Abstract
The ammonium transport (Amt) proteins are a highly conserved family of integral membrane proteins found in eubacteria, archaea, fungi and plants. Genetic, biochemical and bioinformatic analyses suggest that they have a common tertiary structure comprising eleven trans-membrane helices with an N-out, C-in topology. The cytoplasmic C-terminus is variable in length but includes a core region of some 22 residues with considerable sequence conservation. Previous studies have indicated that this C-terminus is not absolutely required for Amt activity but that mutations that alter C-terminal residues can have very marked effects. Using the Escherichia coli AmtB protein as a model system for Amt proteins, we have carried out an extensive site-directed mutagenesis study to investigate the possible role of this region of the protein. Our data indicate that nearly all mutations fall into two phenotypic classes that are best explained in terms of two distinct effects of the C-terminal region on AmtB activity. Residues within the C-terminus play a significant role in normal AmtB function and the C-terminal region might also mediate co-operativity between the three subunits of AmtB.
Introduction
The movement of ammonium across biological membranes is an important process in nearly all organisms from bacteria to man and it is effected by a family of integral membrane proteins that is conserved throughout all domains of life (von Wirén & Merrick [Citation2004]). (In this paper we use the term ammonium to refer to both the protonated (NH4+) and the unprotonated (NH3) forms, and ammonia to refer specifically to NH3). The Amt (ammonium transport) proteins are found in bacteria, archaea, fungi and plants, and in animals they are represented by the closely related Rhesus (Rh) proteins (Marini et al. [Citation2000a], Liu et al. [Citation2001], Winkler [Citation2006]). In Escherichia coli the single Amt protein, AmtB, is encoded by the second gene in the glnK amtB operon. GlnK is a member of the PII family of signal transduction proteins and interacts with AmtB to regulate the activity of the transporter in response to the intracellular nitrogen status (Javelle et al. [Citation2004], Coutts et al. [Citation2002], Durand & Merrick [Citation2006]). The genetic linkage of amtB and glnK is found in nearly all eubacteria and archaea suggesting that control of ammonium uptake by GlnK is probably ubiquitous (Thomas et al. [Citation2000a], Javelle & Merrick [Citation2005]).
The Amt proteins have a conserved core of 11 trans-membrane helices (TMH) with the C-terminus always being intracellular (Thomas et al. [Citation2000b], Marini & Andre [Citation2000]). In E. coli the structural gene amtB encodes a preprotein with a signal peptide that is cleaved off to produce the mature AmtB (Thornton et al. [Citation2006]) the topology of which is shown in (online version only).
AmtB is a stable trimer in the cytoplasmic membrane and retains this structure when purified and reconstituted in 2D crystals (Blakey et al. [Citation2002], Conroy et al. [Citation2004]). The X-ray crystal structure of E. coli AmtB shows that the 11 TMH (M1 to M11) form a right-handed helical bundle around a central channel (Khademi et al. [Citation2004], Zheng et al. [Citation2004]). The structure of helices M1 to M10 reflects a quasi-twofold axis that relates the structural context of the M1 to M5 region to that of M6 to M10. The M11 helix is a 50 Å-long straight helix that surrounds the lipid-accessible face of each monomer. A hydrophobic channel is located at the centre of each monomer and is construed to be the route through which substrate is conducted. It is proposed that the protein binds NH4+ within a periplasmic vestibule; that the ion is then deprotonated and that NH3 is conducted across the membrane and subsequently reprotonated on the cytoplasmic side of the membrane (Khademi et al. [Citation2004], Zheng et al. [Citation2004], Winkler [Citation2006], Lin et al. [Citation2006]). Hence the Amt proteins are proposed to function as ammonia channels and recent in vivo analysis of the mode of action of E. coli AmtB supports this model (Javelle et al. [Citation2005]).
Amt polypeptides are typically 400–450 amino acids in length though in some cases, e.g. Caenorhabditis elegans Amt3 and Synechocystis Amt3, they are predicted to be considerably larger owing to the fact that the cytoplasmic C-terminal region (CTR) is significantly extended. In the CTR a core region of 22 residues, located immediately downstream of the last TMH, M11, is highly conserved in bacteria, archaea, fungi and plants (see in online version only). The structure of the CTR was not resolved in the X-ray structures of E. coli AmtB (Khademi et al. [Citation2004], Zheng et al. [Citation2004]) but it was subsequently determined in the X-ray structure of another Amt protein, Archaeoglobus fulgidus Amt-1 (Andrade et al. [Citation2005]).
The precise role of the CTR is presently unknown. Deletion of the region from E. coli AmtB reduces the apparent activity of the protein to around 30% that of the wild-type and also prevents association of AmtB with GlnK (Coutts et al. [Citation2002]). Similar reductions in activity are observed when the CTR is deleted from the AmtB protein of the thermophile Aquifex aeolicus (Soupene et al. [Citation2002]) and the Mep2 protein of Candida albicans (Biswas & Morschhauser [Citation2005]). In a variety of fungi, including Saccharomyces cerevisiae, C. albicans and Ustilago maydis, the switch to a filamentous growth form that is observed on nitrogen-limited media is dependent on one of the Amt proteins (Lorenz & Heitman [Citation1998], Biswas & Morschhauser [Citation2005], Smith et al. [Citation2003]). The signalling pathway that is implicated in this developmental switch has not yet been characterized, but in S. cerevisiae and C. albicans deletion of the CTR from the relevant Amt protein (Mep2) prevents induction of filamentous growth suggesting that this region may be necessary for interaction with downstream components of the pathway (Biswas & Morschhauser [Citation2005], Van Nuland et al. [Citation2006]).
Certain mutations within the CTR of Amt proteins from S. cerevisiae, Aspergillus nidulans and Lysopersicon esculentum lead to a completely inactive protein that in some cases demonstrates epistasis over a wild-type protein expressed in the same cell (Marini et al. [Citation2000b], Ludewig et al. [Citation2003], Monahan et al. [Citation2002]). In each case these mutations convert a conserved glycine residue (homologous to G393 in E. coli AmtB – see ) to an aspartate residue. This phenotype has been interpreted as evidence for the oligomeric state of the Amt proteins in these organisms, but the reason for the epistatic nature of these particular mutations is unknown. In order to gain further insights into the role of the CTR of Amt proteins we have characterized a large number of mutant variants of the CTR of E. coli AmtB both with respect to ammonia channel activity and to interaction with GlnK.
