Abstract
Colicins are toxic proteins produced by Escherichia coli that must cross the membrane to exert their activity. The lipid insertion of their pf domain is linked to a conformational change which enables the penetration of a hydrophobic hairpin. They provide useful models to more generally study insertion of proteins, channel formation and protein translocation in and across membranes. In this paper, we study the lipid-destabilizing properties of helices H8 and H9 forming the hydrophobic hairpin of colicin E1. Modelling analysis suggests that those fragments behave like tilted peptides. The latter are characterized by an asymmetric distribution of their hydrophobic residues when helical. They are able to interact with a hydrophobic/hydrophilic interface (such as a lipid membrane) and to destabilize the organized system into which they insert. Fluorescence techniques using labelled liposomes clearly show that H9, and H8 to a lesser extent, destabilize lipid particles, by inducing fusion and leakage. AFM assays clearly indicate that H8 and especially H9 induce membrane fragilization. Holes in the membrane are even observed in the presence of H9. This behaviour is close to what is seen with viral fusion peptides. Those results suggest that the peptides could be involved in the toroidal pore formation of colicin E1, notably by disturbing the lipids and facilitating the insertion of the other, more hydrophilic, helices that will form the pore. Since tilted, lipid-destabilizing fragments are also common to membrane proteins and to signal sequences, we suggest that tilted peptides should have an ubiquitous role in the mechanism of insertion of proteins into membranes.
| Abbreviations | ||
| pf | = | pore-forming |
| SIV | = | Simian Immunodeficiency Virus |
| AFM | = | Atomic Force Microscopy |
| ColE1 | = | Colicin E1 |
Introduction
Colicin E1 belongs to a group of proteins including colicin A, Ia, N and E9. They are toxic proteins produced by and against Escherichia coli and closely related bacteria Citation[1]. They bind to specific receptors of the outer membrane of sensitive cells. They are translocated to the cytoplasmic membrane where they form ion channels. These pore-forming toxins are organized in three domains Citation[2], Citation[3]: the N-terminal and central domains are involved in the transport through the E. Coli outer membrane. The C-terminal domain is responsible for the pore-forming (pf) activity in the inner membrane. In the periplasm, the C-domain undergoes a conformational change and inserts spontaneously into the cytoplasmic membrane, forming an ion channel Citation[3–5]. The latter translocates monovalent ions, dissipating the cationic gradients of the cell and increasing ATP consumption Citation[6], Citation[7]. This mechanism finally leads to host cell death.
The crystal structures of the active pore-forming domain of colicin E1, A, Ia and N (for the two latter, in the complete intact colicin structure) have been solved (for a review see Citation[3]). They all share the same signature of ten helix-bundle. Helices have an average length of 12 residue-long and helices 8 and 9 form a hydrophobic hairpin buried inside the bundle. Helices are too short to span the membrane (currently 20 residues), the longest being H8 and H9 (17 residue-long). Helix elongation appears thus required for channel formation. Kinetics studies have shown that unfolding, helix extension and insertion are closely coupled though sequential Citation[3], Citation[5], Citation[8], Citation[9]. Lipid insertion is not required for helix elongation. However, it is established that unfolding precedes insertion Citation[3]. It should also be noted that unfolding results in forming a conformationally mobile helical array which expands at the membrane surface much more than across the membrane Citation[9]. Solid State NMR data Citation[10] and crystal structure (PDB ID: 2i88) Citation[11] of the channel-forming C-domain of colicin E1 have suggested that the initial interaction with the membrane induces a configuration in which the hydrophobic hairpin is almost parallel to the surface of the membrane. The hairpin then inserts into the lipids while the other helices are spread on the surface Citation[10], Citation[11].
It is however not clear which helices form the transmembrane channel since there is no structural data on the lipid-bound state of pf domain. It is thought that the monomeric closed channel is made of only two transmembrane segments, the hydrophobic hairpin.
The open channel should be formed by at least four helices, notably the hydrophobic hairpin Citation[12], Citation[13]. The recent study of Sobko et al. also suggests that the channel is formed by a toroidal pore containing phospholipids in addition to colicin α helices Citation[14].
Sequence analysis of the colicin E1 pore forming domain reveals the presence of two tilted peptides corresponding to the hydrophobic hairpin H8 and H9. Tilted peptides are short protein fragments able to destabilize membranes Citation[15], Citation[16]. When those peptides are modelled as α-helices, they present an asymmetric distribution of hydrophobic residues which is responsible for an oblique orientation at a hydrophobic/hydrophilic interface, such as lipid/water Citation[15].
Tilted peptides were notably found in viral fusion proteins, such as the gp32 fusion protein of the SIV (simian immunodeficiency virus) where they are involved in the first steps of the fusion between the host cell and the viral membrane Citation[17]. They were also detected in proteins involved in lipid metabolism, in signal sequences, in membrane proteins. Citation[18–20].
Many tilted peptides were experimentally evidenced. Most were shown to induce liposome fusion in vitro when used as isolated fragments Citation[21–25].
Neutron diffraction experiments have shown that the SIV tilted peptide is oriented at an angle of 55° in model membranes Citation[26]. In the same way, the NMR structures of the HA2 fusion peptide was shown to insert tilted in model membranes at the active pH Citation[27].
In this paper, we study the lipid-destabilizing properties of colicin E1 H8 and H9 by molecular modelling and biophysical techniques including liposome assays and atomic force microscopy. A potential role in the membrane insertion of the pf domain is proposed.
Material and methods
Material
Egg phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), cholesterol (CHOL), sphingomyelin (SM), dipalmitoylphosphatidylcholine (DPPC), dioleylphosphatidylcholine (DOPC) and dioleylphosphatidic acid (DOPA) were purchased from Sigma (St Louis, USA). Egg phosphatidylethanolamine (PE) is from Lipid Products (Nr. Redhill, Surrey, UK). Octadecyl rhodamine chloride (R18) is from Molecular Probes (Eugene, OR, USA).
