and for human CLN3, the protein defective in Batten disease. Proc Natl Acad Sci USA 2003; 100: 15458–15462
- Kyttälä A, Ihrke G, Vesa J, Schell MJ, Luzio JP. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol Biol Cell. 2004; 15: 1313–1323
- Letourneur F, Cosson P. Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J Biol Chem 1998; 273: 33273–33278
- Lonka L, Kyttälä A, Ranta S, Jalanko A, Lehesjoki AE. The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Hum Mol Genet 2000; 9: 1691–1697
- Mallabiabarrena A, Jimenez MA, Rico M, Alarcon B. A tyrosine-containing motif mediates ER retention of CD3-epsilon and adopts a helix-turn structure. EMBO J 1995; 14: 2257–2268
- Michelsen K, Yuan H, Schwappach B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep 2005; 6: 717–722
- Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, Cutler DF. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res 2004; 298: 399–406
- Nikonov AV, Snapp E, Lippincott-Schwartz J, Kreibich G. Active translocon complexes labeled with GFP-Dad1 diffuse slowly as large polysome arrays in the endoplasmic reticulum. J Cell Biol 2002; 158: 497–506
- Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J Cell Sci 2002; 115: 619–628
- Parker AK, Gergely FV, Taylor CW. Targeting of inositol 1,4,5-trisphosphate receptors to the endoplasmic reticulum by multiple signals within their transmembrane domains. J Biol Chem 2004; 279: 23797–23805
- Quitsch A, Berhörster K, Liew CW, Richter D, Kreienkamp HJ. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J Neurosci 2005; 25: 479–487
- Rayner JC, Pelham HR. Transmembrane domain-dependent sorting of proteins to the ER and plasma membrane in yeast. EMBO J 1997; 16: 1832–1841
- Santavuori P, Vanhanen SL, Autti T. Clinical and neuroradiological diagnostic aspects of neuronal ceroid lipofuscinoses disorders. Eur J Paediatr Neurol 2001; 5(Suppl A)157–161
- Sato M, Sato K, Nakano A. Endoplasmic reticulum localization of Sec12p is achieved by two mechanisms: Rer1p-dependent retrieval that requires the transmembrane domain and Rer1p-independent retention that involves the cytoplasmic domain. J Cell Biol 1996; 134: 279–293
- Schutze MP, Peterson PA, Jackson MR. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J 1994; 13: 1696–1705
- Sevier CS, Weisz OA, Davis M, Machamer CE. Efficient export of the vesicular stomatitis virus G protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that includes both tyrosine-based and di-acidic motifs. Mol Biol Cell 2000; 11: 13–22
- Siintola E, Partanen S, Strömme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki A-E, Tyynelä J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006; 129: 1438–1445
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 1997; 277: 1802–1805
- Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Brück W, Saftig P, Gärtner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet 2006; 78: 988–998
- Teasdale RD, Jackson MR. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu Rev Cell Dev Biol 1996; 12: 27–54
- Tyynelä J. Neuronal Ceroid-Lipofuscinoses. Lysosomes, P Saftig. Springer, New York 2005; 82–99
- Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature 1995; 376: 584–587
- Wang X, Matteson J, An Y, Moyer B, Yoo JS, Bannykh S, Wilson IA, Riordan JR, Balch WE. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J Cell Biol 2004; 167: 65–74
- Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet 2002; 70: 537–542
- Yang M, Ellenberg J, Bonifacino JS, Weissman AM. The transmembrane domain of a carboxyl-terminal anchored protein determines localization to the endoplasmic reticulum. J Biol Chem 1997; 272: 1970–1975
- Zarei MM, Eghbali M, Alioua A, Song M, Knaus HG, Stefani E, Toro L. An endoplasmic reticulum trafficking signal prevents surface expression of a voltage- and Ca2 + -activated K + channel splice variant. Proc Natl Acad Sci USA 2004; 101: 10072–10077
- Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci USA 2001; 98: 2431–2436