Materials and methods
Strains and plasmids
The plasmids used in this study are listed in (Online version only) and in all cases they were present in strain GT1000, a Δ(glnK amtB) derivative of strain ET8000 (Coutts et al. [Citation2002]). Previous studies using this system have shown that it behaves very similarly to a wild-type E. coli strain (Thomas et al. [Citation2000b], Javelle et al. 2004). However as a consequence of engineering an extra BglII site at the 3′-end of the amtB gene the ‘wild-type’ allele previously used (amtB1) actually encodes two extra residues (D407 and L408) at the C-terminus of AmtB (Coutts et al. 2002). Whilst our studies suggested that these additional residues had no major effect on AmtB activity, for this study we modified the plasmid to ensure that the wild-type allele (carried on pSTUART1 or pSTUART2) was identical in its coding properties to the native E. coli gene. All the constructs described here are derivatives of these plasmids.
pSTUART1 was constructed by replacing the PvuI-BglII fragment of pGC2 by a PvuI-BamHI cut PCR product generated using primers 5′-GGGCCATCAGTTGCTGG-3′ and 5′CCGGATCCTTACGCGTTATAGGCATTCTCGCCGTGGCTGTTGACGTCCAGGCCTTCTCGCTC-3′ with pGC2 as template thereby introducing novel StuI and AatII sites into amtB and removing the D407 and L408 codons (altered bases are shown in bold type and novel restriction sites are underlined). However the StuI site in pSTUART1 is subject to dcm methylation. pSTUART2, in which the methylation site is suppressed, was therefore constructed by replacing the PvuI-AatII fragment of pSTUART1 with a PCR product generated using primers 5′-GGGCCATCAGTTGCTGG-3′ and 5′-GTTGACGTCAAGGCCTTCTCGCTC-3′ and pGC2 as template. The plasmids pESVC1 to 22 were all essentially constructed in the same manner, namely by replacement of the PvuI-AatII fragment in pSTUART2 (plasmids 15 to 18) or the PvuI-BamHI fragment in pSTUART1 (all other plasmids) with an appropriately cut PCR product derived using a common 5′ primer (5′-GGGCCATCAGTTGCTGG-3′) and a 3′ primer carrying either a point mutation or a deletion. For plasmids pESVC1 to pESVC18 the PCR template was a wild-type amtB gene and for pESVC19 to pESVC22 the template used was pESVC14. For plasmid pESVC23 the PCR product was generated by overlap PCR using external primers 5′-GGGCCATCAGTTGCTGG-3′ and 5′-GATGTCGGCGATATAGGCG-3′ and internal primers 5′-GTTGGTCTGGCTGTACCGGAAG-3′ and 5′-CTTCCGGTACAGCCAGACCAAC-3′. In this case the PvuI-BamHI fragment was cloned into pSTUART1.
To create plasmid pESVHA1 (coding for the HA-tagged version of AmtB, AmtBHA1) a PCR product was produced by overlap PCR using external primers 5′-CAGGAACGACATATGAAGATAG-3′ and 5′-CACGGTACCACCCGCGAAATCCAGCGCGCCGTGTG-3′, and internal primers 5′-TACCCGTATGATGTGCCGGATTACGCGGAACTGACGGCGGTGATGGGC-3′ and 5′-CGCGTAATCCGGCACATCATACGGGTAGATGTTTTTCAGCATCAACCAG-3′ (the HA-coding sequence is in italics), and used to replace the PstI-KpnI fragment of pAJ2008 (Javelle et al. [Citation2004]). To create plasmids pESVHA2 and pESVHA4 (coding for C-terminal variants of AmtBHA1) the PvuI-BamHI fragment of pESVHA1 was replaced with the equivalent fragments of pESVC2 and pESVC10 respectively.
For co-expression studies, amtB alleles were introduced in the ET8000 and GT1000 chromosomes by the λ-integration method (Simons et al. [Citation1987]) which results in single-copy integration of the target allele at the λ-attachment site (attλ). A pglnK amtBG393D expression cassette was created by replacing the PvuI-BamHI fragment of pESV2 (Javelle et al. [Citation2004]) with the equivalent fragment of pESVC14. The expression cassette was first moved as a HindIII-SalI fragment into pBlueScript IIKS, from where it was eventually cloned into the single BamHI site of pRS552 (Simons et al. [Citation1987]) to give pESV103 in which the cassette is oriented so that transcription of amtBG393D runs towards the downstream lac operon. The expression cassette was moved into the lambdoid vector λRS45 by in vivo recombination in strain MC1061 transformed with pESV103 (Simons et al. [Citation1987], Casadaban & Cohen [Citation1980]). Cell lysates containing the recombinant phage were used to infect either ET8000 or GT1000 giving the integration strains ESV103 and ESV101 respectively. Strain ESV102, whose chromosome contains the pglnK amtB8 expression cassette at attλ, was created as described for ESV101 except pESV2 was used as the donor of the cassette. To create ESV100 λRS45 was recombined with empty pRS552 and the resulting phage was integrated into the chromosome of ET8000. In all cases integration was verified by PCR.
[14C]methylammonium transport assays
Methylammonium (MA) uptake and assimilation rates were determined as described previously (Thomas et al. [Citation2000b]). [14C]MA hydrochloride (2.15 Gbq/mmol) was obtained from Amersham Biosciences. Absolute assimilation rates in washed assays were calculated from the slope of a linear regression of the uptake data over a 10 minute time-course; uptake was linear throughout the duration of the assay. Unwashed assays were modified from the method of Jayakumar and Barnes (Jayakumar & Barnes, Jr. [Citation1983]) as described previously (Javelle et al. [Citation2005]). When L-methionine sulphone (MSF) was added to unwashed assays, a final concentration of 200 µM was added 4 min. before initiation of the assay by addition of 20 µM [14C]MA. Both washed and unwashed assays were performed at 22°C.