Peptides (H8: AADAGVSYVVALLFSLL and H9: IWGIAIVTGILCSYIDK; sequences correspond to the potential TM segments as described in the P02978 entry of the Uniprot bank) were synthesized by conventional solid phase peptide synthesis, using Fmoc for transient NH2-terminal protection and were characterized using mass spectrometry. Peptide purity lies between 80% and 85%, as indicated by analytical HPLC. N and C extremities were acetylated and amidated, respectively.
Methods
Molecular modelling of peptides
Peptides were 3D constructed as α-helices using Hyperchem 6.0 (Hypercube Inc.). Their conformation was minimized by the Polak-Ribiere algorithm in an AMBER force field with a gradient delta inferior to 0.1Kcal/(Å mol).
Membrane insertion
We inserted peptides into an implicit bilayer using the IMPALA (Integral Membrane Protein and Lipid Association) method developed by Ducarme et al. Citation[28]. It simulates the insertion of any molecule (protein, peptide or drug) into a bilayer by adding energy restraint functions to the usual energy description of molecules Citation[19], Citation[29], Citation[30].
The lipid bilayer is defined by C(z), which represents an empirical function describing membrane properties. This function is constant in the membrane plane (x- and y-axes) but varies along the bilayer thickness (z-axis) and more specifically, at the lipid/water interface corresponding to the transition between lipid acyl chains (no water = hydrophobic core) and the hydrophilic aqueous environment:1 α is a constant equal to 1.99; z0 corresponds to the middle of polar heads and z is the position in the membrane.
Two restraints simulate the membrane, one the bilayer hydrophobicity (Epho), and the other, the lipid perturbation (Elip).
The hydrophobicity of the membrane is simulated by Epho:2
Where N is the total number of atoms, S(i) the accessible surface to solvent of the i atom, Etr(i) its transfer energy per unit of accessible surface area and C(z) the zi position of atom i.
The perturbation of the bilayer by insertion of the molecule is simulated by the lipid perturbation restraint (Elip):3 where alip is an empirical factor fixed at 0.018 kcal.mol−1Å−2.
The environment energy (Eenv) applied on the peptide that inserts into the membrane becomes equal to:4
Systematic analysis
A systematic procedure is performed to insert and orient the peptide into the membrane. During this process, the peptide systematically crosses the force field of the membrane from -40 to +40 Å with respect to the membrane centre by step of 1Å. For each position along the z-axis, 2000 random orientations are tested. Amongst these 2000 positions, the orientation of minimal energy is selected. At the end of the systematic analysis, the procedure selects the position and the orientation of minimum energy among all selected minima.
Angular dynamics optimization
In order to analyze structural variations of the peptide inserted in the membrane, we have used the angular dynamics procedure previously defined to simulate the protein folding and described elsewhere Citation[30], Citation[31].
In the simulations, the total energy (Etot) is the sum of the intramolecular energy of the peptide (Eintra) and of the energy due to the membrane environment (Eenv). Etot is distributed at each step of the calculation on the peptide torsion k-axis. This total energy is equal to:
The total energy associated with each torsion axis (E(k)) is therefore represented by the sum of:
The torsion energy of the k axis (E(k)tor);
The intramolecular interaction energies (corresponding to the Van Der Waals energy Evdw, the electrostatic energy Eelec and the hydrophobic energy Epho_in) between atoms i and j, divided by the number of axes between these atoms;
The energy in the membrane for the atoms i and j divided by the number of atoms of the system minus 1 (N-1) and by the number of axes between atoms i and j.
The energy E(k) allows to calculate an angular dynamics that gives rise to an acceleration of torsion axes. During the dynamics, the length of atomic bonds and the value of valence angles are kept constant, only torsion angles are modified. All calculation of energies, angular acceleration and rotational velocity are described in detail in Lins et al. Citation[30]. Calculations are performed on an Intel® Pentium® 4, CPU 3.80GHz, 4.00 Go of RAM.
Liposome preparation
Large unilamellar vesicles (LUV) were prepared by the extrusion technique Citation[32] using an extruder (Lipex Biomembranes Inc, Vancouver, Canada). In brief, dry lipid films which are mixtures in weight of 26.6% PC, 26.6% PE, 26.6% SM and 20.2% cholesterol or 49.5% DOPC, 50% DPPC and 0.5% DOPA were hydrated for 1h at 37°C with a Tris buffer (10 mM Tris, 150mM NaCl, 0.01% EDTA 1 mM NaN3 pH 8). The resulting suspension was submitted to 5 successive cycles of freezing and thawing and thereafter extruded 10 times through 2 stacked polycarbonate filters (pore size 0.08 µm) under a nitrogen pressure of 20 bars.
To obtain small unilamellar vesicles (SUV), the hydrated film (same composition as for LUV) was sonicated (High Intensity Ultrasonic Processor, Sigma Chemical Co., St Louis, Missouri, USA) for 5 min at 50 W. A 10,000 g centrifugation for 2 min (Biofuge Pico, Van der Heyden, Heraeus, Germany) eliminates Titanium deposit and residual multilamellar vesicles. The phospholipid concentrations were determined by phosphorus analysis Citation[33].
Lipid-mixing experiments
Mixing of liposome membranes was followed by measuring the fluorescence increase of R18, a lipid soluble probe, occurring after the fusion of labelled and unlabeled liposomes, as described Citation[25]. Labelled liposomes were obtained by incorporating R18 in the dry lipid film at a concentration 6.3% of the total lipid weight. Labelled and unlabeled liposomes were mixed at a weight ratio 1:4 and a final concentration of 50 µM in 10mM Tris 150 mM NaCl 0.01% EDTA 1 mM NaN3 pH 8. Incubation of labelled and unlabelled vesicles in buffer alone did not modify the fluorescence intensity. Fluorescence was recorded at room temperature (λexc: 560 nm, λem: 590 nm) on an LS-50B PerkinElmer fluorimeter.
