
ABSTRACT
The present study describes the isolation and pheno-genotypic analysis of anaerobic Clostridium sartagoforme strain AAU1 from buffalo rumen, having major role in cellulose and chitin degradation. The isolate was cultured between 37°C and 42°C (optimum 40°C) temperature with an optimum pH of 7.5 in Hungate's medium supplemented with cellulose. Whole genome sequencing was performed using ion torrent PGM platforms. Local BLAST showed highest phylogenetic relation with C. sartagoforme DSM 1292 strain based on comparison of the 16S rDNA sequences. Comparison of sequences coding for housekeeping genes phosphoglycerate kinase (pgk) and gyrase B (gyrB) confirmed its taxonomic position within Clostridium genus. The fermentation ability analysed biochemically using AN-Biolog plate showed that the strain utilized more than 30 different substrates as metabolite precursors. Pathway analysis based on Rapid Annotation using Subsystem Technology reveals the organism to be highly active in the production of volatile fatty acids (VFAs). Genomic analysis confirmed the role of bacteria in determining ruminant host physiology. VFAs produced by the isolate play a prominent role in animal health maintenance by contributing a major proportion of the ruminants’ daily energy requirement. A deeper understanding of enzymes encoded allows further industrial applications and modulation of the rumen for enhanced farming applications.
1. Introduction
Rumen microflora is one of the most explored complex community and their vital role in several functions such as digestion of dietary feed substrates of host, inhibition of pathogenic microbes, gastrointestinal development, host immune, and improvement of animal products quality has led to concomitant increase in the study of rumen bacteria (Hristov et al. Citation2012).
Clostridia, rod-shaped Gram-positive, spore-forming anaerobic bacteria are found to inhabit soil, sewage, and marine sediments, as well as both animal and human intestines. Recently, advancement in genome sequencing due to the development of next-generation sequencing technologies has revealed the genetic basis of various functional abilities of microbes. Genome is sequenced without cloning, using one of the next-generation sequencing techniques which is followed by the data analyses by comparing the sequences to known sequence databases. Genus Clostridium contains about 110 species, out of which whole genomes of approximately 59 species are currently available in the NCBI GenBank database.
Several Clostridia species have been utilized for numerous applications. Several species of clostridia are used industrially for the production of alcohols and commercial solvents. Researchers have also employed anaerobic Clostridial strains that selectively proliferate within tumours as a part of bacteriolytic anti-cancer therapies (Bettegowda et al. Citation2006). Since these necrotic tissues exist only in tumours, anaerobic bacteria such as Clostridium novyi, Clostridium perfringens, and Clostridium sordellii are reported to be promising candidates to treat these conditions.
The presence of diverse categories of microflora provide the edge to ruminant animals for consumption and dependence on lignocellulosic biomass to obtain their energy requirements through volatile fatty acids (VFAs) (viz. acetate, propionate, butyrate, etc.) generated from the degradation and anaerobic fermentation of fibrous materials. Researchers have reported Clostridium strains to possess the potential to synthesize various organic solvents such as butanol, acetate, butyrate, lactate, propionate, and ethanol as by-products via various fermentation pathways (Dabrowski et al. Citation2012).
In addition, certain fibre-degrading Clostridia are the true inhabitants of rumen such as Clostridium lochheadii, Clostridium polysaccharolyticum, and Clostridium longinucleatum (Hungate Citation1957). These Clostridia are not very important fibre degraders as their numbers in the rumen liquor are less than a million cells per gram of rumen content, but the cellulolytic activities of enzymes are sufficiently high to degrade significant quantity of fibre in the diet. Clostridium sartagoforme, one of the Clostridium species, is cultivable on glucose, N-acetylglucosamine, glucosamine, galactose, starch, hemicelluloses, and xylan.
Here we studied and analysed the genomic sequence of C. sartagoforme AAU1 strain, the first strain of the species to be completely sequenced, to our knowledge. The anaerobic bacterium was isolated during study of microbial metagenome profile of the Surti buffalo rumen sample. The DNA isolated from the organism was processed for whole genome sequencing by 454 GS-FLX titanium and ion torrent PGM as per the manufacturer's protocol. Functional annotation was performed to get a better insight into the genetic basis of metabolic properties of the strain that confer various benefits to host metabolism and physiology. The sequence also offered an opportunity to compare moderately related Gram-positive bacteria genomes (Clostridium beijerinckii and Clostridium botulinum) and to study the gene sequences not previously represented in the collection of complete genomes like those involved in metabolic pathways like VFA generation and anaerobic utilization of cellulose, hemi-cellulose, and chitin-like biopolymers.
