Abstract
Membrane proteins represent a significant fraction of all genomes and play key roles in many aspects of biology, but their structural analysis has been hampered by difficulties in large-scale production and crystallisation. To overcome the first of these hurdles, we present here a systematic approach for expression and affinity-tagging which takes into account transmembrane topology. Using a set of bacterial transporters with known topologies, we tested the efficacy of a panel of conventional and Gateway™ recombinational cloning vectors designed for protein expression under the control of the tac promoter, and for the addition of differing N- and C-terminal affinity tags. For transporters in which both termini are cytoplasmic, C-terminal oligohistidine tagging by recombinational cloning typically yielded functional protein at levels equivalent to or greater than those achieved by conventional cloning. In contrast, it was not effective for examples of the substantial minority of proteins that have one or both termini located on the periplasmic side of the membrane, possibly because of impairment of membrane insertion by the tag and/or att-site-encoded sequences. However, fusion either of an oligohistidine tag to cytoplasmic (but not periplasmic) termini, or of a Strep-tag II peptide to periplasmic termini using conventional cloning vectors did not interfere with membrane insertion, enabling high-level expression of such proteins. In conjunction with use of a C-terminal Lumio™ fluorescence tag, which we found to be compatible with both periplasmic and cytoplasmic locations, these findings offer a system for strategic planning of construct design for high throughput expression of membrane proteins for structural genomics projects.
Introduction
Membrane proteins account for 20–30% of open reading frames (ORFs) in genomes sequenced to date and perform many key biological functions ranging from signal transduction to solute transport Citation[1]. They also represent the majority of current drug targets Citation[2]. A particularly large class of such proteins are membrane transporters, which represent between 2 and 16% of ORFs in prokaryote and eukaryote genomes Citation[3]. Much has been learnt about the function of these proteins by examining the functional effects of mutating individual residues. However, for a detailed understanding of the molecular mechanisms of permeant recognition and translocation, and to facilitate drug design, high resolution 3-dimensional structures are of paramount importance. Unfortunately, only about 120 unique membrane protein structures are currently present in the Protein Data Bank, in comparison to >10,000 independent structures for soluble proteins.
The current paucity of structural information on membrane proteins stems in part from the difficulty in producing sufficiently large amounts for structural analysis. A wide range of commercial vectors and affinity tags have been explored in attempts to obtain high yields of correctly-folded membrane proteins in Escherichia coliCitation[4]. Typically, the location and nature of the affinity tag greatly affect expression levels, and multiple constructs have to be explored in order to achieve suitable levels of protein production Citation[4]. To facilitate high throughput screening of multiple constructs, Gateway recombinational cloning methods have been employed Citation[5]. In addition, to monitor membrane insertion versus inclusion body formation Citation[6] and for pre-crystallisation screening of membrane protein monodispersity in detergent solutions Citation[7], fusions to green fluorescent protein have been utilised. However, a sizeable proportion of membrane proteins tested to date have proven recalcitrant to expression – for example in a recent study of 47 E. coli membrane proteins, although many were well-expressed and could be purified, 21 were expressed at low levels or not at all Citation[5].
Experience in expressing a range of bacterial membrane proteins in our laboratory suggested that success might be influenced by a combination of the transmembrane topology of the target membrane protein and the choice of affinity tags and their locations. Experimental assessment of topology, and in particular of the location of the membrane protein termini, has recently been achieved on a genomic scale, using a combination of C-terminal tagging with alkaline phosphatase and green fluorescent protein and a hidden Markov model topology predictor Citation[8], Citation[9]. However, until now this information has not been exploited for the design of expression strategies. In the present study we have therefore investigated a set of bacterial membrane proteins of differing topologies, to see whether exploiting information on topology during design of tagging and cloning strategies can improve the likelihood of achieving high level expression. Our findings suggest that using the predicted topology of membrane proteins to tailor the choice and location of affinity tags used can greatly increase the chance of successful expression and purification of these proteins. Moreover we demonstrate the exploitation of a fluorescent tagging procedure that, in contrast to those used to date, can be used to monitor the expression of membrane proteins with divergent topologies. In combination, these approaches should allow the more efficient, high throughput production of membrane proteins on a genomic scale, thus opening up the way to a better understanding of these biologically and clinically important molecules.
Materials and methods
Materials
L-[G-3H] glutamic acid (50 Ci/mmol) was obtained from GE Healthcare UK Limited. Ni2 + -nitrilotriacetate (Ni-NTA) agarose and Strep-Tactin resin were obtained from Qiagen, Ltd., UK, and IBA GmbH, Germany, respectively. Lauryldimethylamine N-oxide (LDAO) was from Fluka AG (Sigma-Aldrich) and n-dodecyl-β-D-maltopyranoside (DDM) from Melford Laboratories Ltd., UK. Genomic DNA was isolated by standard procedures from E. coli MG1665-K12 and from a clinical isolate of Haemophilus influenzae kindly provided by Dr D. Holland, University of Leeds. Oligonucleotide primers, Lumio™ Green detection kits and the E. coli strains DB3.1™ and TOP10 were obtained from Invitrogen Ltd, UK. E. coli strains BL21 and Tuner™(DE3)pLysS were from Novagen (Merck Biosciences Ltd., UK), and strain BL21(DE3) was from Stratagene Europe.
