
ABSTRACT
Endothelin-1 (ET-1) is relevant to the pathogenesis of pulmonary arterial hypertension (PAH) in several species, including broiler chickens. In ET-1-induced vasoconstriction, L-type voltage-operated calcium channels (VOCCs) play a crucial role. In this study, the effect that ET-1 and the calcium channel blocker (CCB) nifedipine have on ET-1-induced vascular contraction in pulmonary artery rings of both, non-pulmonary hypertensive chickens (NPHC) and pulmonary hypertensive chickens (PHC), was evaluated. A significant increase response in ET-1 was observed for PHC vascular rings with respect to those of NPHC (p < .05). Incubation with 1 μM nifedipine did not completely reverse the contraction induced by 1 × 10−7 M ET-1 in both NPHC and PHC rings, suggesting the participation of additional calcium sources procured via influx through L-type VOCC. Nifedipine reduced, to a higher degree, the contraction induced by ET-1 in NPHC (50%) when compared with that observed in PHC rings (30.7%). Thus, the ET-1-induced vasoconstriction processes are more efficient in PHC, and the influx of calcium into the cell through the L-type VOCC is presumably only a fraction of the necessary calcium for ET-1-induced vasoconstriction. It is suggested that blocking calcium uptake through alternative pathways (receptor-operated calcium channels and storage-operated calcium channels) may produce a vasodilation effect of considerable therapeutic significance in PHC.
1. Introduction
Chronic hypoxia causes disturbances in calcium homeostasis regulatory mechanisms, which is a key to understanding the pathogenesis of pulmonary arterial hypertension (PAH). In this process, the involvement of ionic channels and endothelial dysfunction play a determining role. An increase in endothelin-1 (ET-1) synthesis seems to be critical to the changes in endothelial function. ET-1 is considered to be one of the most relevant vasoconstrictors in the physiopathological process of PAH (Shimoda, Sham, Shimoda et al. Citation2000; Weigand et al. Citation2006). This peptide is also involved in other vascular pathologies, such as subarachnoid haemorrhage, cerebral vasospasm (Kawanabe et al. Citation2002), atherosclerosis, preeclampsia (Okatani et al. Citation1995), hepatic cirrhosis, and cardiac failure (Kawanabe & Nauli Citation2005). Higher ET-1 expression is found in the lungs of mammals and chickens with PAH than in subjects free of disease (Giaid et al. Citation1993; Michel et al. Citation2003; Gómez et al. Citation2007).
ET-1 induces vasoconstriction through pathways that increase the concentration of free cytosolic Ca2+, ([(Ca2+)i]), and determining the channels permeable to this cation is worthwhile. To date, two ET-1 receptors with therapeutic potential have been identified: ETA and ETB. The ETA receptor is located in vascular smooth muscle cells (VSMC) where it induces vasoconstriction and mitosis. The ETB receptor is located in both endothelial and VSMC, where it activates vasodilatation and vasoconstriction, respectively (Yanagisawa et al. Citation1988; Pollock Citation1998). There are two subtypes of the ETB receptor, denoted as ETB1 and ETB2. The ETB1 receptor is present in the endothelium and is responsible for the liberation of vasodilatation factors, such as nitric oxide (NO) and prostacyclin (PGI2), whereas ETB2 is localized in VSMC and has vasoconstrictor activity (Warner et al. Citation1993; Gray et al. Citation1994; Sudjarwo et al. Citation1994). In the normal pulmonary vasculature, ETA and ETB receptors are differentially distributed. ETA receptors predominate in the proximal arteries and ETB ones in the small peripheral pulmonary arterioles (Michel et al. Citation2003). In certain cardiovascular diseases, ETB2 receptors are overexpressed while the number of ETB1 receptors is reduced, which is directly related to an increase in contraction and subsequent decrease in vasodilatation (Dupuis et al. Citation2000; Channick et al. Citation2004; Opitz & Ewert Citation2006).