2. Materials and methods
2.1. Strain and culture conditions
C. sartagoforme AAU1 was isolated from Surti buffalo rumen liquor for rumen metagenomic studies. The sample was collected at the College of Veterinary Science and Animal Husbandry, Anand. The anaerobic culture techniques as described by Hungate were used for all incubations. The rumen liquor was enriched in modified liquid Hungate medium (Hungate Citation1966) supplemented with cellulose powder, peptone, and yeast extract. The medium was pre-gassed with N2:H2 (80:20) in roll tubes and 50 mL anaerobic bottles and autoclaved at 15 psi, incubated under anaerobic conditions after post gassing with N2:CO2 (80:20) and inoculation of ruminal liquor followed by sealing with butyl rubber stoppers and aluminium cap seals maintaining positive pressure in the roll tubes.
Several dilutions (diluents contained: 0.5% NaCl and 0.2% Cysteine HCl, pre-gassed with N2:H2 (80:20) and autoclaved at 15 psi for 15 min) were prepared from the enriched samples for isolation and 0.1 mL from each dilution was streaked on modified Hungate medium containing cellulose agar in plates in anaerobic glove box (DonWhitely, UK) and incubated in anaerobic glove box maintaining strict anaerobic conditions by purging N2:CO2 (80:20) into glove box. Subsequently, to obtain a pure culture three sequential transfers were carried out from well-isolated colony obtained from the dilution. The isolated cultures were incubated at 40 ± 2°C and the colony morphology was studied.
The ruminal liquor was streaked on plates containing Hungate medium supplemented with cellulose powder and pre-gassed with N2:H2 (80:20), incubated under anaerobic conditions after post gassing with N2:CO2 (80:20) and sealing it with butyl rubber stoppers and aluminium cap seals. The anaerobic culture techniques as described by Hungate were used for all incubations. Several dilutions were prepared from the enriched samples for isolation and 0.1 mL from each dilution was streaked on Hungate Agar medium containing cellulose in anaerobic bottles. The cultures were incubated at 40 ± 2°C and the colony morphology was studied. Subsequently, to obtain a pure culture three sequential transfers were made from well-isolated colony after dilution.
Hungate medium used for enrichment and subsequent isolation of the culture contained (per 1000 mL) K2HPO4 0.5 g, KH2PO4 0.2 g, (NH4)2SO4 0.5 g, NaCl 1.0 g, MgSO4 0.05 g, CaCl2 0.05 g, NaHCO3 5.0 g, cysteine hydrochloride 0.5 g, crystalline cellulose powder 10.0 g, peptone 0.5 g, yeast extract 0.4 g, Clarified Rumen fluid 1.5%, Agar 3%, and pH 7.5.
2.2. Whole genome sequencing and assembly
Whole genome sequencing of the Clostridium strain was performed using 454 GS-FLX (Roche) and Ion Torrent PGM platforms as per manufacturer's instructions. The sequencing reads generated were subjected to hybrid assembly by GS De Novo Assembler V.2.6 to obtain the consensus contigs. Nucleotide sequence data are available in the GenBank database under the accession number ASRV00000000.
2.3. Bioinformatic analysis: 16S rRNA and housekeeping genes
Homology studies were carried out by uploading the assembled contigs in RDP-Ribosomal Database Project Classifier. Also, the results were verified by 16S rRNA local BLAST of the assembled contigs performed against the 16S rDNA database of 7545 sequences which were downloaded from NCBI via ftp. Genes encoding housekeeping proteins gyrB (Clostridium) (gyrase B) and pgk (Clostridium) (phosphoglycerate kinase) in the bacteria were compared with the database of the respective housekeeping genes created by sequences downloaded from NCBI via ftp. Sequences coding for 16S rRNA gene of Clostridium were aligned with that of C. sartagoforme AAU1 using ClustalW. Subsequently, an evolutionary distance matrix was generated from these nucleotide sequences in the data set using Maximum Composite Likelihood method. Phylogenetic analysis was performed using the neighbour-joining method by MEGA (Molecular Evolutionary Genetics analysis) version 6.06 (Tamura et al. Citation2007).
2.4. Fatty acid methyl ester analysis
Fatty acid methyl ester (FAME) analysis was carried out by gas–liquid chromatography (Sherlock Microbial Identification System [MIS]; MIDI, Inc.). The obtained chromatographic data of the isolate were analysed using the Sherlock software version 6.0B (S/N; 160277) MIDI, with the SMOORE6 method.
2.5. Characterization and substrate utilization profile
The carbon utilizations of the isolate were investigated biochemically using AN-Biolog® micro plate assay to determine its substrate fermenting potential. Suspension of isolate in anaerobic diluents (as per the manufacturer's instructions) was used to inoculate AN-Biolog plates (150 μL per well), which were then incubated in polythene bags containing Anaerobic Gas Pack at room temperature. The OD595 was measured in a microtitre plate reader after 72 h of incubation. Growth, colony, and morphological characteristics of the isolate were monitored according to methods prescribed in Bergey's Manual (Krieg et al. Citation1994). API 20A kit (BioMérieux API®, USA) was used to study the carbon utilization profile of the rumen isolate C. sartagoforme AAU1. Strict anaerobic conditions were maintained using gas phase N2:CO2 (80:20) and the glove box was maintained anaerobic with N2:H2:CO2 (80:10:10) during incubation.