Vector construction
Genetic manipulation was carried out in the E. coli strains TOP10 or DB3.1™ as appropriate and constructs were verified by DNA sequencing. For construction of expression vectors encoding the transporters detailed in Supplementary (online version only), ‘touchdown’ PCR Citation[10] was used to amplify the corresponding genes either from the NupG expression vector pGJL25 Citation[11] or from genomic DNA, using the primers listed in Supplementary (online version only). Expression vectors were then generated by conventional cloning or recombinational cloning using the Gateway™ system (Invitrogen). A destination vector for the latter process was created by first inserting Linker 1 (Supplementary ), encoding a TEV protease site followed by Lumio™ and His8 tags, into the BamHI and HindIII sites of pTTQ18 Citation[12] to generate the plasmid pMR1. This was then Gateway™-adapted according to the manufacturer's instructions using cassette RFB (Invitrogen), yielding the destination vector pLMPDEST-01. Entry clones encoding bacterial transporters were generated by recombination of the cognate PCR products, bearing 25 bp attB sequences, with the vector pDONR221 (Invitrogen). In the case of NupG, the desired amplification product was obtained in a single PCR procedure, using the primers, NupG-attF and NupG-attR, bearing the attB sequences (Supplementary ). For the remaining transporters a 2-stage PCR procedure with template-specific primers (e.g., UhpT-attF and -attR, Supplementary ) followed by adapter primers (attB1 adapter-F and -R, Supplementary ) was used to incorporate the attB sequences into the amplification products. Expression constructs (e.g., pLMP-01-NupG) bearing a C-terminal TEV site plus Lumio™ and His8 tags (ENLYFQGCCPGCCHHHHHHHH) were generated by recombination between the PCR products and pLMPDEST-01. A version of the NupG expression vector lacking the attB1 site was produced by restriction of pLMP-01-NupG at the flanking EcoRI sites, followed by self-ligation of the 5869 bp fragment, yielding pLMP-01-NupG-E.
Supplementary Table I. Transport proteins examined in this study.
Supplementary Table II. Oligonucleotides used in this study.
For production of expression constructs by conventional cloning procedures, EcoRI and PstI sites introduced at the 5′ and 3′ ends of the transporter ORFs were used to insert products amplified using the primers listed in Supplementary (e.g., ZitB-RGSH6-F and -R) into the corresponding sites of a series of pTTQ18 derivatives (Supplementary ). For C-terminal tagging with an RGSH6 sequence, the desired insert was used to replace the E. coli UraA ORF in the construct pTTQ18-RGSH6-UraA Citation[13]. pMR2, for N-terminal tagging of proteins with a His8 sequence, was generated by insertion of Linker 2 (Supplementary ) into the EcoRI site of pTTQ18. For C-terminal attachment of His8 or tetracysteine (CCPGCC; Lumio™) tags, pMR4 and pMR5 were generated by insertion of Linkers 3 or 4 respectively (Supplementary ) into the PstI and HindIII sites of the vector. pTTQ18-Strep II, for C-terminal attachment of a SerAla linker followed by the Strep-tag II sequence (WSHPQFEK) Citation[14], was generated by insertion of Linker 5 into the same sites. pTTQ18-Strep II*-MntH was generated from the pTTQ18-Strep II-derived construct pTTQ18-Strep II-MntH by using QuickChange™ (Stratagene) mutagenesis and primers MntH-Strep-F2 and MntH-Strep-R2 (Supplementary ) to introduce a longer linker (GGGGGSA) before the Strep-tag II sequence.
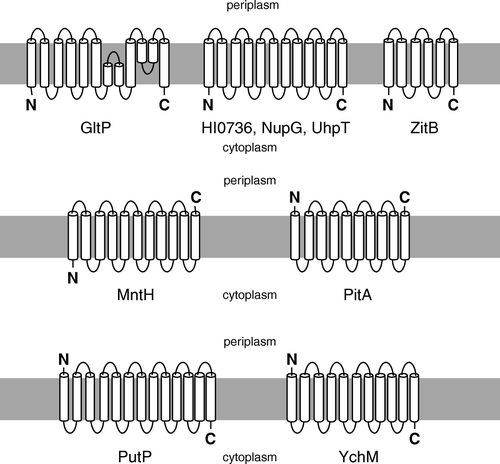
Figure 1. Diagrammatic representations of the putative membrane topologies of the proteins examined in this study. References in support of these topologies are listed in Supplementary (online version only).
Expression and detection of recombinant transporters
For expression experiments, constructs were transformed into E. coli host strains BL21, BL21(DE3) or Tuner(DE3)pLysS as indicated. Cultures were grown at 37°C in a shaking incubator (200 rpm) in Luria Bertani (LB) medium or M9 minimal medium supplemented with 0.2% (w/v) casamino acids plus 0.2% (w/v) glycerol, in the presence of carbenicillin (100 µg/ml). When they reached an A600nm of 0.5–1 expression was induced by the addition of isopropyl-β-D-thiogalactoside (IPTG) to a final concentration of 0.05–1 mM and the cells were harvested by centrifugation after a further 3 h.
Small scale membrane preparations were carried by the water lysis method Citation[15]. For large scale membrane preparations cells were resuspended in 20 mM Tris, 1 mM EDTA, pH 7.5 containing 10% glycerol. Cells were lysed by 2–3 passages at 4°C through a cell disruptor (Constant Systems Ltd.) at 20–30 kpsi and the resulting mixed membrane vesicles (inner and outer membranes) collected by centrifugation at 100,000 g for 2h. Following washing in 20 mM Tris, pH 7.5 by the same procedure, membrane vesicles were stored at −70°C.
Protein concentration was determined using the bicinchoninic acid assay (Perbio Science UK Ltd). After SDS-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were detected by Coomassie blue staining or, for constructs bearing a Lumio™ tag, use of a Lumio™ in-gel detection kit. His- or Strep II-tagged proteins on western blots were detected using HisProbe™-HRP (Perbio Science UK Ltd) or Strep-tactin-HRP conjugate (IBA GmbH, Germany) respectively.
Measurement of glutamate uptake by bacterial cells
Measurements of glutamate uptake by E. coli BL21 cells harbouring pLMP-01-GltP or pTTQ18-RGSH6-GltP, following induction of expression for 30 min with 0.1 mM IPTG, were performed at 25°C as previously described, except that 3H-labelled glutamate (final concentration 50 µM; 10 µCi/ml) was used as permeant Citation[16]. Uptake was measured over a period of 15 sec, in order to estimate the initial rate of transport. Results shown are mean±standard deviation for triplicate measurements.