Cell fractionation
Cell fractionation and ammonium shock treatment were carried out according to procedures described previously (Coutts et al. 2002).
Western blotting
Western blotting was performed as described previously (Coutts et al. [Citation2002]) using appropriate antibodies: anti-E. coli AmtB (Javelle et al. 2004); anti-His (Qiagen); anti-haemagglutinin (αHA) from Roche; anti-E. coli GlnK, raised in rabbits against GlnK protein purified as described previously (Javelle et al. 2004). For disassembly of trimeric AmtB into the monomeric species membrane fractions of E. coli cell cultures were diluted to 0.5 or 1 mg/ml total protein in saline phosphate and protein sample buffer 3X containing SDS and β-mercaptoethanol at a final concentration of 2% w/v and 5% v/v respectively. Samples were frozen at −80°C, and allowed to thaw slowly on ice the following day. One to 3 cycles of freeze/thaw assured virtually complete disassembly of AmtB.
Modelling the CTR of E. coli AmtB
The CTR of E. coli AmtB was manually aligned with the equivalent region of A. fulgidus Amt-1 as shown in Andrade et al. ([Citation2005]). Using the structure of the CTR of A. fulgidus Amt-1 (PDB code 2B2F) as a template, a model for the CTR structure of E. coli AmtB was then generated with Swiss-Model (Schwede et al. [Citation2003]). No optimizing of threading energy was necessary. Using Coot (Emsley & Cowtan [Citation2004]), a complete model of the E. coli AmtB trimer was then reconstructed from the derived model of the E. coli CTR and the 3D model of AmtB (PDB code 1U7G) (Khademi et al. [Citation2004]).
Results
Effects on AmtB activity of alterations in the C-terminal region
The very conserved nature of the ‘core’ region of the C-terminal region of Amt proteins () suggests that specific residues within the CTR, and/or the structure adopted by this region of the protein, play an important role in the function of the ammonia channel. In order to investigate this we constructed a variety of deletions and point mutations that remove or alter residues within the CTR ().
When assessed by Western blotting with an anti-AmtB antiserum, the results indicated that (in all but three cases that were not analysed) AmtB was always expressed in significant amounts and was always correctly targeted to the bacterial inner membrane ( and ). However, nearly every modification of the CTR significantly impaired the activity of AmtB (). The only two exceptions were changes to very C-terminal residues, the ΔA406 and N405D mutants, which had essentially wild-type activities. Hence it would appear that the integrity of the complete CTR is indeed very important for full activity of AmtB.
Figure 1. Expression levels of AmtB in selected variants. (A) Western blots of membrane fractions from wild-type and a selection of AmtB variants. 5µg of protein was loaded in each lane. Estimated molecular weight of major band is indicated to the left of the panel. (B) Activity, in a washed assay, of each of the variants shown in panel A, expressed as a % of the wild-type.
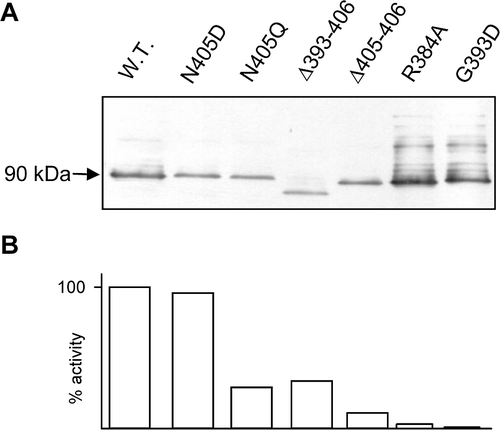
Table I. Relative [14C]-accumulation rates of GT1000 cells expressing AmtB CTR variants.
Two other alterations to N405, namely N405Q and N405A (both residues that occur naturally at position 405 in other AmtB proteins) reduced activity to around 30% of the wild-type level (). Furthermore deletions varying in length from just two residues (Δ405–406) to 23 residues (Δ383–406) all had a similar phenotype, i.e., a reduced but significant level of AmtB activity varying from 12% to 43% with a mean of 25% (). We defined the group of mutants with this phenotype as Class I mutants. This group also includes another single point mutation (Y404G) and three variants carrying multiple changes, i.e., N405A, ΔA406; G393D,Y404G; G393D,Δ(399–406). In essence all Class I mutants have a phenotype comparable to the complete deletion of the CTR, i.e. to AmtB4 (Coutts et al. [Citation2002]) or to Δ(383–406), suggesting that the phenotypes of all of these variants reflect a form of the protein that is functionally equivalent to one lacking the CTR.
A second phenotypic class of mutants was also identified based on their AmtB activity. Class II mutants had activities of 6% or less, with a mean of just 3%, when compared to wild-type and they all involved changes to essentially invariant CTR residues, i.e. R384 and G393. Whilst inactive Amt proteins carrying glycine to aspartate changes in the homologue of G393 have been selected independently both in S. cerevisiae and in A. nidulans (Marini et al. [Citation2000b], Monahan et al. [Citation2002]), our data indicate that a variety of changes to this glycine residue, either to positively or negatively charged residues or to a relatively small residue, i.e. alanine, all inactivate the protein. The potential trans-dominant nature of such changes will be addressed later. Of particular interest is the fact that whereas a single G393D mutation produces an essentially inactive (1%) AmtB, a double mutant in which disruption of the conserved tyrosine residue (Y404G) is combined with G393D markedly relieves the phenotype to give 17% activity (). A similar effect is seen with the G393D,Δ(399–406) double mutant which has 38% activity. Hence it would appear that the ‘inactive’ phenotype of the G393D allele is dependent on the most C-terminal residues of the CTR being in the native state.