2.6. Functional annotation and coding sequences prediction
The assembled contigs of the bacterial species genome were uploaded on the RAST-Rapid Annotation using Subsystem Technology server V. 4.0 (http://rast.nmpdr.org/rast.cgi) (Aziz et al. Citation2008) for gene prediction and annotation. The output result files obtained were further studied for interpretation of bacterial features participating in metabolic pathways and other functions.
2.7. CAZyme (carbohydrate-active enzyme) analysis
The carbohydrate-active enzyme coding genes of C. sartagoforme AAU1 genome were annotated using the CAZymes Analysis Toolkit (CAT) (Park et al. Citation2010) based on the CAZy database classification (Lombard et al. Citation2014). Enzyme annotation is performed by BLAST search against non-redundant enzyme coding sequences (CDS) in the CAZy database followed by their correspondence with the CAZy families and protein family domains.
2.8. Genome features and metabolic pathways for the production of VFAs
The C. sartagoforme genome was mined for the presence of genes involved in the metabolic pathways for propionate and butyrate production and observed genes were compared with corresponding metatranscriptome data of the rumen fluid sample from which the isolate was obtained. Other unique features playing an important role in the microbe's life cycle and its adaptation to the host environment were also taken into consideration based on the RAST annotation results.
3. Results
3.1. Genome assembly statistics
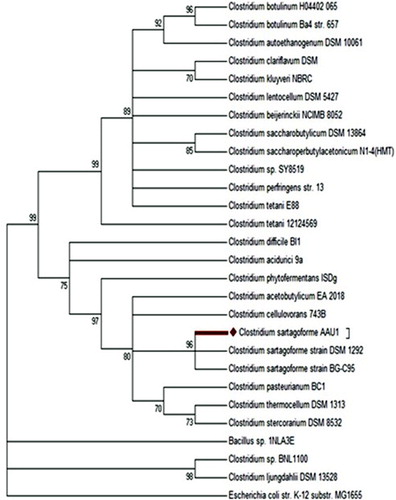
Genome assembly of the 732778 reads obtained from the whole genome sequencing of Clostridium isolate formed a total of 344 contigs giving a genome size of C. sartagoforme to be of 3.9 Mb and GC content of 27.9% with the assembly coverage of 24.0X. The classification of the isolate was studied using RDP Naïve Bayesian rRNA Classifier V.2.4 which identified the strain as belonging to the Clostridium genus as follows: Domain: Bacteria; Phylum: Firmicutes; and Genus: Clostridium. The classification of the Clostridium genus was reconfirmed by studying two housekeeping genes with reference gene sequences. Comparison showed an average of about 81% identity for the housekeeping genes when compared with the reference sequences coding for the pgk and gyrB genes. Local BLAST results showed that the sequenced strain covered 1470 bp length of the anaerobic C. sartagoforme strain DSM 1292 16S ribosomal RNA with a per cent identity of 99%. Phylogenetic analysis showed the strain to be closely related to C. sartagoforme strain DSM 1292 based on 16S gene sequences ().
Figure 1. Phylogenetic tree based on 16S rRNA gene sequences of genus Clostridium: the bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analysed. Branches corresponding to partitions reproduced in less than 70% bootstrap replicates are collapsed.
3.2. FAME analysis
In our isolate the dominant cellular fatty acids from cells grown in PYG medium at 40°C were C16:0, C14:0, C16:0, 15:0 anteiso 30H FAME or 16:1 cis 7 DMA and C14:0. The characteristic difference in FAME was shown with C16:0, 15:0 anteiso 30H FAME, or 16:1 cis 7 DMA, which was approximately 7-fold higher than all other isolates compared ().
Table 1. Comparative FAME profiles: (1) C. beijerinckii, (2) C. botulinum, (3) C. butyricum, (4) C. perfringens, and (5) C. sporogenes.
3.3. Characterization and substrate utilization profile: biochemical assay
Clostridium species showed the ability to ferment various carbohydrates, amino acids, organic acids, esters, and lipid moieties as substrates on the AN Biolog® microplate assay. When compared with the RAST functional annotation, the corresponding sequences coding for the genes involved in utilizing these molecules as substrate was observed (). While the gene sequences coding for pathways involving the utilization of substrates that gave a negative result in the biochemical test was also found to be absent in the genome of C. sartagoforme AAU1. Some metabolic characteristics were studied in roll tubes and hydrolysis of starch, casein, and tributyrin was performed on solid media in anaerobic glove box (Don Whitely). The closest identity of the isolate is C. sartagoforme but differences were observed at the physiological level. The differentiating biochemical characteristics between the two were utilization potential of arabinose, glycerol, and inositol and non-utilization of lactose, mannitol, melibiose, and salicin ().