Purification of His- and Strep II-tagged forms of the manganese transporter MntH
Mixed membrane vesicles (18 mg protein) prepared from IPTG-induced bacteria harbouring pMR2-MntH or pTTQ18-Strep II*-MntH were solubilized by incubation on ice for 1.5 h in 6 ml 20 mM Tris-Cl pH 7.9, containing 100 mM NaCl, 20% (v/v) glycerol, 4 mM 2-mercaptoethanol (βME), 5 mM imidazole and 1% (w/v) DDM, or 100 mM Tris-Cl pH 8.0, containing 150 mM NaCl, 20% (v/v) glycerol, and 1% (w/v) DDM, respectively. Insoluble material was removed by centrifugation at 100 000×g for 1 h at 4°C and then the supernatant was gently mixed for 3 h at 4°C with 0.9 ml Ni-NTA agarose or Strep-Tactin resin respectively. In the case of pMR2-MntH, the Ni-NTA agarose was pre-equilibrated with 20 mM Tris-Cl pH 7.9, containing 100 mM NaCl, 10% (v/v) glycerol, 2 mM βME, 10 mM imidazole and 0.05% (w/v) DDM. After packing into a column the agarose was washed with 40 column volumes of 20 mM Tris-Cl pH 7.9, containing 300 mM NaCl, 10% (v/v) glycerol, 2 mM βME, 30 mM imidazole and 0.05% (w/v) DDM and then His-tagged MntH eluted in 20 mM Tris-Cl pH 7.9, containing 100 mM NaCl, 10% (v/v) glycerol, 2 mM βME, 500 mM imidazole and 0.1% (w/v) DDM. In the case of pTTQ18-Strep II*-MntH, the Strep-Tactin resin was pre-equilibrated with 100 mM Tris-Cl pH 8.0, containing 150 mM NaCl, 10% (v/v) glycerol and 0.05% (w/v) DDM. After packing into a column the resin was washed with 5 column volumes of the equilibration buffer and then Strep II-tagged MntH eluted using the same buffer except for the presence of 0.1% (w/v) DDM and 5 mM desthiobiotin.
Purification and characterization of the E. coli glutamate/aspartate transporter GltP
Mixed membrane vesicles (30 mg protein) prepared from IPTG-induced bacteria harbouring pLMP-01-GltP were solubilized by incubation for 1 h on ice in 6 ml 20 mM Tris-Cl pH 7.9, containing 100 mM NaCl, 20% (v/v) glycerol, 2 mM βME, 5 mM imidazole and 0.5% (w/v) LDAO. Insoluble material was removed by centrifugation at 100,000 g for 1 h at 4°C and then the supernatant was gently mixed for 2 h at 4°C with 0.9 ml Ni-NTA agarose equilibrated with 20 mM Tris-Cl pH 7.9, containing 100 mM NaCl, 20% (v/v) glycerol, 2 mM βME, 10 mM imidazole and 0.1% (w/v) LDAO. The resin was then packed into a column and washed with 40 column volumes of wash buffer (20 mM Tris-Cl pH 7.9, containing 200 mM NaCl, 10% (v/v) glycerol, 2 mM βME, 30 mM imidazole and 0.1% (w/v) LDAO) before elution of the His-tagged GltP protein using the same wash buffer except for the presence of 150 mM imidazole.
The identity of the eluted protein was confirmed by N-terminal protein sequencing using a Procise 494 gas phase/liquid pulse sequencer (Applied Biosystems). Circular dichroism (CD) and Fourier transform infrared (FTIR) spectroscopy measurements on the purified protein were performed in 10 mM potassium phosphate, pH 7.4, containing 0.05% (w/v) LDAO with or without 10% (v/v) glycerol respectively, essentially as described previously Citation[11].
Results
Generation of expression vectors
To test the effects of different cloning and affinity-tagging approaches on the expression of membrane proteins of differing topologies, a panel of expression vectors was constructed, as described in Materials and methods and detailed in Supplementary (online version only). These were all derivatives of the expression vector pTTQ18, in which the polylinker region is flanked by the strong hybrid trp-lac (tac) promoter and the rrnB transcription terminator Citation[12]. The presence of the lacIq allele of the lac repressor gene within the vector ensures maximal repression of the promoter in the absence of IPTG. We and others have successfully used this vector to express multiple examples of major facilitator superfamily transporters at levels up to ∼25% of the inner membrane protein of E. coliCitation[4], Citation[11], Citation[15]. Typically, yields of protein expressed under the control of the tac promoter in this vector are higher than those obtained with vectors where expression is under the control of the T7 promoter Citation[15]. In the present project, vectors were constructed for recombinational cloning (pLMPDEST-01, encoding a C-terminal TEV-Lumio™-His8 tag) and for cloning by conventional restriction and ligation. In the latter category, pTTQ18-RGSH6-UraA and pMR4 were employed to add C-terminal RGSH6 and SH8 affinity tags respectively to proteins encoded by inserted DNA, while pMR2 was employed to add a N-terminal MNCSH8 affinity tag. The vector pTTQ18-Strep II was constructed for C-terminal addition of a second type of affinity tag, the Strep-tag II peptide Citation[14], preceded by a SerAla linker. Finally, the vector pMR5 was constructed in order to add a C-terminal Lumio™ tag to encoded proteins to facilitate detection of their expression.
Investigating the effects of recombinational cloning on the expression of NupG
Recombinational cloning using the Gateway™ system (Invitrogen) offers advantages of speed, efficiency and flexibility for production of constructs encoding water-soluble proteins Citation[17]. However, a potential disadvantage in the context of membrane proteins is that this method results in the addition of at least 25 nucleotides, corresponding to the attB1 and attB2 sites, between the coding region of interest and any N- or C-terminal tags respectively. These sites encode peptides with a net positive charge (TSLYKKAG and NPAFLYKVV), and additional residues may unavoidably be incorporated as a result of Gateway adaptation of conventional expression vectors. While such sequences appear to have little effect on expression levels for water-soluble proteins, it was unclear whether they would have any effect on membrane proteins, where charge distribution is of key importance for membrane insertion during synthesis Citation[18].
To examine the effects of the att site-encoded peptides on membrane protein expression, we first employed the E. coli nucleoside transporter NupG, a 12-transmembrane (™) helix protein with cytoplasmic termini (), which had previously been successfully expressed at high levels in our laboratory using the pTTQ18-derived construct pGJL25 Citation[11].