Leakage of liposome vesicle contents
The HPTS (8-hydroxypyrène -1,3,6-trisulfonic acid)/DPX (p-xylylenebis(pyridinium] bromide) assay of Ellens et al. Citation[34] was used to monitor vesicle leakage. The assay is based on the quenching of HPTS by DPX. HPTS and DPX are both encapsulated in the aqueous phase of the same liposomes. Leakage of vesicles was followed by measuring the dequenching of HPTS released into the medium. Fluorescence was recorded at room temperature (λexc: 360 nm, λém: 520 nm) on a LS-50B PerkinElmer fluorimeter.
Core-mixing experiments
The mixing of liposome contents was monitored using the core-mixing assay of Kendall and McDonald Citation[35]. Liposomes (LUV) were prepared as described above in 10 mM Tris-HCl buffer, 150 mM NaCl, 1 mM NaN3 (pH 8.0) and containing calcein at 0.8 mM and CoCl2 at 1.0 mM or EDTA at 20 mM. Untrapped solutes were removed by one elution on a Sephadex G-75 column with 10 mM Tris-HCl, 150 mM NaCl 1 mM NaN3 buffer, pH 8.0. In a standard experiment, calcein, Co 2 + - and EDTA-containing vesicles were mixed at 1:1 molar ratio in a 10 mM Tris-HCl buffer, pH 8.0 (150 mM NaCl, 1mM NaN3). When peptides were added, the calcein fluorescence was monitored at room temperature (λexc: 490 nm, λém: 520 nm) as a function of time on a LS-50B PerkinElmer fluorimeter. Co 2 + (0.4 mM chelated with citrate at 1:1 mol/mol) was present in the medium to avoid fluorescence due to leakage of vesicle contents. The maximum fluorescence was determined in presence of Triton X-100 0.5% (EDTA 10mM).
Infrared spectroscopy (FTIR) measurements
Attenuated Total Reflection (ATR) infrared spectroscopy was used to determine the secondary structure of the peptides alone and bound to lipids. Spectra were recorded at room temperature on a Brüker Equinox 55 equipped with a liquid nitrogen-cooled Mercury-Cadmium-Telluride (MCT) detector at a resolution of 2 cm−1, by averaging 512 scans. Free peptide samples (20 µg peptide) dissolved in TFE and the lipid-bound peptides (see preparation below) were spread out on a germanium ATR plate (50×20×2 mm-Aldrich Chimica, with an aperture of 45° yielding 25 internal reflections) and slowly dried under a stream of N2. Reference spectra of a germanium plate were automatically recorded after purge for 15 minutes with dry air and substracted to the recently run sample spectra. The plate was sealed in a universal sample holder and rehydrated by flushing the holder with N2 satured with D2O for 3 h at room temperature.
Peptide/lipid sample preparation
A dried mixed film made of 20 µg peptide and 100 µg lipids (same composition as for liposome preparation) was rehydrated with 100µl of a 10mM Tris 150 mM NaCl pH 8 buffer. Phospholipid concentration was determined as mentioned above.
Secondary structure determination
Vibrational bands, especially the amide I band (1600–1700 cm−1), are sensitive to the secondary structures of proteins. The C = O vibration is representative of 80% of the amide I band. This band accounts for all secondary structures which have different vibration values. The combination of resolution-enhancement methods with curve-fitting procedures allow to assign quantitatively different secondary structures such as α-helix, β-sheets and unordered structures. Each band was assigned according to the frequency of its maximum. The areas of all bands assigned to a given secondary structure were then summed and divided by the sum of all areas. This gives the relative ratio of each secondary structure. The bands are assigned as follows: α-helix: 1662–1645 cm−1, β-sheets: 1689–1682 cm−1 and 1637–1613cm−1, random 1644.5–1637 cm−1, β-turns: 1682–1662 cm−1. It should be noted that the proteins spread on the plate are deuterated to avoid an overlap of α-helix and random-coil structures, as previously described Citation[36], Citation[37].
AFM experiments
Preparation of supported lipid bilayers
Supported lipid bilayers composed of DOPC/DPPC/DOPA 495:500:5 (mol/mol/mol) were prepared using the vesicle fusion method Citation[38], Citation[39]. To this end, lipids were dissolved in chloroform at 1 mM final concentration. The mixture of these lipids was then evaporated under nitrogen and dried in a dessicator under vacuum for 2 h. Multilamellar vesicles (MLV) were obtained by resuspending the lipidic dried film in calcium-containing buffer (10 mM Tris, 150 mM NaCl, 3 mM CaCl2, pH 7.4) at 1 mM final lipid concentration. To obtain small unilamellar vesicles (SUV), the suspension was sonicated to clarity (4 cycles of 2 min) using a 500 W probe sonicator (Fisher Bioblock Scientific, France; 35% of the maximal power; 13 mm probe diameter) while keeping the suspension in an ice bath. The liposomal suspension was then filtered on 0.2 µm nylon filters (Whatman Inc., USA) to eliminate titanium particles.
Freshly cleaved mica squares (16 mm2) were glued onto steel sample pucks (Veeco Metrology LLC, Santa Barbara, CA, USA) using Epotek 377 (Gentec Benelux, Waterloo, Belgium). A total of 2 ml of the SUV suspension were then deposited onto the mica samples and the SUVs were allowed to adsorb and fuse on the solid surface for 1 h at 60°C. Subsequently, samples were rinsed five times with 2 ml of EDTA-containing buffer (10 mM Tris, 150 mM NaCl, 5 mM EDTA pH 7.4) and slowly cooled to room temperature.