The MA uptake data () were derived using a ‘washed assay’. This assay is used routinely for assessment of Amt protein activity in a wide variety of organisms. However we recently showed that, at least in E. coli, this assay is effectively a coupled assay of AmtB activity and subsequent MA assimilation into methylglutamine, mediated by glutamine synthetase (GS) (Javelle et al. [Citation2005]). Consequently the washed assay will reflect the ammonium transport activity of an AmtB variant providing that the nature of the apparent coupling between AmtB and GS is unaffected by the modification of AmtB. To investigate whether the data in were likely to be affected by effects on coupling of uptake and assimilation we repeated our analysis on a selection of the AmtB variants using the alternative ‘unwashed assay’ which assesses AmtB-dependent [14C]MA uptake into cells prior to GS-dependent assimilation (Javelle et al. [Citation2005]). To ensure that the calculated AmtB activity is essentially independent of GS-dependent assimilation, assays were also carried out after pre-incubation with the GS inhibitor L-methionine sulphone (MSF), which effectively prevents MA conversion to methylglutamine without having any significant effect on AmtB-dependent uptake (Javelle et al. [Citation2005]).
The activities of all the strains tested were essentially identical with or without MSF (). Furthermore the AmtB activities calculated using this assay were very similar to those using the washed assay (Table ). In both cases the Class I mutants had activities ranging from 21% to 31% of the wild-type activity, confirming that the reduced level of activity observed for this class of mutants in the washed assay is indeed a reflection of the uptake activity of these AmtB variants and not a defect in coupling of AmtB to MA assimilation. The Class II variants tested showed activities of 1–3% in both assays.
Table II. AmtB activities of representative variants measured in the ‘unwashed’ [14C]MA uptake assay.
CTR mutations affect AmtB activity and not stability
For mutants from both classes very significant levels of AmtB protein were detected () and consequently activity did not appear to correlate with levels of protein in the membrane. However in order to deduce conclusively that the reduced activity of around 30% seen in Class I mutants is not a function of reduced levels of AmtB in the cell membrane we wished to obtain an accurate in vivo quantification.
Firstly, we needed an appropriate antibody assay and our polyclonal anti-AmtB antibody was not suitable because the CTR is potentially a major epitope on the protein and modifications to this could quite possibly change the reactivity to the antibody. Indeed as changes to the CTR could potentially alter many aspects of the cytoplasmic face of the protein we chose to engineer AmtB so as to introduce a defined epitope on the periplasmic face. We used the haemagglutinin (HA) epitope that has been used previously for extracytoplasmic tagging of integral membrane proteins both with the E. coli KdpD protein (Facey & Kuhn [Citation2003]) and the human reduced folate carrier (Liu & Matherly [Citation2002]). The epitope (sequence YPYDVPDYA) was introduced between residues I86 and E87 in the first periplasmic loop (see ) to give AmtBHA1.
Introduction of the HA tag had no significant effect on AmtB activity. Comparison, using washed assays, of a plasmid encoding AmtBHA1 (pESVHA1), with a plasmid encoding wild-type AmtB (pSTUART1) in host strain GT1000 gave 93% of the wild-type activity. However detection of AmtBHA1 in whole cell extracts or membrane fractions showed that the protein behaved very differently to the wild-type. The wild-type AmtB trimer is extremely stable and is the predominant species when detected in cell extracts or after protein purification (Blakey et al. [Citation2002], Conroy et al. [Citation2004], Durand & Merrick [Citation2006]). By comparison AmtBHA1, which could be efficiently detected by either an anti-AmtB or an anti-HA antibody, appeared as multiple bands after electrophoresis on SDS-PAGE with the predominant species being the monomer (). As AmtBHA1 is not significantly impaired in activity, it is most likely that the presence of the additional residues in the first periplasmic loop somewhat destabilizes the protein in such a way that the trimer is no longer stable during electrophoresis. This is consistent with the multiple bands seen in the Western blot (). Nevertheless this behaviour poses problems for quantification of the protein and we therefore developed a method, based on repeated freeze-thawing of samples in the presence of β-mercaptoethanol, whereby the protein is essentially completely reduced to the monomeric species. This procedure allows quantification of any AmtB variant by comparison against a dilution series of the wild-type protein ().
Figure 2. Detection of AmtBHA1, a HA-tagged derivative of AmtB, by western blotting. (A) Whole cell lysates (W), cytoplasmic (C) and membrane (M) fractions of GT1000(pGC2)-AmtB1, and GT1000(pESVHA1)-AmtBHA1, probed with αAmtB antiserum. (B) The same samples as in A, probed with αHA antibody. Estimated molecular weights of major bands are indicated to the left of the panels.
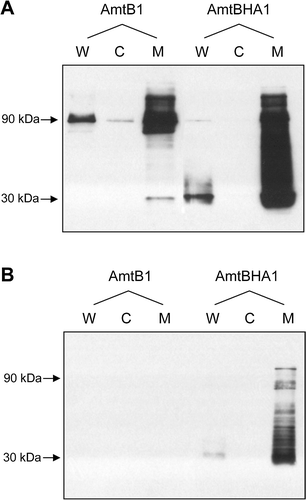
Figure 3. Titration of the in vivo levels of AmtB variant proteins. Membrane fractions from GT1000(pESVHA1), GT1000(pESVHA2) and GT1000(pESVHA4) were freeze-thawed to disassemble AmtB into the monomer. Serial 2-fold dilutions of those preparations were probed with the αHA antibody. Amounts of protein loaded are indicated below the control in panel A. Estimated molecular weights of major bands are indicated to the left of the panels.
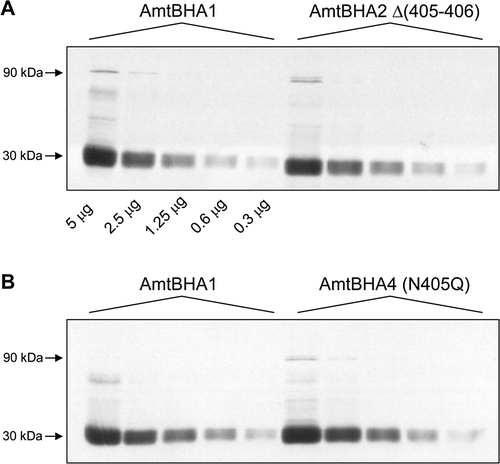
We then generated derivatives of two Class I AmtB variants that carried the HA epitope at the same location. These variants, Δ(405–406) (amtBHA2) and N405Q (amtBHA4) had the same activities in a washed assay as their untagged counterparts (data not shown). Membrane fractions from GT1000 carrying either wild-type, AmtBHA2 or AmtBHA4 were then processed by freeze thawing to denature the Amt proteins followed by analysis by western blotting of serial 2-fold dilutions on 10% SDS-PAGE gels. Serial dilution of the wild-type membrane fraction exhibited no signal saturation after a four minute exposure of the film indicating a reliable titration curve for the AmtB monomer (). Furthermore, both AmtBHA2 and AmtBHA4 had a titration profile indistinguishable from wild-type. Hence as they have 12% and 28% activity respectively we conclude that the reduced activity that characterizes Class I mutants reflects a genuine change in the ammonium conducting activity of the variants and is not a consequence of a reduced level of fully active protein.