The corresponding Gateway™ expression vector, pLMP-01-NupG, was constructed using the destination vector pLMPDEST-01 as described in Materials and methods. Recombinational cloning introduced peptides with sequences RSTSLYKKAGWNS and NPAFLYKVVDDP flanking the N- and C-terminal regions of NupG respectively. A second construct, pLMP-01-NupG-E, lacking the N-terminal attB1-encoded sequence, was produced by removal of a short EcoRI fragment from pLMP-01-NupG. Expression of NupG from the two constructs produced by recombinational cloning was compared with that from pGJL25, an expression construct created by conventional cloning and thus lacking both att-encoded sequences. Following induction with IPTG, all three constructs yielded approximately the same amounts of membrane-associated NupG, as revealed both by Coomassie blue staining and Western blotting ().
Figure 2. Comparison of NupG expression levels yielded by constructs generated by recombinational and conventional cloning. Membranes (20 µg) from non-induced and 1 mM IPTG-induced cultures of E. coli strain Tuner(DE3)pLysS harbouring the indicated constructs were subjected to SDS-PAGE followed by (A) Coomassie blue staining or (B) Western blotting and staining with HisProbe™–HRP. pMR1 is vector lacking an insert, pLMP-01-NupG and pLMP-01-NupGE were produced by recombinational cloning, and pGJL25 was produced by conventional cloning. The mobilities of marker proteins of known molecular mass are shown on the right and the position of the overexpressed monomeric form of NupG is arrowed.
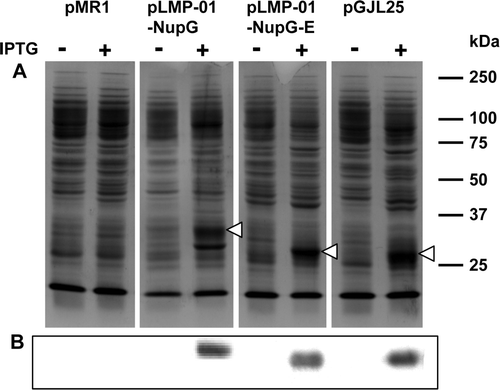
Hence neither the N- nor the C-terminal att-encoded sequences had a marked effect on the expression level of the protein or its membrane insertion. The NupG proteins encoded by pGJL25 (431 residues, predicted Mr 47 911) and pLMP-01-NupG-E (451 residues, predicted Mr 50 238) migrated anomalously fast (apparent Mr ∼28 000), probably reflecting their hydrophobic natures, as previously reported Citation[11]. That encoded by pLMP-01-NupG (464 residues, predicted Mr 51 718) migrated with an apparent size of ∼33 kDa, presumably because of the presence of the 13 additional residues, three of which are positively charged, incorporated into the N-terminal region of the protein as a result of recombinational cloning.
Use of recombinational cloning to produce expression constructs for membrane proteins of differing topologies
To investigate the general applicability of recombinational cloning methods for membrane protein expression, a further six bacterial transporters were next tested. These transporters, listed in Supplementary , were representatives of six different, widely-distributed protein families which have physiologically important homologues in humans. Four of these (UhpT, ZitB, HI0736 and GltP) resembled NupG in possessing cytoplasmic N- and C-termini (), a characteristic shared by the majority of bacterial polytopic membrane proteins Citation[8]. However, in addition they included examples of proteins in which one (MntH) or both (PitA) termini were located on the periplasmic side of the membrane (). Constructs were prepared by using pLMPDEST-01 and the Gateway™ approach, and expression tested as described in the Materials and Methods. Substantial expression, at levels readily detected by Coomassie blue staining of SDS-polyacrylamide gels of membrane samples, was achieved for ZitB, HI0736 and GltP (A).
Figure 3. Use of recombinational cloning for expression of transporters from three different protein families. Membranes (20 µg) from IPTG-induced cultures of E. coli strain Tuner(DE3)pLysS (A, B) or BL21 (C-F) harbouring the indicated constructs were subjected to SDS-PAGE. Proteins were detected using Coomassie blue (A, C, E), Lumio™ Green (B) or by staining Western blots with HisProbe™–HRP (D, F). Constructs designated ‘-R’ were produced by recombinational cloning, while those designated ‘-C’ were produced by conventional cloning. The mobilities of marker proteins of known molecular mass are shown on the left and the positions of the overexpressed monomeric forms of the proteins are indicated by arrowheads.
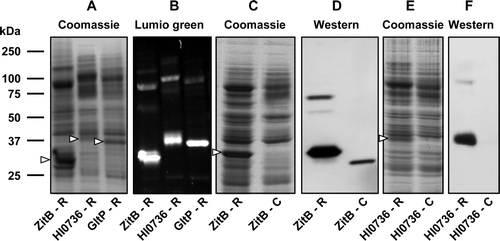
The identity of the bands ascribed to the transporters was confirmed using the in-gel Lumio™ detection assay (B). No similar bands were observed in membranes prepared from cells harbouring the control vector pMR1 (data not shown). A low level of expression, detectable only by western blotting for the His8 tag, was observed for UhpT but no expression was detectable for either PitA or MntH, despite the testing of a wide range of cell culture conditions (data not shown).
In the case of ZitB and HI0736, direct comparison was made between constructs produced by recombinational cloning and those produced by conventional cloning procedures using pTTQ18-RGSH6-UraA. Interestingly, much higher expression levels were yielded by the Gateway™ constructs. Coomassie blue staining of SDS-polyacrylamide gels of membrane samples revealed that the two proteins were expressed at levels equivalent to ∼22% and ∼6% of the total membrane protein in the case of the Gateway constructs, whereas expression from the conventionally constructed vectors was undetectable (C, 3E). The identity of the expressed proteins was confirmed by staining for the oligohistidine tags, which revealed a low level of expression from the conventionally-constructed ZitB expression vector but none from that encoding HI0376 (D, 3F).