The contractile mechanism induced by ET-1 begins with the activation of the ETA receptors located in VSMC, in turn inducing the activation of phospholipase C through a G protein to produce 1,4,5 inositol triphosphate (IP3). This metabolite produces an increase in [(Ca2+)i] through the liberation of Ca2+ from intracellular deposits, such as the sarcoplasmic reticulum. Liberation is accomplished through the opening of store-operated calcium channels (SOCCs) (Takuwa et al. Citation1990; Ohlstein et al. Citation1996). Subsequently, VSMC membrane depolarization occurs via a process initiated by the inactivation of the voltage-dependent potassium channels (Kv), activated by Ca2+ (KCa2+) or ATP (KATP) (Goto et al. Citation1989; Nelson & Quayle Citation1995; Shimoda et al. Citation1998; Al-Jiffry et al. Citation2001). These ionic changes induce modifications in the membrane potential, in turn allowing the activation of voltage-operated calcium channels (VOCCs), which is responsible for the sustained increase in [(Ca2+)i].
The above may explain why ET-1-induced vasoconstriction is considered biphasic and concentration dependent. Thus, concentrations above 10−9 M lead to an increase in [(Ca2+)i] through the liberation of Ca2+ from intracellular deposits, which induces a short and transient contraction phase, and also through the activation of VOCC, which induces a higher, sustained increase in [(Ca2+)i]. Thus, a contraction phase with a longer duration and higher magnitude is produced (Takuwa et al. Citation1990; Komuro et al. Citation1997; Minowa et al. Citation1997; Zhang et al. Citation1998). In contrast, lower ET-1 concentrations only induce the sustained phase; that is, the contraction in this case is mainly dependent on extracellular Ca2+ (Komuro et al. Citation1997; Kawanabe et al. Citation2002). This result implies the involvement of the L-type Ca2+ channels, which are involved in the main pathway for the Ca2+ required to trigger muscular contraction. These channels are characterized by a high threshold, slow inactivation, and acute sensitivity to dihydropyridine-type (DPH) Ca2+ channel blockers (CCBs) (Katz Citation1997; Lipscombe et al. Citation2004).
Although the L-type VOCC is the main Ca2+ uptake pathway to trigger an ET-1-induced contraction, the involvement of the Ca2+ channels activated through stimuli independent of cellular depolarization was also observed. These are Ca2+ storage-operated channels (SOCCs), also known as capacitative Ca2+ entry channels. Their opening occurs via a reduction in the Ca2+ concentration in the ER lumen after the activation of IP3 receptors and ryanodine receptors. Another type of Ca2+ channel that activates ET-1 is the receptor-operated Ca2+ channel (ROCC) (Takuwa et al. Citation1990; Ohlstein et al. Citation1996), located in the plasma membrane. Their opening occurs via the binding of its agonist (diacylglycerol through protein kinase-C [PKC]) to its receptor (Miwa et al. Citation2005).
Thus, the role of ET-1 in the physiopathology of PAH is a crucial one and includes the ability of this substance to generate a strong, sustained contractile stimulus through the activation of L-type VOCCs. Based on this finding and the results of previous studies showing a higher expression of ET-1 in broiler chickens with PAH (Gómez et al. Citation2007), we wanted to determine the physiological response of the pulmonary vasculature to this substance. In this study, the in vitro effect of ET-1 on the contractility of pulmonary artery rings from both pulmonary hypertensive chickens (PHC) and non-pulmonary hypertensive chickens (NPHC) was investigated. In addition, the contractile response of the pulmonary artery rings to the DPH-type CCB, ‘nifedipine’, induced by ET-1 was studied to understand the potential changes in the behaviour of the pulmonary vasculature affected by PAH.
2. Materials and methods
2.1. Drugs and solutions
The nifedipine and ET-1 were procured from Sigma-Aldrich (St. Louis, MO, USA). ET-1 was diluted (10−7 M) in distilled water and stored at −20°C, according to the manufacturer's recommendations. The nifedipine was diluted in dimethyl sulphoxide (DMSO) to yield a 1 micromolar (µM) solution after the addition to the tissue organ bath and was kept refrigerated at 4°C until use. The quantities of DMSO added to the tissue organ bath did not change the contractile response of the vascular rings. Sodium lights were used to protect the solutions containing nifedipine from photodegradation. The composition of the Krebs–Henseleit solution was the following, in mM: 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3 and 5.6 glucose.