Alterations in the CTR affect the interaction of AmtB with GlnK
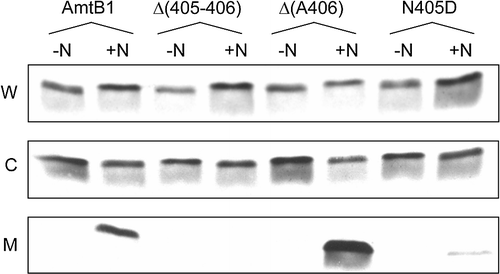
GlnK is sequestered to the inner membrane in an AmtB-dependent fashion following addition of ammonium to a nitrogen-limited culture and we have shown that GlnK binds specifically to AmtB (Coutts et al. 2002, Javelle et al. [Citation2004], Durand & Merrick [Citation2006]). Furthermore deletion of the AmtB CTR prevents GlnK sequestration indicating that the CTR is required, either directly or indirectly, for the GlnK-AmtB interaction (Coutts et al. 2002). We therefore characterized representatives of each of the classes of CTR variant for their ability to promote GlnK association. Nearly all of the CTR variants tested failed to show membrane localization of GlnK following an ammonium shock. The only exceptions to this were the two variants with essentially wild-type activity, Δ(A406) and N405D, and of these N405D showed a very impaired level of sequestration ().
Figure 4. Interaction of AmtB CTR variants with GlnK. Whole cell lysates (W), cytoplasmic (C) and membrane (M) fractions were probed with a αGlnK antiserum. -N: no NH4Cl added; +N: after addition to the culture of 30 mM NH4Cl for 15 min. The AmtB variants co-expressed with GlnK are indicated above the lanes. Data for representative variants are shown. Similar assays were carried out with Δ(399–406), R384A, G393D, G393R, Y404G, N405A and N405Q all of which showed no GlnK sequestration.
Previous studies have demonstrated that mutations that impair AmtB function can result in a reduced rate of deuridylylation of GlnK in response to ammonium shock although this effect is most pronounced in response to a ‘low’ shock, e.g. addition of 50 µM ammonium for 30 sec and is much less pronounced with a ‘high’ shock, i.e. 30 mM ammonium for 15 min (Javelle et al. 2004). Although a high ammonium shock was used for the experiments shown in we checked the deuridylylation response of GlnK and found that it was comparable to wild-type in each of the variants (data not shown).
Andrade et al. ([Citation2005]) recently proposed a structural model for the interaction between A. fulgidus Amt-1 and an A. fulgidus PII protein, GlnB-1. Whilst this model is not supported by any experimental data, it suggests that the CTR makes up a considerable part of the Amt-1/GlnB-1 interaction surface. Our observations on the effects of AmtB CTR mutants on GlnK interaction are consistent with this proposal.
The G393D mutation does not produce a dominant negative phenotype in E. coli AmtB
As described earlier, it has been reported in a number of organisms that a change of the conserved glycine residue in the cytoplasmic CTR to an aspartate residue can result in an inactive protein that shows post-translational inhibition of a wild-type Amt protein expressed in the same cell (Marini et al. [Citation2000b], Monahan et al. [Citation2002], Ludewig et al. [Citation2003]). These data have been interpreted as being due to formation of heteromeric complexes between wild-type and mutant monomers and we therefore wanted to examine whether a similar phenomenon would occur in E. coli AmtB.
This approach required the co-expression of AmtBG393D with wild-type AmtB. To achieve this we integrated a single copy of amtBG393D into the genome of the wild-type strain ET8000 at the attλ site using a λ phage-based integration system (Simons et al. [Citation1987]). This integrated gene copy is some 330 kb from the natural glnK amtB operon in the E. coli genome. The resultant strain, ESV103, carries wild-type amtB and amtBG393D both in single copy and both expressed from the natural glnK amtB promoter. The control for this was strain ESV100 in which the λ vector alone was integrated at the attλ site. We used the same approach to introduce both the wild-type (amtB8) and amtBG393D alleles at the attλ site in strain GT1000, which carries a deletion of the normal chromosomal copy of amtB, giving ESV102 and ESV101 respectively.
All these strains were assayed for Amt activity with the washed assay and all assays were repeated between two and four times. Using ET8000 as the reference (100% activity) we found that both positive controls, i.e. with just the λ vector present (ESV100) and with a single copy of amtB at the attλ site (ESV102), had 100% activity. The negative control, i.e. with amtBG393D alone at the attλ site (ESV101), was expressed at normal levels as judged by Western blotting but had 0% activity. Finally the strain carrying both the wild-type (amtB8) and amtBG393D genes (ESV103) had 59% of wild-type activity suggesting that there is a small transdominant effect.
Modelling the CTR of E. coli AmtB
Our results strongly suggest that the CTR of AmtB plays a crucial role in the function and the activity of the protein. Unfortunately in both E. coli structures published so far (Khademi et al. [Citation2004], Zheng et al. [Citation2004]) the CTR was disordered and the structure was not defined beyond residue P386. However, the structure of the AmtB paralogue Amt-1 of A. fulgidus revealed a defined structure for its CTR (Andrade et al. [Citation2005]) and the high sequence identity (50%) between the CTRs of these two proteins allowed us to model the CTR of E. coli AmtB using the A. fulgidus structure (A). No steric clashes occur in the model and moreover the side chains of the conserved residues can make the same interactions and contacts (see below) are observed in Amt-1. Therefore we believe that we can interpret our model of the CTR with a high degree of confidence.