Alternative strategies for expression of membrane proteins with periplasmic termini
A periplasmic location for the C-terminus of PitA, which is predicted to contain 10 ™ segments, has been inferred from lack of expression of a C-terminally-tagged GFP fusion protein Citation[8], and the extracellular location of both the N- and C-termini has been experimentally demonstrated for the human homologue Pit-2 by epitope tagging Citation[19]. Similarly, MntH has been experimentally demonstrated by gene fusion approaches to possess 11 ™ segments and a periplasmic C-terminus Citation[20]. To investigate whether failure to express either PitA or MntH using constructs produced by recombinational cloning reflected the periplasmic locations of one or both protein termini, a number of alternative expression constructs were tested. For these experiments, conventional cloning strategies were used to fuse the protein coding regions either to a C-terminal Strep-tag II peptide via a Ser-Ala linker (using vector pTTQ18-Strep II, Supplementary , and oligonucleotides designated -Strep-F and -R in Supplementary ) or, in the case of MntH, an N-terminal His8 tag (using vector pMR2, Supplementary , and oligonucleotides MntH-pMR2-F and -R in Supplementary ). It was hypothesised that fusion of the effectively neutral Strep-tag II peptide to a periplasmic terminus of a membrane protein would have a less deleterious effect on biosynthesis than a His-tag, which is likely to be at least partly charged at physiological pH. Similarly, for a protein such as MntH which has a cytoplasmic N-terminus and a periplasmic C-terminus, fusion of a His-tag to the N-terminus was hypothesised to be less deleterious than its fusion to the C-terminus.
Consistent with these hypotheses, and in contrast to the C-terminally His-tagged form of the protein encoded by the Gateway construct described above, a construct encoding PitA fused to a C-terminal Strep-tag II peptide yielded significant expression of the protein, detectable both by Coomassie blue staining (A) and by western blotting (B).
Figure 4. Use of Strep-tag II for detection and purification of proteins bearing periplasmic C-termini. Membranes (20 µg) from 0.1 mM IPTG-induced or non-induced cultures of E. coli strain BL21 harbouring pTTQ18-Strep II-PitA (A, B; PitA bearing a C-terminal Strep-tag II) or pTTQ18-Strep II-MntH (C, D; MntH bearing a C-terminal Strep-tag II) were subjected to SDS-PAGE. Proteins were detected using Coomassie blue (A, C) or by staining Western blots with Strep-tactin-HRP (B, D). Panel E shows purified MntH C-terminally tagged with a Strep-tag II peptide linked via G5SA. The mobilities of marker proteins of known molecular mass are shown on the right and the positions of the overexpressed monomeric forms of the proteins are indicated by arrowheads.
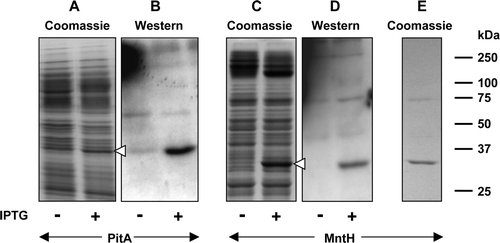
A similar finding was made for MntH bearing a C-terminal Strep-tag II, where expression levels corresponding to ∼13% of total membrane protein were obtained (C, 4D). Surprisingly, despite the fact that it was detectable using Strep-tactin-HRP, the detergent-solubilised Strep-tag II tagged MntH protein was found not to bind to Strep-Tactin resin (data not shown). The C-terminal extramembranous segment of MntH is predicted to be only 4 residues long Citation[20] and so access of the Strep-tag II peptide to its binding site on the Strep-Tactin resin may have been sterically hindered by the protein and/or the surrounding detergent molecules. In an attempt to circumvent this problem, a second construct, pTTQ18-Strep II*-MntH, bearing a longer linker (G5SA) between the protein and the Strep-tag II peptide, was constructed. The encoded protein was found to be expressed at levels similar to those obtained with the pTTQ18-Strep II-derived construct but in contrast to the latter it did bind to the Strep-Tactin resin and could be purified to homogeneity in a yield of ∼ 0.3 mg/litre of culture (E).
As previously described, no expression was detected when recombinational cloning was used to fuse the C-terminus of MntH via an att-site-encoded sequence and a TEV-cleavable linker to Lumio and His8 tags. In order to assess which features of the C-terminal region were responsible for failure of expression, constructs bearing just a C-terminal His8 tag or a Lumio-tag were constructed by conventional cloning procedures using vectors pMR4 and pMR5 respectively (Supplementary ). Once again, no expression was detectable either by Coomassie blue staining or by western blotting from the construct encoding the His8-tagged protein (A, 5B).
Figure 5. Effect of tag type and location on expression of MntH. Membrane samples (20 µg) were prepared from cultures of E. coli strain BL21 harbouring the indicated MntH constructs, before and after induction with 0.1 mM IPTG. MntH-H8: pMR4-MntH, encoding MntH bearing a C-terminal His8 tag; MntH-lumio: pMR5-MntH, encoding MntH bearing a C-terminal Lumio™ tag; H8-MntH: pMR2-MntH, encoding MntH bearing an N-terminal His8 tag. Proteins were detected using Coomassie blue (A, C, E, G), Lumio™ Green (D) or by staining Western blots with HisProbe™–HRP (B, F). Panel G shows purified MntH bearing an N-terminal His8 tag. The mobilities of marker proteins of known molecular mass are shown on the right and the positions of the overexpressed monomeric forms of MntH are indicated by arrowheads.
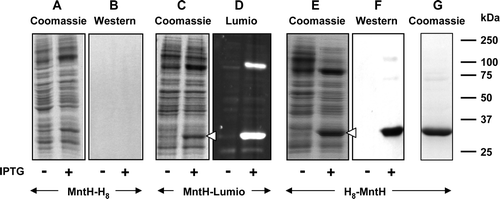
However, the Lumio-tagged protein was expressed at a high level, corresponding to ∼16% of the total membrane protein (C, 5D). The ∼90 kDa band seen in addition to the monomeric form of MntH in D probably represented an oligomeric form of the transporter. In contrast to the C-terminally His8-tagged protein, high levels of expression, corresponding to ∼23% of the total membrane protein (E), were obtained for a pMR2-derived construct encoding a His8 tag fused to the N-terminus of MntH. The identity of the expressed protein was confirmed by western blotting (F). Consistent with its higher level of expression, the N-terminally His-tagged form of the protein could be purified by chromatography in Ni-NTA agarose with a yield (∼1.5 mg/l of culture), substantially greater than obtained for the Strep-tagged protein (G).