2.2. Experimental protocol
A total of 300 one-day-old male broiler chickens from the Cobb 500 genetic line were kept in the Poultry Research Building, located in Bogotá (Colombia), at an altitude of 2638 m above sea level. A storehouse was used, with the temperature automatically set according to the following protocol: days 1 to 6: 32–33°C; days 7 to 13: 29–30°C; days 14 to 20: 27–28°C; days 21 to 27: 24–26°C; days 28 to 34: 21–23°C and days 35 to 41: 19–21°C (Cobb Broiler Management Guide Citation2008). These chickens were not subject to any kind of medical treatment and were kept under standard broiler chicken farm conditions. The experiments were performed after four weeks of age.
2.3. Obtainment of pulmonary artery rings
The animals were subjected to procedures approved by the bioethics committee of the Veterinary Medicine and Animal Husbandry Faculty of the National University of Colombia. One or two pulmonary rings were retrieved from each bird after sacrifice. The cardiac index (CI), which is the right ventricle weight divided by the total ventricular weight and multiplied by 100, was computed for all of the animals (Alexander & Jensen Citation1959). Broilers with IC≤25% were considered to be non-hypertensive, and chickens with IC ≥ 26% were considered to be hypertensive according to the conclusions derived for the Cobb 500 line from previous work performed in Bogota, Colombia (Areiza et al. Citation2011; Vásquez & Hernández Citation2011).
The pulmonary artery was dissected after the animals' sacrifice and deposited in a container in Krebs–Henseleit solution at a constant temperature of 42°C that was kept bubbling with a carbogen gas mixture (5% CO2 and 95% O2). The artery was later sliced into 3–5 mm-long rings, and each ring was attached at both ends with stainless steel wires in a tissue organ bath containing a Krebs–Henseleit solution (20 ml) at a constant temperature of 42°C and kept bubbling with the carbogen gas mixture. One of the wires was attached to the tissue organ bath, and the other wire was attached to an isometric transducer (MLT0202 Panlab, Spain, 2008) that was connected to a data acquisition unit (PowerLab). Labchart 6 software (AD Instruments, Sydney, Australia, 2008) was used for the data acquisition. The arterial rings were stabilized for approximately 30 min, changing the media every 5 min. A basal tension of 1 g (Ramírez et al. Citation2007; Álvarez et al. Citation2012) was obtained after the vascular rings were subjected to a change in the Krebs–Henseleit solution every 5 min, and no changes in the basal tension were observed
2.4. Isometric tension measurements
The vascular rings assembly was carried out, and the set-up was confirmed to be in working order. All pulmonary artery rings were initially subjected to a 40 mM KCl solution to obtain the maximum possible contraction, with a duration ranging from 80 to 100 min and ending once a stable contractile response of the vessel was obtained. This tension was defined as 100% and taken as the reference for the evaluation of the subsequent responses, which were reported as relative quantities. Then, the rings were washed with Krebs–Henseleit solution every 5 mins to obtain vessel relaxation to the basal tension values. The maximum vasoconstriction was determined for the vasoconstrictive substances employed in all the experiments after a minimum lapse of 10 min without changes in the arterial ring pressure.
The protocols employed are described as follows:
Effect of the ET-1 induced contraction in NPHC and PHC pulmonary artery rings
The vasculature rings from NPHC and PHC were initially incubated in 40 mM KCl and washed, and their basal tension stabilized. After resting for 30 min, the rings were incubated with the vasoconstrictor ET-1 at 1 × 10−7 M (Goto et al. Citation1989; Linder & Bendhack Citation2002). Rings from NPHC (n = 15). Rings from PHC (n = 9).
Blocking effect of the L-type VOCC on the contractile response induced by ET-1 in the NPHC pulmonary artery rings
The DPH antagonist nifedipine (1 µM) was used to establish the blocking effect of the L-type VOCC on the ET-1-induced vasoconstriction. The concentration of 1 µM was selected based on several studies (Godfraind Citation1983; Bialecki et al. Citation1998; Weigand et al. Citation2006). After induction of the maximum contractile response with 40 nM KCl and the subsequent stabilization to the basal tension level, two protocols were established. In protocol 1 (n = 15), the vessels were maintained for 30 min without any treatment. In protocol 2 (n = 9), the vessels were incubated with nifedipine (1 µM). Finally, the rings in both protocols were exposed to 10−7 M ET-1 (Goto et al. Citation1989; Linder & Bendhack Citation2002).
Blocking effect of the L-type VOCC on the ET-1-induced contractile response in the PHC pulmonary artery rings
The previous procedure was followed in the PHC rings. Protocol 1 (n = 9). Protocol 2 (n = 10).