Figure 5. Model for the CTR of E. coli AmtB. This model was derived as described in Materials and methods. (A) A single AmtB subunit showing the predicted CTR structure. The two helices of the CTR; from E387 to R391 and from D395 to S398 are labelled H1 and H2 respectively. (B) The cytoplasmic face of the AmtB trimer. Two monomers are shown as ribbons, the molecular surface of the third is shown in white and its cytoplasmic vestibule is indicated by the yellow circle (in on-line Figure). The CTR of each monomer is represented in red (in on-line Figure). (C) Hydrogen bonding (yellow dashed lines in on-line Figure) between Y404 and H196 (2.79 Å) and between G393, Q389 and N397. (D) Potential contacts between the H399, E401 and A403 side chains from one monomer (the core of the monomer has been removed) with the cytoplasmic face of the adjacent subunit of which the molecular surface is shown in yellow (in on-line Figure). This Figure is reproduced in colour in Molecular Membrane Biology on-line.
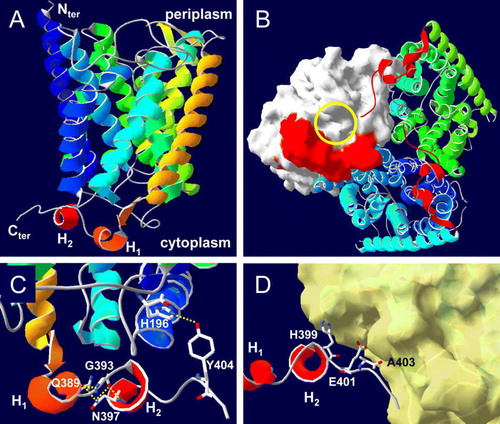
Consistent with secondary structure predictions using the primary amino acid sequence the model predicts that the CTR comprises two single-turn helices, from E387 to R391 and from D395 to S398 interrupted by the motif E392-G393-L394. The very end of the CTR, H399 to A406, extends beyond its own subunit and contacts the cytoplasmic vestibule of a neighbouring subunit of the trimer (A, B, D). The interactions made by the highly conserved residues () within the CTR are highlighted in . R384 forms hydrogen bonds with the backbone carbonyl oxygen atoms of I182 and K184, located in the cytoplasmic loop between M5 and M6. E390 forms hydrogen bonds with the guanidinium group of R384 and the backbone nitrogen of V186. D395 makes a hydrogen bond with the backbone nitrogen of L42 at the base of M2, and Y404 makes a hydrogen bond with the side chains of H196 at the base of M6 (C). G393, which is located between the two short helices of the CTR, is involved in a network of hydrogen bonds with residues in both helices (Q389 and N397) and is consequently critical in shaping the CTR (C). Moreover the psi/phi angles for the G393 are 152/110 in our model (147/103 in Amt-1), making this position unfavourable for any other residue. The side chains of the conserved residues H399, E401 and A403 all make potential contacts (between 2.9 Å and 3.5 Å) with the cytoplasmic face of TMH 6, 8 and 10 of the adjacent subunit in the trimer (D).
Discussion
The cytoplasmic C-terminal region of Amt proteins comprises a conserved core region of 22 amino acids, suggesting that it has a conserved structure. As we do not presently have a crystal structure for the E. coli AmtB CTR we have derived a model based on the A. fulgidus Amt-1 structure. The CTRs of the two proteins are predicted to be structurally very similar and to comprise two short helices separated by three residues including G393 which facilitates a change of orientation between helix1 and helix2. The most C-terminal residues extend outside the envelope of the peptide of which they are a part and make potential contacts with the adjacent subunit. This structural model, together with our observation that deletion of the CTR significantly impairs but does not inactivate E. coli AmtB, suggests that the CTR core region may have an important structural and/or functional role in Amt proteins.
We have now used the E. coli AmtB protein to initiate an investigation of the role of the CTR by analysing the phenotypes of a variety of mutant variants. Our analysis revealed two mutant phenotypes, either Class I, a protein with reduced activity around 25% of the wild–type, analogous to the activity of a complete CTR deletion, or Class II, an essentially inactive protein (typified by changes at the conserved residues R384 or G393). We propose that these two phenotypes reflect two distinct states of the CTR and hence of AmtB.
In the case of the Class I variants it would appear that these essentially phenocopy removal of the CTR, i.e. that this is the null phenotype with respect to the CTR. Indeed wild-type activity requires essentially the complete CTR, only removal of A406 being tolerated. Amongst the conserved residues in the CTR the most C-terminal is Y404, mutation of which produces a Class I phenotype. In both A. fulgidus Amt-1 and in the modelled E. coli AmtB CTR, Y404 makes a hydrogen bond with H196, another highly conserved residue, at the cytoplasmic end of M6. Our data suggest that Y404 is required for maximal Amt activity and that failure to position the CTR precisely may significantly impair activity.
Two explanations can be considered for the observation that all of these Class I variant proteins show a reduced activity, rather than no activity at all (as seen in Class II). In one model each subunit of the trimer acts independently of the other two and the complete CTR is required to facilitate the maximal ammonia conductance of the channel. In this case the reduced activity is a consequence of the effects of the CTR mutation on the activity of the channel in the cognate monomer. In an alternative model the three subunits function in a coordinated fashion and the CTR facilitates co-operativity between the three AmtB monomers. In this case full activity is only achieved in the cooperative state and the reduced activity of the Class I variants reflects a non-cooperative state. Whilst our data don't allow us to distinguish between these two hypotheses, the fact, previously unremarked by Andrade et al. ([Citation2005]), that the CTR makes contacts with the cytoplasmic surface of the adjacent monomer () is consistent with a role for the CTR in mediating inter-subunit co-operativity.