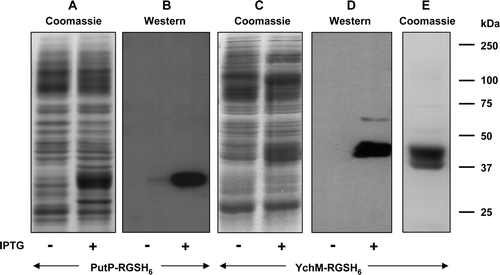
To explore the generality of this approach for tagging membrane proteins with an odd number of ™ segments, we also tested the use of C-terminal His-tagging on proteins from two additional transporter families ( and Supplementary ). These were the putative sulphate transporter YchM from E. coli, which is predicted to resemble MntH in possessing 11 ™ segments but unlike the manganese transporter to have a cytoplasmic C-terminus Citation[8], and the sodium-proline transporter PutP from E. coli, which is predicted to possess 13 ™ segments and is known to have a cytoplasmic C-terminus Citation[21]. Constructs encoding C-terminally RGSH6-tagged versions of both proteins were generated using pTTQ18-RGSH6-UraA. Both proteins were expressed at levels up to ∼20% of the total membrane protein upon IPTG induction, and YchM could be purified to homogeneity by Ni-NTA agarose chromatography with yield of more than 2 mg per litre of culture ().
Figure 6. C-terminal His-tagging of membrane proteins with an odd number of TM segments and a cytoplasmic C-terminus. Membranes were prepared, before and after induction with 0.1 mM or 1 mM IPTG respectively, from cultures of E. coli strain BL21 harbouring constructs encoding PutP-RGSH6 or YchN-RGSH6. Samples (20 µg) were subjected to SDS-PAGE followed by Coomassie blue staining (A, C, E) or Western blotting and staining with HisProbe™–HRP (B, D). Panel E shows purified PutP-RGSH6. The mobilities of marker proteins of known molecular mass are shown on the right.
Functional characterization of GltP produced by recombinational cloning
As described earlier, the E. coli glutamate/aspartate transporter GltP bearing a C-terminal TEV-Lumio™-His8 tag could be expressed at a level equivalent to ∼10% of the membrane protein in E. coli (A). This protein was therefore chosen as an example to investigate the effects of vector construction by recombinational cloning on the function and purification of membrane proteins. The initial rate of [3H]glutamate (50 µM) uptake by IPTG-induced E. coli BL21 cells harbouring the expression construct (pLMP-01-GltP) was 7.7-fold greater than that for cells harbouring a comparable vector, pMR1, lacking an insert (A) and 5.6-fold greater than for un-induced cells (data not shown).
Figure 7. Characterization of the E. coli glutamate/aspartate transporter GltP expressed using recombinational cloning. (A) Comparison of [3H]glutamate uptake by IPTG-induced E. coli harbouring pLMP-01-GltP (recombinational cloning product), the corresponding parental vector pMR1, pTTQ18-RGSH6-GltP (conventional cloning product) or the corresponding parental vector pTTQ18. (B) Coomassie blue-stained SDS-polyacrylamide gel of purified GltP(TEV-Lumio™-His8). The mobilities of marker proteins of known molecular mass are shown on the left and the positions of putative GltP oligomers are indicated by arrowheads. (C) Thermal unfolding of purified GltP(TEV-Lumio™-His8). CD measurements were made during the heating of the protein at a rate of 0.6°C per min. (D) Amide I region of the FTIR spectrum of a hydrated film of GltP(TEV-Lumio™-His8), and bands obtained by deconvolution. The latter were assigned to β-sheet (i & vi), unordered structure (ii), a-helix (iii) and β-turns (iv & v). The dotted line shows the curve fitted using these component bands.
![Figure 7. Characterization of the E. coli glutamate/aspartate transporter GltP expressed using recombinational cloning. (A) Comparison of [3H]glutamate uptake by IPTG-induced E. coli harbouring pLMP-01-GltP (recombinational cloning product), the corresponding parental vector pMR1, pTTQ18-RGSH6-GltP (conventional cloning product) or the corresponding parental vector pTTQ18. (B) Coomassie blue-stained SDS-polyacrylamide gel of purified GltP(TEV-Lumio™-His8). The mobilities of marker proteins of known molecular mass are shown on the left and the positions of putative GltP oligomers are indicated by arrowheads. (C) Thermal unfolding of purified GltP(TEV-Lumio™-His8). CD measurements were made during the heating of the protein at a rate of 0.6°C per min. (D) Amide I region of the FTIR spectrum of a hydrated film of GltP(TEV-Lumio™-His8), and bands obtained by deconvolution. The latter were assigned to β-sheet (i & vi), unordered structure (ii), a-helix (iii) and β-turns (iv & v). The dotted line shows the curve fitted using these component bands.](/cms/asset/7b70dd02-03ba-40fe-acd2-c83357718a5b/imbc_a_224312_f0007_b.gif)
Moreover, the rate of glutamate uptake attributable to pLMP-01-GltP (i.e., corrected for endogenous uptake seen in cells harbouring pMR1) was 1.2-fold greater than that seen in cells harbouring a construct (pTTQ18-RGSH6-GltP) produced by conventional cloning procedures (A). These results demonstrated that addition of the att-encoded sequences at the N- and C-termini of the GltP protein as a result of recombinational cloning had not interfered with its proper folding and insertion into the membrane.