Table 2. AN-Biolog result and corresponding genes coding for various enzymes involved in substrate utilization pathways.
Table 3. Comparison of phenotypic characteristics with nearest neighbours: (1) C. sartagoformea, (2) C. butyricuma, (3) C. sporogenesa, (4)C. perfringensa, (5) C. botulinuma, and (6) C. beijerinckiia.
3.4. Functional annotation and CDS prediction
RAST annotation showed the presence of a total of 4017 CDS functionally classified into 375 subsystems and 83 RNAs. A large proportion of the CDS were observed to be classified into the carbohydrate and the amino acid metabolism of the bacteria. A total of 457 CDS were observed in the strain AAU1 genome that participated in the carbohydrate metabolism. The features were further classified into enzyme coding for subcategories such as the central carbohydrate metabolic pathways (88), amino sugars (27), di- and oligosaccharides (155), one-carbon metabolism (27), organic acids (4), fermentation (42), sugar alcohols (36), carbohydrates – no subcategory (2), polysaccharides (11), and monosaccharides (65). Functional comparison of the carbohydrate subsystem with C. beijerinckii NCIMB 8052, C. perfringens strain 13, and C. botulinum E3 strains (shown to be the closest neighbours via RAST server) showed many enzymes utilizing various carbohydrate substrates unique to the sartagoforme species providing valuable information to the microbial database about newly whole genome-sequenced Clostridium (). We elucidated the chitin degradation pathway in more detail for the presence of enzyme encoding sequences in the organism and observed the presence of seven CDS involved in the same (). The organism converts chitin to N-acetyl glucosamine oligomers and further converts to hexose monosaccharides, rather than converting chitin to chitosan molecule; it possesses the enzyme machinery for direct conversion.
Figure 2. Chitin degradation pathway: number of CDS present in the genome (gi|507135708|gb|ASRV00000000) for the enzymes involved and corresponding contig number of the CDS.
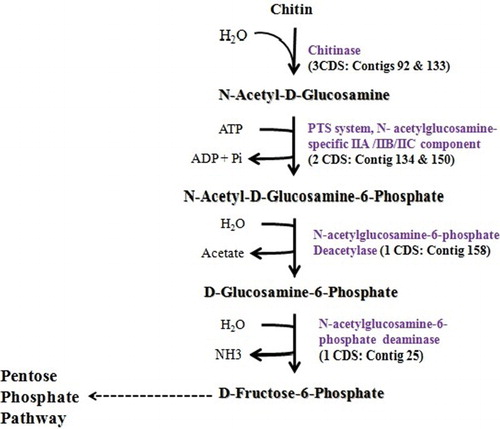
Table 4. Carbohydrate and amino acids utilizing enzymes unique to C. sartagoforme compared to C. beijerinckii NCIMB 8052, C. perfringens strain 13, and C. botulinum E3 strains.
Apart from the carbohydrate subsystem, a total of 277 CDS in amino acids and derivatives and 159 in protein metabolism were observed in the sartagoforme genome. The features were further classified into enzyme coding for subcategories such as glutamine, glutamate, aspartate, asparagine; ammonia assimilation (25), histidine metabolism (13), arginine; urea cycle, polyamines (60) lysine, threonine, methionine, and cysteine (88), amino acids and branched-chain amino acids (22), aromatic amino acids and derivatives (32), proline and 4-hydroxyproline (6), alanine, serine, and glycine (29) in amino acids and derivatives and protein folding (11), selenoproteins (1), protein biosynthesis (91), protein processing and modification (24), and protein degradation (32). Functional comparison of the protein subsystem with C. beijerinckii NCIMB 8052, C. perfringens strain 13, and C. botulinum E3 strains also depicted unique enzymes utilizing various amino acids and derivative substrates specific to C. sartagoforme when compared with the nearest species in the Clostridium genus ().
Several other features in the genome include a 776 bp segment coding for CodY gene found to be involved in various metabolic pathways. Other than carbohydrates and protein, many enzymes unique to the AAU1 strain when compared with the nearest species such as the 3-ketoacyl-CoA thiolase, molybdopterin-binding domain and pseudouridine-5’ phosphatase, glycerol-3-phosphate cytidylyltransferase (GCT), lipoprotein releasing system ATP-binding protein LolD and transmembrane protein LolC, CDP-glycerol:poly(glycerophosphate) glycerophosphotransferase, and dipeptide transport system permease protein DppC were found in various subcategories including the cofactors, vitamins, prosthetic groups, pigments, cell wall and capsule, and membrane transport pathway.