3. Statistical analysis
Comparisons among the treatments were performed using Student's t-test. The differences were considered statistically significant at p < .05 (*) and highly significant at p < .01 (**). The values are given as the mean ± standard deviation. The StatView 5.0 software was used for the statistical analysis (San Francisco, CA, EEUU 2007).
4. Results
4.1. ET-1-induced contraction in the NPHC pulmonary artery rings
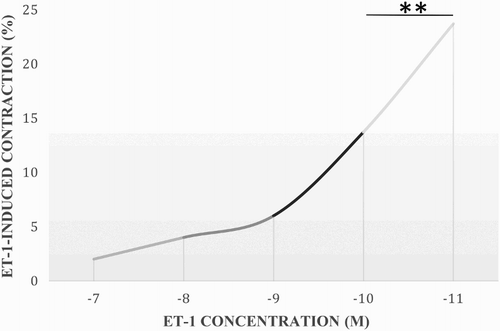
A dose–response curve was created to determine the concentration of ET-1 necessary to induce a significant vessel contraction of rings obtained from NPHC. The concentrations ranged from 1 × 10−11 to 1 × 10−7 M. The latter one induced a constant contraction that was reproducible and significant (). The difference between the concentrations of 10−7 and 10−8 M was highly significant (p < .01).
Figure 1. ET-1 dose–response curve. The contraction in the pulmonary arteries of NPHC was induced with concentrations ranging from 10-11 to 10-7 M. Results are expressed as a percentage of the maximum contractile response induced by 40 mM KCl. **Statistical difference between treatments (p < .01).
4.2. Comparison of the effect caused by the ET-1-induced contraction in the NPHC and PHC pulmonary artery rings
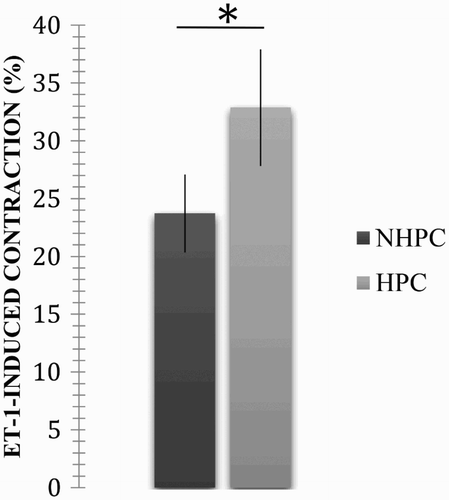
A concentration of 10−7 M was used to compare the response in the rings from the NPHC and PHC because it produced a highly reproducible contraction. An average contraction of 23.72 ± 6.75% was obtained in the rings from the NPHC. This response was statistically significant (p < .05; p = .0324) with respect to that observed in the rings from the PHC (average: 32.87 ± 10.07%, error: 3.36; see ).
Figure 2. Effect caused by the ET-1-induced contraction in the NPHC and PHC pulmonary artery rings. The results are expressed as a percentage of the maximum contractile response induced by 40 mM KCl. *A statistically significant difference between treatments (p < .05).
4.3. Effect of incubation with nifedipine on ET-1 induced contractile response in the NPHC pulmonary artery rings
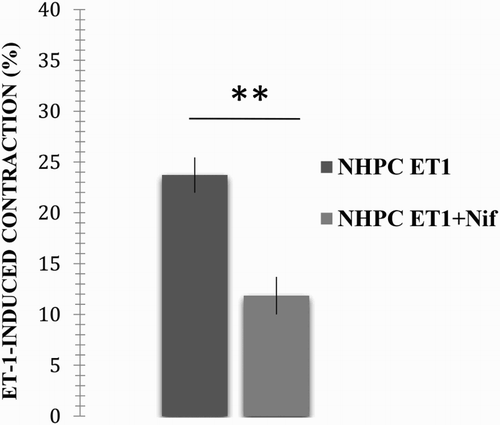
The rings from the NPHC were incubated for 30 min with the L-type VOCC antagonist, nifedipine, to determine the blocking effect of the L-type channels on the ET-1-induced contraction. The rings were incubated before the ET-1-induced contraction, and the response was compared with the ET-1-induced contraction in the absence of nifedipine. ET-1 induced an average contraction of 23.72 ± 6.75% (error: 1.74). When compared with the average contraction obtained in the presence of DPH (11.85 ± 5.57%, error: 1.86), the difference was highly significant (p < .01, p = .00014; see ).