To characterize the expressed protein in more detail, it was purified to apparent homogeneity by chromatography of LDAO-solubilized membrane proteins on Ni-NTA agarose. The purified protein migrated as a major band of apparent size ∼37 kDa on SDS-PAGE (B), additional bands at ∼75 kDa and 110 kDa probably representing dimeric and trimeric forms of this natively trimeric protein Citation[22]. N-terminal sequencing of the purified protein yielded the sequence, MNCTS, corresponding to that expected for the desired construct, and detection of the His8 tag by blotting confirmed that the expressed protein was full-length: its faster migration on SDS-PAGE than predicted for its size of 52.2 kDa probably reflected the hydrophobic nature of the sequence. Confirmation that the purified protein was correctly folded was provided by spectroscopic investigation of the LDAO-solublized transporter. Analysis of the CD spectrum (not shown) indicated that the protein was predominantly α-helical in structure, the helix content of approximately 66% being similar to that of 68% derived from the crystal structure of the glutamate transporter GltPh from Pyrococcus horikoshiiCitation[23]. The two proteins exhibit 34% sequence identity and so are likely to have very similar folds. Analysis of the purified GltP protein by FTIR spectroscopy similarly provided evidence for an α-helix content of approximately 66%: the amide I region of the spectrum could be reproduced well by a fit of six components, dominated by one at 1657 cm−1 indicative of α-helix (D) Citation[24], Citation[25]. The conclusion that the structural integrity of GltP was conserved during its purification was supported by the observation by CD spectroscopy of a profound change in ellipticity at 222 nm, indicative of disruption of secondary structure, upon heating the protein to 90°C (C).
Discussion
In the present study of E. coli and H. influenzae transporters from nine different families we have shown that the topology of membrane proteins is an important consideration when designing expression and purification strategies. For four of the five proteins tested in which both termini were cytoplasmic, substantial levels of expression, ranging from 6–22% of total membrane protein, were yielded by expression vectors constructed by Gateway™ recombinational cloning. These high levels of expression may reflect the use of vectors incorporating the tac promoter, which we have typically found to yield higher levels of membrane protein expression than for corresponding T7 promoter-based constructs. Interestingly, for two of these proteins, ZitB and HI0736, much higher levels of expression were obtained than for expression vectors employing the same tac promoter but constructed by conventional restriction and ligation. A possible explanation is that the presence of the att-site-encoded peptides inhibits proteolytic degradation of the proteins. However, at least in the case of GltP (A) and NupG (data not shown), the presence of these regions did not impair protein function. The expressed proteins could readily be purified by virtue of an attached C-terminal His-tag, in amounts ranging from 0.8 to ∼2 mg per l of culture (B and data not shown). Moreover, possession of a C-terminal His tag, in combination with N-terminal sequencing, provided confirmation that the purified proteins were full length, despite their anomalous mobilities on SDS-PAGE. Because the majority of polytopic membrane proteins are predicted to have cytoplasmic termini Citation[8], use of this recombinational cloning approach should be widely applicable for production of the amounts of protein required for crystallography from 10–25 litre-scale fermentations.
Supplementary Figure 1. Graphical representation of pTTQ18-based expression vectors employed in this study, illustrating the encoded tag sequences and key restriction sites. ptac, tac promoter; CmR, chloramphenicol resistance marker; ccdB, gene resulting in lethality towards most E. coli strains because of the interaction of its protein product with DNA gyrase; attR1 & attR2, modified λ phage recombination sites; MCS, multicloning site; UraA, E. coli uracil transporter gene.
In contrast to the successful use of recombinational cloning for expression of membrane proteins possessing cytoplasmic termini, we found that in the examples tested recombinational cloning grossly impaired expression of proteins in which one or both termini were located on the periplasmic side of the bacterial inner membrane. The same was true for proteins in which a periplasmic terminus was fused to an oligohistidine tag ( and data not shown). One simple explanation for these findings might be that the presence of the charged att-site-encoded peptides and/or oligohistidine sequences interferes with the proper translocation of the adjacent segments of the protein across the membrane during biosynthesis: the charge distribution across ™ segments is known to have a profound effect on their orientation Citation[8], Citation[18]. However, attachment of the effectively neutral Strep-tag II peptide to the periplasmic C-terminus of MntH or PitA appeared to be much less deleterious, allowing substantial levels of expression and enabling protein purification by Strep-Tactin chromatography (). Alternatively, for proteins in which the two termini were located on opposite sides of the membrane, attachment of an oligohistidine tag to the cytoplasmic terminus allowed abundant expression and purification ( and ).
In the light of our findings, we propose a generic strategy for high throughput expression of membrane proteins for structural genomics projects. The first stage is prediction or experimental determination of topology Citation[8]. For those proteins bearing cytoplasmic termini, the method of choice would then be to introduce a C-terminal oligohistidine tag for purification, ideally by recombinational cloning. For proteins in which both termini are periplasmic, a C-terminal Strep-II purification tag would instead be used, while for proteins in which one terminus is cytoplasmic, this would be fused to an oligohistidine tag. While the presence of a periplasmic terminus precludes use of recombinational cloning, other approaches such as ligation-independent cloning offer a route to high throughput, robotic construction of expression vectors in these cases. Such approaches are currently being implemented in our laboratory as part of the BBSRC-funded UK Membrane Protein Structure Initiative. Incorporation of the Lumio™ tag into such vectors similarly opens up the way to high throughput screening of membrane protein expression: in contrast to tagging with GFP, which yields fluorescence only when attached to cytoplasmic termini Citation[26], we have found that attachment of the Lumio™ tag to both cytoplasmic and periplasmic termini is compatible with protein insertion into the membrane and high level expression (B and 5D). While we have so far used this tag only for in-gel fluorescence detection, it should be possible to detect and quantify fluorescence in detergent-solubilized cell lysates from strains of bacteria lacking SlyD, which interacts with the Lumio™ reagent, thereby enabling high throughput screens of membrane protein expression.
In conclusion, by consideration of protein topology, we have been able to implement an improved, generic methodology for high level expression, detection and purification of bacterial polytopic membrane proteins. Using the suite of tac-promoter based vectors developed in this study, it has been possible readily to express and purify proteins from multiple families of transporters with differing topologies in amounts up to ∼2 mg per litre of culture. We believe that these approaches should be widely applicable in future structural investigations of this biologically important but technically challenging class of proteins.
Acknowledgements
This work was supported principally by grants from Invitrogen Corporation, the Biotechnology and Biological Sciences Research Council (Membrane Protein Structure Initiative; Grant BBS/B/14418) and the University of Leeds. We are indebted to Drs Jon Chesnut, Toni Kudlicki and David Odelson of Invitrogen Corporation for helpful discussions. MR is grateful to the Islamic Development Bank (IDB) for a PhD studentship. We thank Dr J. Keen and Ms D. Ashworth of the University of Leeds for assistance with protein and DNA sequencing respectively.