The Class II variants have been typified in previous studies of Amt/Mep proteins by substitution of the conserved glycine in the CTR (G393 in E. coli AmtB) by an aspartate residue (Marini et al. [Citation1997], Monahan et al. [Citation2002], Ludewig et al. [Citation2003]). As mentioned above, this glycine is positioned at the turn between the two short helices that comprise the major structural elements of the CTR. Inspection of the model CTR structure shows very clearly that no residue other than glycine could be accommodated at this position and consequently substitution of any alternative residue, not just aspartate, will disrupt the wild-type structure. Hence it is not apparent from our analysis that the phenotype of an aspartate substitution should be in any way distinct from any other replacement, and indeed in E. coli AmtB it is not, as all five substitutions that we analysed had identical phenotypes. Furthermore this phenotype is not confined to changes of G393 because we observe the same phenotype when the conserved R384 residue is substituted. This is again predictable from the modelled structure, as R384 is part of an intricate hydrogen bonding network that is disrupted by the R384A substitution.
The phenotype of these Class II variants is distinct from that of Class I, suggesting that either the CTR adopts an alternative structure that inactivates the transport function of the protein or the variant CTR induces a conformational change in the rest of AmtB leading to its inactivation. It is conceivable that these variants phenocopy a transient state adopted by the wild-type protein as part of its normal transport cycle. If so it might be expected that a Class II phenotype would require essentially the full length CTR. This appears to be the case, in that the Δ(405–406) deletion has almost no suppressive effect when combined with G393D, whereas the G393D, Δ(399–406) double mutant reverts to a Class I phenotype and has 38% of wild-type activity. Mutation of the conserved tyrosine (Y404G) also significantly suppresses G393D suggesting that this residue may be required for the Class II phenotype. These phenotypes suggest a situation where, in G393D and similar mutants, the CTR retains the significant contacts to the rest of the protein that are mediated by the highly conserved C-terminal residues, H399, E401, A403 and Y404 (see C and D) but is distorted by the substitution of G393 in such a way that it prevents the normal functioning of the channel.
The epistatic nature of some Amt/Mep mutations that introduce an aspartate residue in place of the conserved glycine in the CTR has been taken as an indication of the oligomeric state of Amt proteins in fungi and plants (Marini et al. [Citation1997], Monahan et al. [Citation2002], Ludewig et al. [Citation2003]). There is no quantitative methylammonium uptake data to assess the degree of epistasis in the eukaryotic systems. However, the very marked ammonium-dependent growth inhibition phenotypes imply that there must be relatively free exchange of subunits between different Amt oligomers and also suggest that the presence of one mutant subunit within an oligomer (e.g. within a trimer) could potentially inactivate the other subunits of the protein. By contrast we did not observe a very marked transdominant effect of the equivalent G393D substitution in E. coli AmtB. This may reflect the extraordinary stability of the E. coli AmtB trimer (Blakey et al. [Citation2002]) and the consequent lack of significant subunit mixing. The observed epistasis of this substitution in other systems does however support the notion that the subunits of Amt oligomers may indeed function cooperatively. Whilst the current models for Amt function do not require or entertain such cooperative features there is also presently no empirical evidence that a single Amt subunit is competent to mediate conductance of ammonia.
This paper was first published online on iFirst on 20 December 2006.
Supplementary data: Severi et al. The conserved carboxy-terminal region of the ammonia channel AmtB plays a critical role in channel function (for online version only)
Supplementary Table I. Plasmid-encoded AmtB variants.
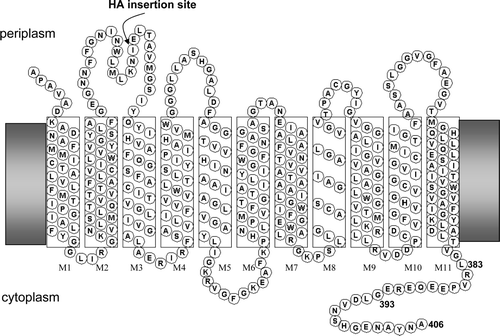
Supplementary Figure 1. Topology of E. coli AmtB. Position of insertion of haemagglutinin (HA) epitope in some variants is indicated.
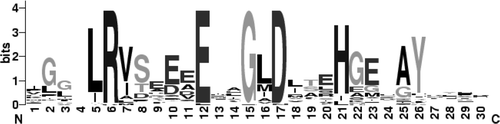
Supplementary Figure 2. Sequence logo for the CTR of Amt proteins. The logo was generated using WebLogo (Crooks et al. 2004) from a CLUSTALW alignment of 51 Amt proteins including 27 from bacteria, 7 from archaea, 4 from fungi, 9 from plants, and 4 from C. elegans. L5 in the logo alignment is equivalent to L383 in E. coli AmtB and the highly conserved glycine residue, G15 in the logo, is G393.
E.S. acknowledges a postgraduate fellowship from the John Innes Foundation. A.J. and M.M. acknowledge generous support from the BBSRC (UK). We thank Jeremy Thornton for the anti-GlnK antibodies and Anne Durand, Gavin Thomas and Fritz Winkler for helpful comments on the manuscript.