Figure 3. Effect caused by incubation with nifedipine on the ET-1-induced contractile response in the NPHC pulmonary artery rings. Vascular rings from the NPHC were contracted in the presence of 1 × 10−7 M ET-1, and the response after incubation with and without 1 µM nifedipine was compared. The results are expressed as a per cent of the maximum contractile response induced by 40 mM KCl. **A highly significant difference between treatments (p < .01).
4.4. Effect caused by incubation with nifedipine on the ET-1-induced contractile response in the PHC pulmonary artery rings
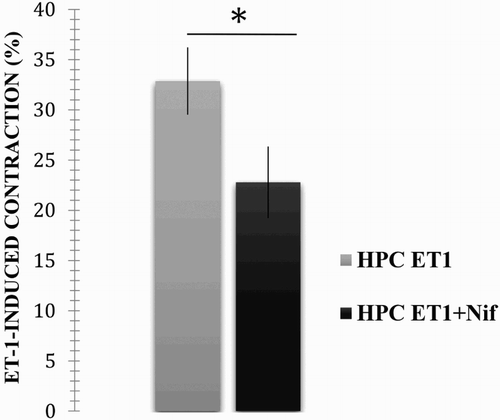
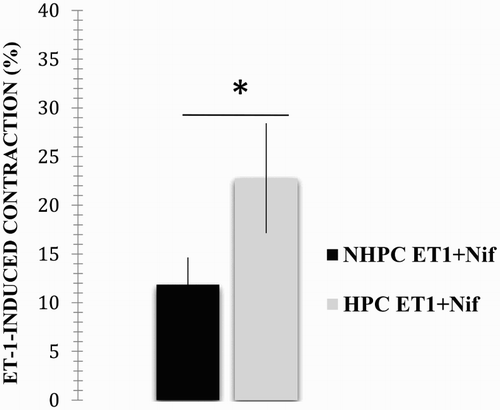
The following results were obtained when using the same methodologies of the previous protocol. In the absence of nifedipine, ET-1 induced an average contraction of 32.87 ± 10.07%, error: 3.36. This response was highly decreased in the presence of nifedipine (average: 22.78 ± 11.28 %, error: 3.57; p < .05, p = .05; see ), as in the case of the healthy chickens.
Figure 4. Effect caused by incubation with nifedipine on the ET-1-induced contractile response of PHC pulmonary artery rings. The vascular rings from the PHC were contracted in the presence of 10−7 M ET-1, and the response after incubation with and without 1 µM nifedipine was compared. The results are expressed as a per cent of the maximum contractile response induced by 40 mM KCl. *A significant difference between treatments (p < .05).
4.5. Effect caused by incubation with nifedipine on the ET-1-induced contractile response in the NPHC and PHC pulmonary artery rings
The contractile responses of the rings from the NPHC and PHC were compared to determine any differences in the response induced by nifedipine in the contraction induced by 10−7 M ET-1. The presence of antagonists in the rings from NPHC resulted in an average contraction of 11.85 ± 5.57% (error: 1.86). With respect to the response by the PHC rings (average contraction 22.78 ± 11.28%, error: 3.57), the difference is clear and statistically significant (p < .05, p = .0175) when the average is compared with that obtained from the rings of non-hypertensive animals ().
Figure 5. Effect caused by incubation with nifedipine on the ET-1-induced contraction in the pulmonary artery rings of NPHC and PHC. The results are expressed as a per cent of the maximum contractile response induced by 40 mM KCl. *A statistically significant difference (p < .05).
5. Discussion
This study is likely one of the first to examine and compare the responses in PHC and NPCH pulmonary artery rings to ET-1. Moreover, the effect of nifedipine in vasodilatation is described for the first time in the pulmonary artery rings from broiler chickens, adding to our understanding of the crucial roles played by ET-1 and the L-type VOCC in the physiopathology of PAH.
The results from the current study showed statistically significant differences in comparing the response in the vascular rings from NPHC and PHC caused by an ET-1-induced contraction, which is higher in PHC. These findings are similar to those reported by Barman (Citation2007), who employed isolated pulmonary arteries from rats with PAH induced after placement in hypoxia chambers for 12 weeks.