Atomic Force Microscopy
Supported bilayers were investigated using a commercial AFM (NanoScope IV MultiMode AFM, Veeco Metrology LLC, Santa Barbara, CA) equipped with a 125 µm×125 µm×5 µm scanner (J-scanner). Topographic images were recorded in contact mode using oxide-sharpened microfabricated Si3N4 cantilevers (Microlevers, Veeco Metrology LLC, Santa Barbara, CA) with spring constant of 0.01 N/m (manufacturer specified), with a minimal applied force (< 500 pN) and at a scan rate of 5–6 Hz. Images were obtained at room temperature (23–25°C) either in Tris/EDTA buffer (10 mM Tris, 150 mM NaCl, 5 mM EDTA, pH 7.4) or after incubation in Tris/EDTA buffer containing 10 µM peptide.
Results
Modelling
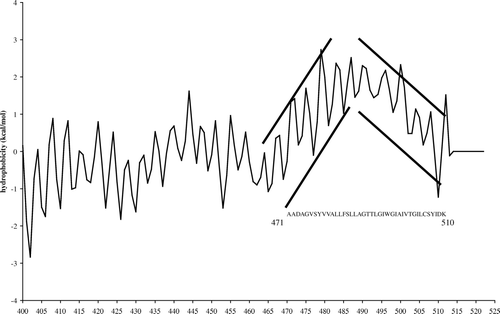
The sequence analysis of colicin E1, performed as previously described Citation[15], revealed the presence of two potential tilted peptides. As an example, shows the Jahnig profile of the C-end of the ColE1 pf sequence. The 470–510 domain presents successive increasing and decreasing oscillating curves; the sequence corresponds to the hydrophobic helical hairpin H8 and H9. This kind of increasing or decreasing profile is one of the main features of tilted peptides Citation[15].
Figure 1. Jahnig profile of the Cter domain (400–522) of colicin E1. The increasing oscillating curve corresponding to the H8 helix (residues 471–487) and the decreasing oscillating curve corresponding to H9 (residues 494–510) are indicated.
The peptides were 3D constructed as α-helices and their lipid insertion was simulated using IMPALA. Briefly, this method uses restraints to mimic membrane properties and allows us to test the insertion of any molecule into a simplified bilayer. Peptide stability during the insertion was analysed by an angular dynamics as described in Methods. shows the most stable position and conformation in the modelled membrane for H8 and H9. The peptides are mainly helical: the N and C extremities of both peptides are slightly destructured but the core (8–10 residues) is helical. The helix axis forms an angle of 45 and 65° with the membrane plane for H8 and H9, respectively. The mass centre of H8 is at the level of the lipid headgroup/acyl chain interface (13 Å from the bilayer centre) and H9 is more embedded in the membrane, its mass centre being at 9 Å from the centre, as for other tilted peptides Citation[19].
Figure 2. Most stable position of H8 (A) and H9 (B) in the IMPALA membrane after the angular dynamics. Mid plane = bilayer centre (z = 0); first upper (beneath) plane = lipid acyl chain/polar headgroup interface at 13.5 Å from the centre; second upper (beneath) plane = lipid/water interface (z = 18 Å).

FTIR experiments
In the modelling approach we predicted that H8 and H9 are helical. To experimentally check the peptide structure, the conformation of the peptide alone and in the presence of lipids is analyzed by ATR-FTIR (). The peptides are around 50% helical, in agreement with the conformation predicted by calculations.
Table I. Conformation of the H8 and H9 peptides in 100% TFE and in the presence of lipids.
Liposome fusion
Since most tilted peptides discovered up to now induce liposome fusion in vitro due to their membrane-destabilizing activity, H8 and H9 were tested for fusogenic properties and compared to the SIV (Simian Immunodeficiency Virus) fusion peptide as control. The induction of vesicular lipid mixing by the different peptides was tested with liposomes (LUVs and/or SUVs) made of PC/PE/SM/chol and of DOPC/DPPC/DOPA. The latter is used in the AFM experiments. R18-labelled and R18-free liposomes were mixed and increase of fluorescence intensity due to the dequenching of the probe is indicative of lipid fusion. H8 and H9 induce fusion with the two lipid compositions (). Extent of the fusion depends on the liposome type and on the peptide concentration; this parameter appears especially important for H8 (). For both peptides, the rate of lipid mixing is nevertheless lower than with the SIV peptide (, ).
Figure 3. Time course of lipid mixing induced by H8 and H9 peptides. Liposomes (SUV) are a mixture of 26.6% PC, 26.6% PE, 26.6% SM and 20.2% cholesterol (black symbols-called NC SUV below) or of 49.5% DOPC, 50% DPPC and 0.5% DOPA (open symbols-called DOPC SUV below). Peptides are added at 150 µM (corresponding to a peptide to lipid molar ratio of 1/10) to a mixture (1:4 w/w ratio) of R18-labelled and unlabelled SUVs. Increase of the R18 relative fluorescence is followed at room temperature. The SIV tilted peptide at 150 µM is used as positive control; the contribution of TFE (1.6% final concentration) is substracted from the curves. 100% fusion is obtained by adding 2% Triton X-100 to liposomes. ♦ H9/NC SUVs; ◊ H9/DOPC SUV; + H8/NC SUV; * H8/DOPC SUV; • SIV/NC SUV; ○ SIV/DOPC SUV.
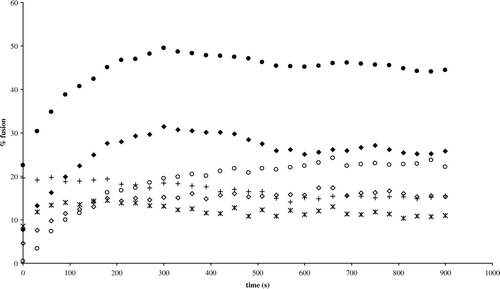
Table II. Percentage of lipid mixing induced by H9 and H8 in the presence of LUV or SUV (PC/PE/SM/chol as described in methods). 100% corresponds to the fusion induced by the SIV tilted peptide in the same conditions.