3.5. CAZyme (carbohydrate-active enzyme) analysis
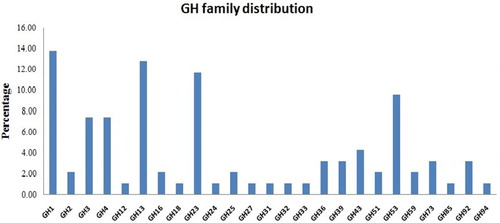
The glycoside hydrolase (GH) protein superfamily was the most highly represented CAZyme superfamily in the genome with 25 families within it, while the Polysaccharide Lyase class was the least represented with only two families identified within it.
Among the 25 families of the GH class, results depict that the GH1, GH3, GH4, GH13, GH23, and GH53 family CAZymes occupied about 50% of the total GH hits (). The CAZyme analysis profile further supports the results revealed by the RAST CDS prediction that the organism possesses the potential to digest various carbohydrate substrates.
Figure 3. GH superfamily classification and relative abundance of its families.
3.6. Genome features and metabolic pathways for the production of VFAs
In our study, we also studied the metabolic pathways for propionate and butyrate production in the C. sartagoforme genome and compared with corresponding results from rumen metatranscriptome data ( and ). The results showed that the genome comprised a total of seven CDS involved in conversion of 2-hydroxy butyrate to propanoate. also depicts the presence of nine CDS encoding enzymes of the butyrate production pathway to be present in the whole genome sequence.
Figure 4. Butanoate metabolism: number of CDS present in the genome (gi|507135708|gb|ASRV00000000) for the enzymes involved, corresponding contig number, and the percentage of that enzyme CDS observed in buffalo rumen metatranscriptome data.
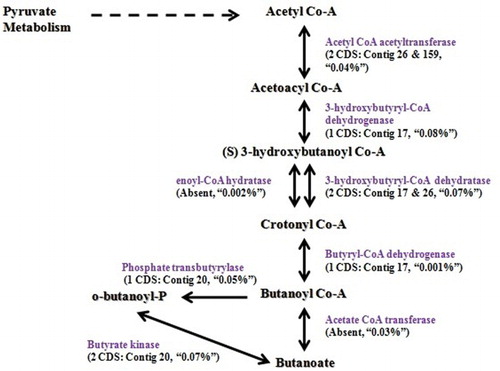
Figure 5. Propanoate metabolism: number of CDS present in the genome (gi|507135708|gb|ASRV00000000) for the enzymes involved, corresponding contig number, and the percentage of that enzyme CDS observed in buffalo rumen metatranscriptome data.

4. Discussion
4.1. Genome assembly and phylogenetic analysis
The genome size and GC content were in the range as observed for the Clostridia species. And 16S-based phylogenetic analysis revealed it to be most close to C. sartagoforme thus confirming the species. The genomic analysis and sequence deposited in repository helped increase the rumen microbe genome database and would help further unravel the potentials of such novel, hitherto difficult to cultivate rumen anaerobes. Housekeeping genes, such as the rpoB gene encoding RNA polymerase subunit B, gyrB gene encoding the subunit of DNA gyrase, and recA, involved in homologous recombination offer possibilities of using them as phylogenetics markers for the identification of new bacterial species from complex microbial communities (Drancourt & Raoult Citation2005). pgk has been identified as one of the most stable reference genes to identify closely related bacterial species (Smuts & Lastovica Citation2011). The gyrB gene encodes the subunit B protein of DNA gyrase and is distributed universally among bacterial species (Huang Citation1996). Thus, we considered these two housekeeping genes to confirm the placement of the isolate within the Clostridium genus.
4.2. FAME
The use of cellular fatty acid composition for taxonomy and identification of Clostridia has been investigated previously. In Clostridia, the presence and relative amounts of fatty acids with a chain length of 10–20 carbon atoms can distinguish easily among C. perfringens, Clostridium sporogenes, and Clostridium bifermentans (Lewis et al. Citation1967).
4.3. Substrate utilization
Microorganisms, especially bacteria are known for their potential of utilizing large variety of organic substances as substrates (Muniz et al. Citation2014; Nathani et al. Citation2014). The anaerobic isolate from the rumen environment was found to be utilizing large variety of substrates. The genome sequence reveals the active presence of genes coding for the enzymes playing role in the utilization of substrates by the bacterium as per the Biolog results.