The higher reactivity observed in rings from hypertensive subjects may be due to an increase in the sensitivity of their vasculature to ET-1, which could be related to changes in the number and distribution of the ETA and ETB receptors that are present in cardiovascular pathologies such as PAH. Such changes include the following: the overexpression of ETA receptors in VSMS (McCulloch et al. Citation1998; Weigand et al. Citation2006); the overexpression of the ETB2 receptor in small peripheral pulmonary arterioles (Channick et al. Citation2004; Opitz & Ewert Citation2006); and the decreased expression of the ETB1 receptor, which is directly associated with the reduction of endothelium-dependent vasorelaxation due to a reduction in the synthesis of vasodilators, such as NO and PGI2 (Adnot et al. Citation1991; Inagami et al. Citation1995; Mam et al. Citation2010)
In addition to ET-1, serotonin (5-HT), angiotensin II (Ang II), noradrenaline, and histamine are considered to be substances that induce an increase in the reactivity of the pulmonary vasculature under chronic hypoxic conditions (Shimoda, Sham, Sylvester Citation2000). The increase in the vasoreactivity of hypoxic PAH could also be due to abnormalities found in pulmonary VSMC, such as the following: increased [(Ca2+)i] at resting levels (Shimoda, Sham, Shimoda et al. Citation2000), the inhibition of K+ (Kv) channels due to the reduced expression of their alpha units (Wang et al. Citation1997); and the decreased activity of the K+ (KCa) channels (Peng et al. Citation1997). The change in K+ channels due to hypoxia may explain the increased effectiveness of agonists such as ET-1 in the pulmonary arteries of animals subjected to chronic hypoxia than in individuals exposed to normoxia (Shimoda, Sham, Sylvester Citation2000).
In contrast to the results obtained in this study, Bialecki et al. (Citation1998) and Itoh et al. (Citation1999) reported a lower response in the pulmonary artery rings of rats exposed to chronic hypoxia. The behaviour of the pulmonary vasculature in those rodents could be due to factors such as the change in the number and affinity of receptors or the metabolism of the ET-1 agonist. Some authors have discussed a possible change in the activation of ETA receptors and/or an increase in the response mediated by the ETB1 receptor (Mam et al. Citation2010). Differences among species cannot be overlooked in this context. Contradictory results were also found in numerous studies using systemic hypertension models with contracted arterial rings (De Carvalho et al. Citation1990; Lin et al. Citation1990; Laurant & Berthelot Citation1996; Linder & Bendhack Citation2002). Disparities in the results among the different analysed models may be due to the different types of PAH induction, the species studied, and particular vasculature batches, as differences can even be found within the same model.
The contractile mechanism induced by ET-1 implies the involvement of certain cationic channels that are permeable to Ca2+. When activated, these channels produce an increase in the [(Ca2+)i] through the Ca2+ influx from the cell exterior and through the liberation of intracellular deposits, such as from the ER. The Ca2+ channels indirectly activated by ET-1 are the VOCCs. However, other Ca2+ channels, which open independent of voltage change and are DPH-resistant, are also involved, and they are the SOCCs and ROCCs, some of which can be activated by ET-1 (Kawanabe et al. Citation2002; Miwa et al. Citation2005). When the ET-1-induced vasocontractile response was taken into account, the effects caused by DPH-type CCBs, such as nifedipine, were studied in the pulmonary vasculature of species, including the rat (Weigand et al. Citation2006), pig (Janssen et al. Citation2004), and human (Hall et al. Citation2009). In the current study, 1 μM nifedipine was added to the incubation of the NPHC and PHC pulmonary artery rings. The inactivation of the VOCCs due to nifedipine did not completely cancel the response by ET-1 in both NPHC and PHC, which may be explained by ET-1-induced contractile mechanisms, in which the observed [(Ca2+)i] increase not only arises from the extracellular Ca2+ uptake through the L-type VOCC but also from other Ca2+ sources that are independent of changes in the membrane potential. These alternative sources are also resistant to nifedipine, as in the case of Ca2+ liberation from the ER through the activation of SOCCs and the case of Ca2+ influx from the cell exterior activated by binding of an agonist (ET-1) to its receptor through ROCCs (Miwa et al. Citation2005).