To rule out that the fluorescence increase is due to liposome aggregation instead of fusion, leakage and core-mixing assays were carried out. The HPTS/DPX assays indicated that H8 and H9 are able to destabilize PC/PE/SM/chol SUVs, inducing a HPTS release of 45% and 32% for H8 and H9 at a 0.1 peptide/lipid (P/L) molar ratio after 15 min, respectively. For the core-mixing assays, the H8 and H9 peptides induce 35 and 20% fusion, respectively (in both assays, 100% is after liposome lysis by detergent). This is similar to what is observed for the SIV peptide at the same P/L ratio (40% leakage and 28% core-mixing).
AFM experiments
DOPC/DPPC/DOPA bilayers were incubated in the presence of either colicin H8 or H9 peptides and imaged in real-time by AFM.
As shown in A and A', the topographic image obtained for a native DOPC/DPPC/DOPA bilayer revealed the coexistence of two phases, as observed for DOPC/DPPC supported bilayers Citation[40]. The phase diagrams of DPPC and DOPC Citation[38] as well as previous AFM studies Citation[40], Citation[41] lead us to believe that the lighter level correspond to DPPC in the gel phase and the darker level to DOPC in the fluid phase. The height between the two phases was 1.1±0.1 nm and resulted from a difference in the thickness and mechanical properties of the DOPC and DPPC films Citation[38], Citation[42]. A third phase attributable to PA-enriched domains was never seen in the images, presumably because the amount of PA (0.5%) was too low to induce lateral segregation.
Figure 4. Influence of colicin H8 (A, B and C) and H9 (A' to D') peptides on DOPC/DPPC/DOPA bilayers. AFM topographic images (A–C, 15 µm×15 µm; A'–D', 20 µm×20 µm; z- range: 10 nm) of a DOPC/DPPC/DOPA (495/500/5; mol/mol/mol) bilayer recorded in Tris/NaCl/EDTA, pH7.4 (A and A'). Images of the same bilayer was acquired in the presence of a 10 µM peptide solution (Tris/NaCl/EDTA): H8, after 3 (B) and 10 min (C); H9, after 5 (B'), 15 (C') and 120 min (D').
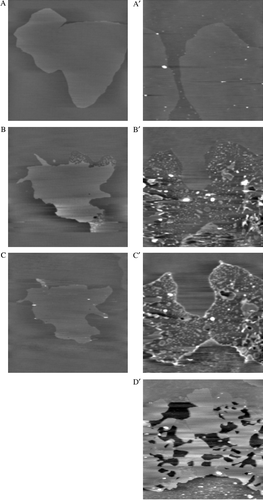
Addition of colicin H8 peptide led to some changes at the boundary of DPPC gel domains (B). As one can see, the overall surface covered by DPPC phase was considerably reduced within 3 min after H8 injection. Some regions (see B, upper right corner of DPPC domain) reveal the formation of depressions of about 1.9±0.1 nm with respect to the remaining DPPC phase. This behaviour was previously observed with SIV peptide Citation[43]. As previously suggested by Rinia et al. Citation[44], it may correspond to interdigitation or local fluidification of DPPC gel phase due to peptide insertion. This modification rapidly evolved and finally disappeared 10 min after H8 injection, the only features remaining were those corresponding to the DPPC domains surrounded by DOPC fluid phase (C).
As shown on B', the addition of H9 induced important morphological changes of the lipid bilayer. First, the complete surface of DPPC domains was converted to a depressed area (0.8±0.1 nm lower than the surrounding DOPC phase). This system progressively evolved to yield new topographic characteristics in DPPC domains (C'). An elevation at the boundary of the depressed DPPC domains appeared. This new level is protruding at 2.4±0.2 nm above the DOPC fluid phase. The elevated structures accumulated with time (data not shown), they were organized in striated domains with the same amplitude and periodicity as previously described for the SIV tilted peptide (periodicity of 64±7 nm and amplitude of 0.67±0.10 nm Citation[43]). One can also notice the apparition of holes in the depressed area. Hole depth corresponded to the bilayer thickness (∼5nm with DOPC as reference). After 120 min, the striated structures finally desorbed from the surface leaving the lipid bilayer with the two initial levels, DOPC and DPPC, and some holes of about 5 nm in depth (D'). These holes are thought to result from local fragilization of the membrane by high concentration of H9 peptides, leading to desorption of peptido-lipidic patches either spontaneously or under the influence of the AFM scanning tip. These results show the difference in the interaction of H8 and H9 peptides with the membrane.
Discussion
Colicins are proteins that must cross the membrane to exert their cytotoxic activity. Therefore, they provide useful examples to more generally study insertion of proteins, channel formation or protein translocation across membranes. The membrane insertion of their pf domain is linked to a conformational change and the subsequent penetration of a hydrophobic hairpin Citation[3]. The mechanism of channel formation remains unclear.
In this paper, we studied the lipid-destabilizing properties of helices H8 and H9 forming the hydrophobic hairpin of colicin E1. Modelling analysis suggests that those fragments are tilted peptides. The latter are characterized by an asymmetric distribution of hydrophobicity when helical. They are able to interact with a membrane and to destabilize the organized system into which they insert. They were found in viral fusion proteins, in membrane proteins and in signal sequences Citation[15], Citation[19].