4.4. Functional annotation and CDS prediction
Researchers have been working to elucidate the metabolic pathways by which solvents are produced by various Clostridium species (Nolling et al. Citation2001). Chitin is the second most abundant organic compound after cellulose. The degradation of chitin is catalysed by an enzymatic system consisting endo- and exo-hydrolases. The endochitinase cleaves chitin to oligomeric intermediates. The oligomers further are converted to monosaccharides as by the action of other microbial enzymes involved as shown in . Rumen comprises of various bacteria able to degrade chitin. Fungal and invertebrate chitin is many a times engulfed by the ruminants making it a considerable part of the dry matter (∼8%) of rumen content (Orpin Citation1975). Partial chitin digestion in the rumen has also been studied and reported in C. sartagoforme species and the enzyme was isolated and characterized (Simunek et al. Citation2001). The results thus confirmed earlier reports that Clostridium is one of the important genera present in the rumen content of different animals which plays an important role in chitin degradation and produced mainly acetate, butyrate, and lactate (Kopecny et al. Citation1996) as the end products.
Tagatose 1, 6-bisphosphate aldolase metabolizes d-tagatose, a hexoketose isomer of d-galactose. Tagatose-6-phosphate pathway is reported in some Gram-positive microorganisms such as Staphylococcus aureus, Streptococcus lactis, Streptococcus cremoris, Lactobacillus casei, and Lactococcus lactis (Yu et al. Citation1990; Kim Citation2004). The tagatose-6-phosphate pathway differs from the central metabolism as it excludes the fructose-1, 6-diphosphate formation (Bond et al. Citation1998). Tagatose is generally regarded as safe approved as animal feed and the presence of the tagatose 1, 6-bisphosphate aldolase CDS in the strain suggests its ability to ferment varied substrates and subsequent potential role in rumen metabolism. The organism also possesses the gene sequences coding for various enzymes participating in the Entner–Dourdoroff pathway along with those participating in the utilization of maltose and mannose. Overall, the genome showed the presence of glycolytic enzymes such as the alpha mannosidase, periplasmic alpha-amylase, and gluconate dehydratase that will be participating in the utilization of a variety of carbohydrate substrates by breaking down the compounds into simpler sugars that can enter the central glycolytic pathway. Apart from the carbohydrate moieties, the results show the presence of about 13 CDS involved in the subsystems such as arginine and ornithine degradation, cysteine biosynthesis, and aromatic amino acids utilization. Uptake of arginine and related metabolites are highly dependent on the transport mechanisms which carry out an important step in the utilization of these molecules for protein synthesis as well as nitrogen sources (Cunin et al. Citation1986). Polyamines are reported to be playing an important role in the growth of microorganisms. As they are cationic in nature they are also associated with nucleic acid and protein metabolism by binding to the negatively charged molecules (Stevens Citation1970). Nitrogen source and the polyamine pathways are reported to be affecting solvent yields significantly in many Clostridium species (Stutz et al. Citation2007).
Presence of adenylyl-sulphate kinase in the genome indicates the probable potential of the microbe to metabolize sulphur and its derivatives. Clostridium kluyveri is reported to incorporate selenium as selenomethionine (Se-Met) via the methionine biosynthesis pathway. It is observed that this incorporation might be linked to an extremely active sulphur metabolism (Seedorf et al. Citation2008).
4.5. CAZyme (carbohydrate-active enzymes) analysis
Animal digestive tracts/rumen compartments are a rich source of diverse microflora, where the nutrient source is major complex carbohydrates. Thus, microorganisms especially bacteria are a prolific source of CAZymes. Here, the results showed that the GH family 1, 13, and 23 are found to be abundant in the bacterial genome. The GH1 includes the enzyme β glucosidase aka the cellobiase and is involved in the hydrolysis of terminal, non-reducing β-d-glucosyl residues with release of β-d-glucose. The presence of abundant levels of this group reflects that C. sartagoforme plays a major role in the digestion of cellulose, a major component in the diet fed to the ruminant. The results also show the presence of GH 12 family coding for endo-1,4-β-d-glucanase enzyme which was earlier classified as cellulase family. Thus, the GH enzyme classification shows the presence of CDS for the first enzyme converting cellulose to cellobiose units in the cellulose utilization pathway on which the organism grew.
4.6. Genome features and metabolic pathways for the production of VFAs
VFAs like propanoate and butyrate are produced by rumen microflora and these are subsequently used in the process of milk production and also play a role in animal health maintenance contributing a major proportion of the daily energy requirement of ruminants (Bergman Citation1990). In our study, results show that the genome comprises CDS to convert 2 hydroxybutyrate to propionate in contrast to the succinate pathway which is usually well represented within the rumen ecosystem as per the metatranscriptome data (Sergeant et al. Citation2014). However, even in the corresponding metatranscriptome data, any CDS for methylmalonyl Co-A epimerase, which is involved in isomerization of (R) methylmalonyl-CoA to (S) methylmalonyl-CoA was found to be absent (unpublished data), suggesting incompleteness of the pathway and highlighting the significance of the pathway observed in the genome. As observed from the results, crotonyl-CoA seems to be the probable major intermediate in the pathway. Results thus confirm the role of organism in the production of VFA, further playing a vital role in animal homeostasis.