References
- Andrade SL, Dickmanns A, Ficner R, Einsle O. Crystal structure of the archaeal ammonium transporter Amt-1 from Archaeoglobus fulgidus. Proc Natl Acad Sci USA 2005; 102: 14994–14999
- Biswas K, Morschhauser J. The Mep2p ammonium permease controls nitrogen starvation-induced filamentous growth in Candida albicans. Mol Microbiol 2005; 56: 649–669
- Blakey D, Leech A, Thomas GH, Coutts G, Findlay K, Merrick M. Purification of the Escherichia coli ammonium transporter AmtB reveals a trimeric stoichiometry. Biochem J 2002; 364: 527–535
- Casadaban MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 1980; 138: 179–207
- Conroy MJ, Jamieson SJ, Blakey D, Kaufmann T, Engel A, Fotiadis D, Merrick M, Bullough PA. Electron and atomic force microscopy of the trimeric ammonium transporter AmtB. EMBO Reports 2004; 5: 1153–1158
- Coutts G, Thomas G, Blakey D, Merrick M. Membrane sequestration of the signal transduction protein GlnK by the ammonium transporter AmtB. EMBO J 2002; 21: 536–545
- Durand A, Merrick M. In vitro analysis of the Escherichia coli AmtB-GlnK complex reveals a stoichiometric interaction and sensitivity to ATP and 2-oxoglutarate. J Biol Chem 2006; 281: 29558–29567
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60: 2126–2132
- Facey SJ, Kuhn A. The sensor protein KdpD inserts into the Escherichia coli membrane independent of the Sec translocase and YidC. Eur J Biochem 2003; 270: 1724–1734
- Javelle A, Merrick M. Complex formation between AmtB and GlnK: an ancestral role in prokaryotic nitrogen control. Biochem Soc Trans 2005; 33: 174–176
- Javelle A, Severi E, Thornton J, Merrick M. Ammonium sensing in E.coli: the role of the ammonium transporter AmtB and AmtB-GlnK complex formation. J Biol Chem 2004; 279: 8530–8538
- Javelle A, Thomas G, Marini AM, Kramer R, Merrick M. In vivo functional characterisation of the E. coli ammonium channel AmtB: evidence for metabolic coupling of AmtB to glutamine synthetase. Biochem J 2005; 390: 215–222
- Jayakumar A, Barnes EM, Jr 1983. A filtration method for measuring cellular uptake of [14C]methylamine and other highly permeant solutes. Anal Biochem 135:475–478.
- Khademi S, O'Connell J, III, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 Å. Science 2004; 305: 1587–1594
- Lin Y, Cao Z, Mo Y. Molecular dynamics simulations on the Escherichia coli ammonia channel protein AmtB: Mechanism of ammonia/ammonium transport. J Am Chem Soc 2006; 128: 10876–10884
- Liu XY, Matherly LH. Analysis of membrane topology of the human reduced folate carrier protein by hemagglutinin epitope insertion and scanning glycosylation insertion mutagenesis. Biochim Biophys Acta 2002; 1564: 333–342
- Liu Z, Peng J, Mo R, Hui C, Huang CH. Rh Type B glycoprotein is a new member of the Rh superfamily and a putative ammonia transporter in mammals. J Biol Chem 2001; 276: 1424–1433
- Lorenz MC, Heitman J. The MEP2 ammonium permease regulates pseudohyphal differentiation in Saccharomyces cerevisiae. EMBO J 1998; 17: 1236–1247
- Ludewig U, Wilken S, Wu B, Jost W, Obrdlik P, El Bakkoury M, Marini AM, Andre B, Hamacher T, Boles E, von Wiren N, Frommer WB. Homo- and hetero-oligomerization of AMT1 NH4+ -uniporters. J Biol Chem 2003; 278: 45603–45610
- Marini A-M, Andre B. In vivo N-glycosylation of the Mep2 high-affinity ammonium transporter of Saccharomyces cerevisiae reveals an extracytosolic N-terminus. Mol Microbiol 2000; 38: 552–564
- Marini A-M, Matassi G, Raynal V, Andre B, Cartron JP, Cherif-Zahar B. The human Rhesus-associated RhAG protein and a kidney homologue promote ammonium transport in yeast. Nat Genet 2000a; 26: 341–344
- Marini A-M, Soussi-Boudekou S, Vissers S, Andre B. A family of ammonium transporters in Saccharomyces cerevisiae. Mol Cell Biol 1997; 17: 4282–4293
- Marini A-M, Springael JY, Frommer WB, Andre B. Cross-talk between ammonium transporters in yeast and interference by the soybean SAT1 protein. Mol Microbiol 2000b; 35: 378–385
- Monahan BJ, Unkles SE, Tsing IT, Kinghorn JR, Hynes MJ, Davis MA. Mutation and functional analysis of the Aspergillus nidulans ammonium permease MeaA and evidence for interaction with itself and MepA. Fungal Genet Biol 2002; 36: 35–46
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 2003; 31: 3381–3385
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 1987; 53: 85–96
- Smith DG, Garcia-Pedrajas MD, Gold SE, Perlin MH. Isolation and characterization from pathogenic fungi of genes encoding ammonium permeases and their roles in dimorphism. Mol Microbiol 2003; 50: 259–275
- Soupene E, Chu T, Corbin RW, Hunt DF, Kustu S. Gas channels for NH3: proteins from hyperthermophiles complement an Escherichia coli mutant. J Bacteriol 2002; 184: 3396–3400
- Thomas G, Coutts G, Merrick M. The glnKamtB operon: a conserved gene pair in prokaryotes. Trends Genet 2000a; 16: 11–14
- Thomas GH, Mullins JG, Merrick M. Membrane topology of the Mep/Amt family of ammonium transporters. Mol Microbiol 2000b; 37: 331–344
- Thornton J, Blakey D, Scanlon E, Merrick M. The ammonia channel protein AmtB from Escherichia coli is a polytopic membrane protein with a cleavable signal peptide. FEMS Microbiol Lett 2006; 258: 114–120
- Van Nuland A, Vandormael P, Donaton M, Alenquer M, Lourenco A, Quintino E, Versele M, Thevelein JM. Ammonium permease-based sensing mechanism for rapid ammonium activation of the protein kinase A pathway in yeast. Mol Microbiol 2006; 59: 1485–1505
- von Wirén N, Merrick M. Regulation and function of ammonium carriers in bacteria, fungi and plants. Trends Curr Genetics 2004; 9: 95–120
- Winkler FK. Amt/MEP/Rh proteins conduct ammonia. Pflugers Arch 2006; 451: 701–707
- Zheng L, Kostrewa D, Bernèche S, Winkler FK, Li X-D. The mechanism of ammonia transport based on the crystal structure of AmtB of E. coli. Proc Natl Acad Sci USA 2004; 101: 17090–17095
Supplementary Data References
- Coutts G, Thomas G, Blakey D, Merrick M. Membrane sequestration of the signal transduction protein GlnK by the ammonium transporter AmtB. EMBO J 2002; 21: 536–545
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res 2004; 14: 1188–1190
- Javelle A, Severi E, Thornton J, Merrick M. Ammonium sensing in E. coli: the role of the ammonium transporter AmtB and AmtB-GlnK complex formation. J Biol Chem 2004; 279: 8530–8538