Dynamic studies show that the H8 and H9 fragments are stable helices obliquely inserted in the IMPALA membrane. The tilted configuration was already mentioned by Tory and Merrill Citation[12], Citation[13] and by Zhakarov and Cramer Citation[9] who have suggested that the hydrophobic hairpin is not in a transbilayer configuration when the channel is in the closed state, but is rather tilted and deeply embedded within the hydrocarbon core. In our calculations, H9 should be able to interact with the second lipid layer, as shown on B.
The hydrophobic fragments remain mainly helical during the lipid insertion, only the N and C ends are locally destructured, as usual for small peptides. In the IMPALA simulations, 9–11 residues on 17 residues are in a classical helical conformation. This corresponds to 55–65% helical structure. Taking the limitations of the FTIR measurements into account (that overestimates β structures and underestimates helical ones), the modelling and structural approaches are in quite good agreement. Fluorescence techniques using labelled liposomes clearly show that H8 and H9 destabilize lipid particles, by inducing fusion and leakage. H9 is more fusogenic than H8 when small particles are used. We have used liposomes containing PC, PE SM and cholesterol that was shown to be effective to test the fusogenic potential of different peptides Citation[45]. It is clear that it is not the composition of bacterial membranes, the target of colicins. The effect of the presence of acidic lipids was tested with bilayers containing 0.5% DOPA, in fusion as well as in AFM experiments. H8 and H9 also induce lipid destabilization with SUVs containing phosphatidylglycerol and cardiolipin (60% PC, 20% PE, 10% PG, 10% CL in weight) (data not shown), indicating that lipid composition does not have a major role in the activity of the peptides.
Usually, the fusion, as detected by the dilution of a lipid-inserted probe, is dose-dependent, i.e., when the concentration increases, the fusion proportionally increases as well. This is the case for SIV and H9, but not for H8. Indeed, when H8 concentration is doubled, its fusogenic activity is multiplied by 3–4, indicating a potentialization effect. For H9, the activity is doubled and thus remains a constant percentage of the SIV effect. This suggests that increasing the quantity of H8 peptide induces a phenomenon that potentializes the dilution of the probe. This is not true when we take results from core-mixing assays into account. No significant difference can be observed for H8, H9 and SIV with the concentration (i.e., core-mixing is dose-dependent for the three peptides). The peculiar effect of H8 could derive from an additive effect on liposome aggregation.
AFM studies on DPPC/DOPC/DOPA membranes show that H8 and H9 are able to insert into lipids. While H8 induces local and temporary modifications in the DPPC phase, the presence of H9 leads to the apparition of holes within the DPPC depressed areas. Holes are thought to arise from membrane fragilization. This could be related to the deeper insertion of H9 into the bilayer, as predicted by the IMPALA simulations.
These results, together with the peculiar effect of H8 in the fusion assays, suggest that H8 and H9 could have different effects on the lipids.
From all those results, one can assume common features for H8 and H9 helices of colicin E1 (especially H9) and the fusion peptide of viruses such as SIV or HA2 concerning their lipid-interacting properties. This is in agreement with the literature since Zhakarov and Cramer Citation[9] and Tory and Merrill Citation[12], Citation[46] have proposed, based on NMR data and Trp fluorescence, that H8 and H9 adopt a tilted orientation with respect to the membrane plane in the closed state channel. This configuration is similar to what is observed for the Influenza Hemagglutinin (HA2) fusion domain Citation[27].
We suggest that the tilted fragments of ColE1 are able to destabilize the membrane and thus to facilitate the insertion of the whole pf domain. This is related to the work of Sobko et al. Citation[47] who have suggested that distortion of the membrane structure by oxidative modifications can facilitate the channel formation.
It is also suggested that the channel forms via a mechanism involving a toroidal lipidic pore Citation[14], Citation[48]. This model supports a direct implication of lipids in the formation of the pore. It was notably demonstrated that, for the whole pf domain, lipids promoting negative curvature (like oleic acid) increase transiently the efficiency of pore formation Citation[44]. Since tilted peptides are thought to promote negative curvature, this is in agreement with our hypothesis that the tilted peptides of colicin E1 help pore formation and notably insertion, i.e., the first steps of the process. Stabilization should notably occur through the presence of positive curvature inducers (such as lysophophatidylcholine) Citation[44]. Furthermore, the formation of the toroidal pore would allow helices forming the pore to occupy a TM configuration with length less than 20 residues, as a consequence of membrane thinning associated with the toroidal pore Citation[48]. It should be noted that the tilted configuration of H8 ands H9 should be transitory, as suggested for tilted peptides from membrane proteins where they are also detected by pairs Citation[18]. Actually, we propose that, after destabilization of the membrane due to tiltedness, the TM configuration of the hydrophobic hairpin (35 residues in total length) in the pore would then be favoured, notably by their mutual interaction, as for the tilted peptides found in the TM proteins Citation[18].
The formation of structures closely related to a toroidal pore is also suggested in the process of membrane fusion induced by viruses such as HA2 Citation[49], Citation[50], again underlying common features between fusion proteins and colicins.
It is also worth to note that apoptotic proteins such as Bax could form a toroidal pore Citation[51], Citation[52]. These proteins have a conformation close to that of the pore forming domain of colicins (notably by the presence of a hydrophobic hairpin), and they are able to switch from a water-soluble form to a lipid-inserted configuration. They also have a tilted peptide in their hydrophobic domain (unpublished data). On the other hand, other colicins (such as colicin A or Ia) with a pf domain similar to that of E1, have a potential tilted fragment too (data not shown).
Since the occurrence of tilted, lipid-destabilizing fragments is common to membrane proteins (for example, bacteriorhodopsin, Citation[18]) and to signal sequences Citation[20], we suggest that tilted peptides could have a ubiquitous role in the mechanism of insertion of proteins into membranes.