The potential to adapt to limiting nutrients is a universal trait in the microbial community most likely for Gram-positive, anaerobic, spore-forming bacterial species like Clostridium. CodY proteins are found in many low G+C content Gram-positive bacteria. They appear to have ability to repress genes whose products are not needed when nutrients are in excess. One interpretation of this effect is that CodY proteins help to regulate distribution of pyruvate and 2-oxoglutarate that are key intermediates in central metabolism (Sonenshein Citation2007). Functions regulated by CodY in various bacteria include carbon metabolism such as the Krebs cycle; synthesis of few amino acids, their uptake, and catabolism; genetic competence; motility; and sporulation. CodY proteins also have species-specific roles, particularly with respect to virulence gene expression (Dineen et al. Citation2007). As one of the reports show Clostridium difficile strains produce two large protein toxins, A and B, when nutrients become limiting and cells enter stationary phase. Gene tcdA and tcdB coding for these toxins lie within a 19.6 kb pathogenicity locus that includes the tcdR, tcdE, and tcdC genes. Expression studies of these genes depict link between nutrient limitation and toxin gene expression which is repressed by the global transcriptional regulator CodY. The strategy is widespread in Gram-positive pathogens and their implication in virulence in several such species due to the presence of CodY homologues in them (Dineen et al. Citation2010).
GCT is an enzyme central to the synthesis of teichoic acids, components of the cell wall in Gram-positive bacteria. CDP-glycerol:poly (glycerophosphate) glycerophosphotransferase and GCT participate glycerophospholipid metabolism with Glycerol-3-phosphate as substrate (Shaw Citation1962). Dpp is involved in peptide transport system and plays a role in initiation of spore-forming process (Sutcliffe & Russell Citation1995). LolC and LolD are ATP-binding cassette transporter proteins participating in the lipoprotein secretion and identified as catalysing the release of outer membrane-specific lipoproteins from the inner membrane of the microbe (Kemperman et al. Citation2003).
Genomic and phylogenetic analyses along with 16S rRNA and housekeeping gene similarities designate the isolate AAU1 phylogenetic placed within the Clostridium genus proximal to C. sartagoforme, corroborated by AN-Biolog and CAZyme profiling.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9:75–99. doi: 10.1186/1471-2164-9-75
- Bergman EN. 1990. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. Apr;70:567–590.
- Bettegowda C, Huang X, Lin J, Cheong I, Kohli M, Szabo SA, Zhang X, Diaz LA, Jr., Velculescu VE, Parmigiani G, et al. 2006. The genome and transcripto mes of the anti-tumor agent Clostridium novyi-NT. Nat Biotechnol. Dec;24:1573–1580. doi: 10.1038/nbt1256
- Bond DR, Tsai BM, Russell JB. 1998. The diversion of lactose carbon through the tagatose pathway reduces the intracellular fructose 1,6-bisphosphate and growth rate of Streptococcus bovis. Appl Microbiol Biotechnol. May;49:600–605. doi: 10.1007/s002530051220
- Cunin R, Glansdorff N, Pierard A, Stalon V. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. Sep;50:314–352.
- Dabrowski S, Zablotna E, Pietrewicz-Kubicz D, Dlugolecka A. 2012. Screening of environmental samples for bacteria producing 1,3-propanediol from glycerol. Acta Biochim Pol. 59:353–356.
- Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol. Oct;192:5350–5362. doi: 10.1128/JB.00341-10
- Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. J Mol Microbiol. Oct;66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x
- Drancourt M, Raoult D. 2005. Sequence-based identification of new bacteria: a proposition for creation of an orphan bacterium repository. J Clin Microbiol. Sep;43:4311–4315. doi: 10.1128/JCM.43.9.4311-4315.2005
- Ghanem FM, Ridpath AC, Moore WE, Moore LV. 1991. Identification of Clostridium botulinum, Clostridium argentinense, and related organisms by cellular fatty acid analysis. J Clin Microbiol. Jun;29:1114–1124.
- Hristov AN, Callaway TR, Lee C, Dowd SE. 2012. Rumen bacterial, archaeal, and fungal diversity of dairy cows in response to ingestion of lauric or myristic acid. J Anim Sci. Dec;90:4449–4457. doi: 10.2527/jas.2011-4624
- Huang WM. 1996. Bacterial diversity based on type II DNA topoisomerase genes. Annu Rev Genet. 30:79–107. doi: 10.1146/annurev.genet.30.1.79
- Hungate RE. 1957. Microorganisms in the rumen of cattle fed a constant ration. Can J Microbiol. Mar;3:289–311. doi: 10.1139/m57-034
- Hungate RE. 1966. In the rumen and its microbes. New York: Academic Press; p. 533.