Related Research Data
References
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol 2001; 305: 567–580
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002; 1: 727–730
- Ren Q, Paulsen IT. Comparative analyses of fundamental differences in membrane transport capabilities in prokaryotes and eukaryotes. PLoS Comput Biol 2005; 1: e27
- Wang DN, Safferling M, Lemieux MJ, Griffith H, Chen Y, Li XD. Practical aspects of overexpressing bacterial secondary membrane transporters for structural studies. Biochim Biophys Acta 2003; 1610: 23–36
- Eshaghi S, Hedren M, Nasser MI, Hammarberg T, Thornell A, Nordlund P. An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins. Protein Sci 2005; 14: 676–683
- Drew DE, von Heijne G, Nordlund P, de Gier JW. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett 2001; 507: 220–224
- Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 2006; 14: 673–681
- Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science 2005; 308: 1321–1323
- Granseth E, Daley DO, Rapp M, Melen K, von Heijne G. Experimentally constrained topology models for 51,208 bacterial inner membrane proteins. J Mol Biol 2005; 352: 489–494
- Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS. “Touchdown” PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 1991; 19: 4008–4008
- Xie H, Patching SG, Gallagher MP, Litherland GJ, Brough AR, Venter H, Yao SY, Ng AM, Young JD, Herbert RB, Henderson PJ, Baldwin SA. Purification and properties of the Escherichia coli nucleoside transporter NupG, a paradigm for a major facilitator transporter sub-family. Mol Membr Biol 2004; 21: 323–336
- Stark MJR. Multicopy expression vectors carrying the lac repressor gene for regulated high-level expression of genes in Escherichia coli. Gene 1987; 51: 255–267
- Hadden, DA. 2002. Molecular mechanisms of nucleoside and nucleobase transporters [thesis]. Leeds(YorkshireUK): University of Leeds.
- Skerra A, Schmidt TG. Use of the Strep-Tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol 2000; 326: 271–304
- Ward A, Sanderson NM, O'Reilly J, Rutherford NG, Poolman B, Henderson PJF. The amplified expression, identification, purification, assay and properties of histidine-tagged bacterial membrane transport proteins. Membrane transport – a practical approach, SA Baldwin. Oxford University Press, Oxford 2000; 141–166
- Henderson PJF, Macpherson AJ. Assay, genetics, proteins and reconstitution of proton-linked galactose, arabinose and xylose transport systems of Escherichia coli. Methods Enzymol 1986; 125: 387–429
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol 2000; 328: 575–592
- Nilsson J, Persson B, von Heijne G. Comparative analysis of amino acid distributions in integral membrane proteins from 107 genomes. Proteins 2005; 60: 606–616
- Salaun C, Rodrigues P, Heard JM. Transmembrane topology of PiT-2, a phosphate transporter-retrovirus receptor. J Virol 2001; 75: 5584–5592
- Courville P, Chaloupka R, Veyrier F, Cellier MF. Determination of transmembrane topology of the Escherichia coli natural resistance-associated macrophage protein (Nramp) ortholog. J Biol Chem 2004; 279: 3318–3326
- Jung H, Rubenhagen R, Tebbe S, Leifker K, Tholema N, Quick M, Schmid R. Topology of the Na+/Proline transporter of Escherichia coli. J Biol Chem 1998; 273: 26400–26407
- Gendreau S, Voswinkel S, Torres-Salazar D, Lang N, Heidtmann H, Detro-Dassen S, Schmalzing G, Hidalgo P, Fahlke C. A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J Biol Chem 2004; 279: 39505–39512
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004; 431: 811–818
- Goormaghtigh E, Cabiaux V, Ruysschaert JM. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell Biochem 1994; 23: 405–450
- Arrondo JL, Goni FM. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog Biophys Mol Biol 1999; 72: 367–405
- Drew D, Slotboom DJ, Friso G, Reda T, Genevaux P, Rapp M, Meindl-Beinker NM, Lambert W, Lerch M, Daley DO, Van Wijk KJ, Hirst J, Kunji E, de Gier JW. A scalable, GFP-based pipeline for membrane protein overexpression screening and purification. Protein Sci 2005; 14: 2011–2017
Supplementary references
- Tolner B, Poolman B, Wallace B, Konings WN. Revised nucleotide sequence of the gltP gene, which encodes the proton-glutamate-aspartate transport protein of Escherichia coli K-12. J Bacteriol 1992; 174: 2391–2393
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004; 431: 811–818
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005; 437: 215–223
- Courville P, Chaloupka R, Veyrier F, Cellier MF. Determination of transmembrane topology of the Escherichia coli natural resistance-associated macrophage protein (Nramp) ortholog. J Biol Chem 2004; 279: 3318–3326
- Xie H, Patching SG, Gallagher MP, Litherland GJ, Brough AR, Venter H, Yao SY, Ng AM, Young JD, Herbert RB, Henderson PJ, Baldwin SA. Purification and properties of the Escherichia coli nucleoside transporter NupG, a paradigm for a major facilitator transporter sub-family. Mol Membr Biol 2004; 21: 323–336
- Harris RM, Webb DC, Howitt SM, Cox GB. Characterization of PitA and PitB from Escherichia coli. J Bacteriol 2001; 183: 5008–5014
- Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science 2005; 308: 1321–1323
- Jung H, Rubenhagen R, Tebbe S, Leifker K, Tholema N, Quick M, Schmid R. Topology of the Na+/Proline transporter of Escherichia coli. J Biol Chem 1998; 273: 26400–26407
- Lloyd AD, Kadner RJ. Topology of the Escherichia coli uhpT sugar-phosphate transporter analyzed by using TnphoA fusions. J Bacteriol 1990; 172: 1688–1693
- Lee SM, Grass G, Haney CJ, Fan B, Rosen BP, Anton A, Nies DH, Rensing C. Functional analysis of the Escherichia coli zinc transporter ZitB. FEMS Microbiol Lett 2002; 215: 273–278
- Saier MH, Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res 2006; 34: D181–186