In conclusion, we have shown that the hydrophobic hairpin of colicin E1 is composed of two tilted fragments, one similar to viral fusion peptides. Those peptides are able to disturb the lipid organization of membranes. We hypothesize that they could be involved in the toroidal pore formation of colicin E1, notably because we demonstrate that they modify the lipid layer by AFM. We then suggest that they could disturb the lipids creating fracture, facilitating the insertion of the other, more hydrophilic, helices that will form the pore.
Acknowledgements
L.L. and R.B. are respectively Research Associate and Research Director at the National Funds for Scientific Research (FNRS) of Belgium. A.T. is Research Director at the Institut National de la Santé et de la Recherché Médicale (INSERM, France). This work was supported by the Interuniversity Poles of Attraction Program-Belgian State, Prime Minister's Office- Federal Office for Scientific, Technical and Cultural Affairs, the National Fund for Scientific Research of Belgium (FNRS) and the Region Wallonne.
Related Research Data
References
- Lazdunski C J. Pore-forming colicins: synthesis, extracellular release, mode of action, immunity. Biochimie 1988; 70: 1291–1296
- Baty D, Frenette M, Lloube R, Geli V, Howard S P, Pattus F, Lazdunski C. Functional domains of colicin A. Mol Microbiol 1988; 2: 807–811
- Zakharov SD, Cramer WA. Colicin crystal structures: pathways and mechanisms for colicin insertion into membranes. Biochim Biophys Acta 2002; 1565: 333–346
- Bullock JO, Cohen FS, Dankert JR, Cramer WA. Comparison of the macroscopic and single channel conductance properties of colicin E1 and its COOH-terminal tryptic peptide. J Biol Chem 1983; 258: 9908–9912
- Zakharov SD, Lindeberg M, Cramer WA. Kinetic description of structural changes linked to membrane import of the colicin E1 channel protein. Biochemistry 1999; 38: 11325–11332
- Cramer WA, Dankert JR, Uratani. The membrane channel-forming bacteriocidal protein, colicin El. Biochim Biophys Acta 1983; 737: 173–193
- Cleveland MV, Slatin S, Finkelstein A, Levinthal C. Structure-function relationships for a voltage-dependent ion channel: properties of COOH-terminal fragments of colicin E1. Proc Natl Acad Sci USA 1983; 80: 3706–3710
- Zakharov SD, Lindeberg M, Griko Y, Salamon Z, Tollin G, Prendergast FG, Cramer WA. Membrane-bound state of the colicin E1 channel domain as an extended two-dimensional helical array. Proc Natl Acad Sci USA 1998; 95: 4282–4287
- Zakharov SD, Cramer WA. Insertion intermediates of pore-forming colicins in membrane two-dimensional space. Biochimie 2002; 84: 465–475
- Kim Y, Valentine K, Opella SJ, Schendel SL, Cramer WA. Solid-state NMR studies of the membrane-bound closed state of the colicin E1 channel domain in lipid bilayers. Protein Sci 1998; 7: 342–348
- Elkins P, Bunker A, Cramer WA, Stauffacher CV. A mechanism for toxin insertion into membranes is suggested by the crystal structure of the channel-forming domain of colicin E1. Structure 1997; 5: 443–458
- Tory MC, Merrill AR. Adventures in membrane protein topology – a study of the membrane-bound state of colicin E1. J Biological Chem 1999; 274: 24539–24549
- Tory MC, Merrill AR. Determination of membrane protein topology by red-edge excitation shift analysis: application to the membrane-bound colicin E1 channel peptide. Biochimica et Biophysica Acta-Biomembranes 2002; 1564: 435–448
- Sobko AA, Kotova EA, Antonenko YN, Zakharov SD, Cramer WA. Effect of lipids with different spontaneous curvature on the channel activity of colicin E1: evidence in favor of a toroidal pore. FEBS Letters 2004; 576: 205–210
- Brasseur R. Tilted peptides: a motif for membrane destabilization (hypothesis). Molec Membrane Biol 2000; 17: 31–40
- Brasseur R, Pillot T, Lins L, Vandekerckhove J, Rosseneu M. Peptides in membranes: tipping the balance of membrane stability. Trends Biochem Sci 1997; 22: 167–171
- Horth M, Lambrecht B, Khim MCL, Bex F, Thiriart C, Ruysschaert JM, Burny A, Brasseur R. Theoretical and functional-analysis of the Siv fusion peptide. Embo J 1991; 10: 2747–2755
- Rahman M, Lins L, Thomas-Soumarmon A, Brasseur R. Are amphipathic asymmetric peptides ubiquitous structures for membrane destabilisation?. J Molec Modeling 1997; 3: 203–215
- Lins L, Charloteaux B, Thomas A, Brasseur R. Computational study of lipid-destabilizing protein fragments: towards a comprehensive view of tilted peptides. Proteins-Structure Function Genetics 2001; 44: 435–447
- Talmud P, Lins L, Brasseur R. Prediction of signal peptide functional properties: a study of the orientation and angle of insertion of yeast invertase mutants and human apolipoprotein B signal peptide variants. Protein Engineering 1996; 9: 317–321
- Lambert G, Decout A, Vanloo B, Rouy D, Duverger N, Kalopissis A, Vadekerckhove J, Chambaz J, Brasseur R, Rosseneu M. The C-terminal helix of human apolipoprotein AII promotes the fusion of unilamellar liposomes and displaces apolipoprotein AI from high-density lipoproteins. Eur J Biochem 1998; 253: 328–338
- Martin I, Dubois MC, Defrise-Quertain F, Saermark T, Burny A, Brasseur R, Ruysschaert JM. Correlation between fusogenicity of synthetic modified peptides corresponding to the NH2-terminal extremity of Simian immunodeficiency virus Gp32 and their mode of insertion into the lipid bilayer – an infrared spectroscopy study. J Virol 1994; 68: 1139–1148