- Kemperman R, Jonker M, Nauta A, Kuipers OP, Kok J. 2003. Functional analysis of the gene cluster involved in production of the bacteriocin circularin A by Clostridium beijerinckii ATCC 25752. Appl Environ Microbiol. Oct;69:5839–5848. doi: 10.1128/AEM.69.10.5839-5848.2003
- Kim P. 2004. Current studies on biological tagatose production using L-arabinose isomerase: a review and future perspective. Appl Microbiol Biotechnol. Aug;65:243–249.
- Kopecny J, Hodrova B, Stewart CS. 1996. The isolation and characterization of a rumen chitinolytic bacterium. Lett Appl Microbiol. Sep;23:195–198. doi: 10.1111/j.1472-765X.1996.tb00063.x
- Krieg NR, Holt JG, Sneath PHA, Staley JT, Williams ST. 1994. Bergey's manual of determinative bacteriology. 9th ed. Baltimore (MD): Williams & Wilkins.
- Lewis VJ, Moss CW, Jones WL. 1967. Determination of volatile acid production of Clostridium by gas chromatography. Can J Microbiol. Aug;13:1033–1040. doi: 10.1139/m67-138
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. Jan;42:D490–D495. doi: 10.1093/nar/gkt1178
- Muniz S, Lacarta J, Pata MP, Jimenez JJ, Navarro E. 2014. Analysis of the diversity of substrate utilisation of soil bacteria exposed to Cd and earthworm activity using generalised additive models. Plos One. 9:e85057. doi: 10.1371/journal.pone.0085057
- Nathani NM, Duggirala SM, Bhatt VD, KaPatel J, Joshi CG. 2014. Genomic analysis of a novel strain of Bacillus nealsonii, isolated from Surti buffalo rumen. Adv Biosci Biotechnol. 5:235–245. doi: 10.4236/abb.2014.53030
- Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, et al. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol. Aug;183:4823–4838. doi: 10.1128/JB.183.16.4823-4838.2001
- Orpin CG. 1975. Studies on the rumen flagellate Neocallimastix frontalis. J Gen Microbiol. Dec;91:249–262. doi: 10.1099/00221287-91-2-249
- Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. 2010. CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. Dec;20:1574–1584. doi: 10.1093/glycob/cwq106
- Seedorf H, Fricke WF, Veith B, Bruggemann H, Liesegang H, Strittmatter A, Miethke M, Buckel W, Hinderberger J, Li F, et al. 2008. The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc Natl Acad Sci USA. Feb 12;105:2128–2133.
- Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. 2014. Extensive microbial and functional diversity within the chicken cecal microbiome. Plos One. 9:e91941. doi: 10.1371/journal.pone.0091941
- Shaw DR. 1962. Pyrophosphorolysis and enzymic synthesis of cytidine diphosphate glycerol and cytidine diphosphate ribitol. Biochem J. Feb;82:297–312. doi: 10.1042/bj0820297
- Simunek J, Hodrova B, Bartonova H, Kopecny J. 2001. Chitinolytic bacteria of the mammal digestive tract. Folia Microbiol. 46:76–78. doi: 10.1007/BF02825892
- Smuts HE, Lastovica AJ. 2011. Molecular characterization of the 16S rRNA gene of Helicobacter fennelliae isolated from stools and blood cultures from paediatric patients in South Africa. J Pathog. 2011:1–9. doi: 10.4061/2011/217376
- Sonenshein AL. 2007. Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol. Dec;5:917–927. doi: 10.1038/nrmicro1772
- Stevens L. 1970. The biochemical role of naturally occurring polyamines in nucleic acid synthesis. Biol Rev Camb Philos Soc. Feb;45:1–25. doi: 10.1111/j.1469-185X.1970.tb01073.x
- Stutz HE, Quixley KW, McMaster LD, Reid SJ. 2007. Co-regulation of the nitrogen-assimilatory gene cluster in Clostridium saccharobutylicum. Microbiology. Sep;153:3081–3090. doi: 10.1099/mic.0.2007/005371-0
- Sutcliffe IC, Russell RR. 1995. Lipoproteins of gram-positive bacteria. J Bacteriol. Mar;177:1123–1128.
- Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. Aug;24:1596–1599. doi: 10.1093/molbev/msm092
- Yu PL, Hodge RA, Li XP. 1990. In vitro expression of Lac-PTS and tagatose 1,6-bisphosphate aldolase genes from Lactococcus lactis subsp. cremoris plasmid pDI-21. Appl Microbiol Biotechnol. Sep;33:677–679. doi: 10.1007/BF00604936