- Martin I, Defrise-Quertain F, Mandieau V, Nielsen NM, Saermark T, Burny A, Brasseur R, Ruysschaert JM, Vandenbranden M. Fusogenic activity of Siv (Simian Immunodeficiency Virus) peptides located in the Gp32 NH2 terminal domain. Biochem Biophys Res Communic 1991; 175: 872–879
- Perez-Mendez O, Vanloo B, Decout A, Goethals M, Peelman F, Vandekerckhove J, Brasseur R, Rosseneu M. Contribution of the hydrophobicity gradient of an amphipathic peptide to its mode of association with lipids. Eur J Biochem 1998; 256: 570–579
- Lins L, Flore C, Chapelle L, Talmud PJ, Thomas A, Brasseur R. Lipid-interacting properties of the N-terminal domain of human apolipoprotein C-III. Protein Engineering 2002; 15: 513–520
- Bradshaw JP, Darkes MJ, Harroun TA, Katsaras J, Epand RM. Oblique membrane insertion of viral fusion peptide probed by neutron diffraction. Biochemistry 2000; 39: 6581–6585
- Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol 2001; 8: 715–720
- Ducarme P, Rahman M, Brasseur R. IMPALA: a simple restraint field to simulate the biological membrane in molecular structure studies. Proteins-Structure Function Bioinformatics 1998; 30: 357–371
- Vogt B, Ducarme P, Schinzel S, Brasseur R, Bechinger B. The topology of lysine-containing amphipathic peptides in bilayers by circular dichroism, solid-state NMR, and molecular modeling. Biophysical J 2000; 79: 2644–2656
- Lins L, Charloteaux B, Heinen C, Thomas A, Brasseur R. De novo’ design of peptides with specific lipid-binding properties. Biophysical J 2006; 90: 470–479
- Brasseur R. Simulating the folding of small proteins by use of the local minimum energy and the free solvation energy yields native-like structures. J Molec Graphics 1995; 13: 312–322
- Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta 1986; 858: 161–168
- Mrsny RJ, Volwerk JJ, Griffith OH. A simplified procedure for lipid phosphorus analysis shows that digestion rates vary with phospholipid structure. Chem Phys Lipids 1986; 39: 185–191
- Ellens H, Bentz J, Szoka FC. H + - and Ca2 + -induced fusion and destabilization of liposomes. Biochemistry 1985; 24: 3099–3106
- Kendall DA, MacDonald RC. A fluorescence assay to monitor vesicle fusion and lysis. J Biol Chem 1982; 257: 13892–13895
- Goormaghtigh E, Cabiaux V, Ruysschaert JM. Secondary structure and dosage of soluble and membrane proteins by attenuated total reflection Fourier-transform infrared spectroscopy on hydrated films. Eur J Biochem 1990; 193: 409–420
- Goormaghtigh E, Raussens V, Ruysschaert JM. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim Biophys Acta 1999; 1422: 105–185
- Giocondi MC, Vié V, Lesniewska E, Milhiet PE, Zinke-Allmang M, Le Grimellec C. Phase topology and growth of single domains in lipid bilayers. Langmuir 2001; 17: 1653–1659
- Reviakine I, Brisson A. Formation of supported phospholipid bilayers from unilamellar vesicles investigated by atomic force microscopy. Langmuir 2000; 16: 1806–1815
- Milhiet PE, Giocondi MC, Baghdadi O, Ronzon F, Le Grimellec C, Roux B. AFM detection of GPI protein insertion into DOPC/DPPC model membranes. Single Molec 2002; 3: 135–140
- Berquand A, Mingeot-Leclercq MP, Dufrene YF. Real-time imaging of drug-membrane interactions by atomic force microscopy. Biochim Biophys Acta 2004; 1664: 198–205
- Dufrene YF, Barger WR, Green JB, Lee GU. Nanometer-scale surface properties of mixed phospholipid monolayers and bilayers. Langmuir 1997; 13: 4779–4784
- El Kirat K, Lins L, Brasseur R, Dufrene YF. Fusogenic tilted peptides induce nanoscale holes in supported phosphatidylcholine bilayers. Langmuir 2005; 21: 3116–3121
- Rinia HA, Boots W, Rijkers DT, Kik RA, Snel MM, Demel RA, Killian JA, van der Eerden JP, de Kruijff B. Domain formation in phosphatidylcholine bilayers containing transmembrane peptides: specific effects of flanking residues. Biochemistry 2002; 41: 2814–2824
- Adam B, Lins L, Stroobant V, Thomas A, Brasseur R. Distribution of hydrophobic residues is crucial for the fusogenic properties of the Ebola virus Gp2 fusion peptide. J Virol 2004; 78: 2131–2136
- Tory MC, Merrill AR. Acrylamide quenching of colicin E1 channel peptide intrinsic fluorescence to probe the membrane-topology of the closed channel. Biophysical J 1999; 76: A120–A120
- Sobko AA, Vigasina MA, Rokitskaya TI, Kotova EA, Zakharov SD, Cramer WA, Antonenko YN. Chemical and photochemical modification of colicin E1 and gramicidin a in bilayer lipid membranes. J Membrane Biol 2004; 199: 51–62
- Zakharov SD, Kotova EA, Antonenko YN, Cramer WA. On the role of lipid in colicin pore formation. Biochim Biophys Acta 2004; 1666: 239–249
- Chernomordik LV, Leikina E, Frolov V, Bronk P, Zimmerberg J. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J Cell Biol 1997; 136: 81–93
- Yang L, Huang HW. Observation of a membrane fusion intermediate structure. Science 2002; 297: 1877–1879
- Epand RF, Martinou JC, Montessuit S, Epand RM, Yip CM. Direct evidence for membrane pore formation by the apoptotic protein Bax. Biochem Biophys Res Commun 2002; 298: 744–749
- Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem 2002; 277: 49360–49365