The greatest decrease in the vascular ring response corresponded to NPHC (50%) compared with the PHC (30.7%) when analysing the effect caused by nifedipine on the ET-1-induced contraction in the arterial rings from the NPHC and PHC. This result strongly suggests the involvement of Ca2+ sources other than those activated due to membrane depolarization. However, the different response in the rings of healthy subjects compared with the hypertensive ones shows a type of over-regulation of voltage-independent channels, such as the previously mentioned SOCCs and ROCCs (Miwa et al. Citation2005). Lin et al. (Citation2004) showed that the exposure to chronic hypoxia over-regulates the expression of certain members of the cationic channel protein family, such as transient receptor potential cation channels (TPRC). Members of this family are essential components of the ROCCs and SOCCs (Gosling et al. Citation2005; Patel et al. Citation2010). The different TPRC types reportedly overexpressed in PAH are the TPCR-1, considered to be a SOCC-type, and the TPRC-6, considered to be a ROCC-type. Therefore, considering that hypoxia increases the expression of certain specific types of TPRC (TPRC-1 and TPRC-6) (Lin et al. Citation2004; Wang et al. Citation2006; Abramowitz & Birnbaumer Citation2009) and that these channels are activated by ET-1, an increase in these types of channels and ET-1 overexpression will clearly play a critical role in an increase in the vascular tone and in the proliferation of VSMC, which are fundamental processes in the development of PAH.
Although the influx of extracellular Ca2+ through the VOCC plays a significant role in the development of the ET-1-induced contraction, the results from the current study provide evidence of the importance of other sources of Ca2+ in the physiopathology of hypoxic PAH in broiler chickens. These findings may support recent efforts to enlist novel pharmacological alternatives in the treatment of PAH, having therapeutic targets other than the fundamental mechanisms in the development of the disease, such as ET-1 receptor antagonists (Itoh et al. Citation1999; Opitz & Ewert Citation2006). However, the use of [(Ca2+)i] inhibitors through the inactivation of ROCCs and SOCCs may have some therapeutic value. Nevertheless, performing additional studies of molecules with this activity to specify the precise involvement of Ca2+ originating from sources different than the L-type channels in hypoxic vasoconstriction will be necessary.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Abramowitz J, Birnbaumer L. 2009. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 23:297–328. doi: 10.1096/fj.08-119495
- Adnot S, Raffestin B, Eddhahibi S, Braquet P, Chabrier P. 1991. Loss of endothelium-dependent relaxant activity in the pulmonary circulation of rats exposed to chronic hypoxia. J Clin Invest. 87:155–162. doi: 10.1172/JCI114965
- Alexander AF, JENSEN R. 1959. Gross cardiac changes in cattle with high mountain disease and in experimental cattle maintained at high altitudes. Am J Vet Res. 20:680–689.
- Al-jiffry BO, Meedeniya ACB, Chen JW, Toouli J, Saccone GTP. 2001. Endothelin-1 induces contraction of human and Australian possum gallbladder in vitro. Regul Peptides. 102:31–39. doi: 10.1016/S0167-0115(01)00302-0
- Álvarez D, Hernández A, Orozco C. 2012. Endothelial hyperpolarizing factor increases acetylcholine-induced vasodilatation in pulmonary hypertensive broilers arterial rings. Res Vet Sci. 92:1–6. doi: 10.1016/j.rvsc.2011.02.004
- Areiza RA, Rivas PC, Hernández A. 2011. A quantitative study of the pulmonary vascular bed and pulmonary weight: Body weight ratio in chickens exposed to relative normoxia and chronic hypobaric hypoxia. J Poult Sci. 48:267–274. doi: 10.2141/jpsa.011030
- Barman SA. 2007. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol. 293:L472–L479. doi: 10.1152/ajplung.00101.2006
- Bialecki RA, Fisher CS, Murdoch WW, Barthlow HG, Stow RB, Mallamaci M, Rumsey W. 1998. Hypoxic exposure time dependently modulates endothelin induced contraction of pulmonary artery smooth muscle. Am J Physiol. 274 (Lung Cell. Mol. Physiol. 18):L552–L559.
- Broiler Management Guide. 2008. Cobb 500. Cobb.Vantress.com.
- Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. 2004. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol. 43:S62–S67. doi: 10.1016/j.jacc.2004.02.042
- De Carvalho MH, Nigro DR, Barbeiro HV, De Oliveira MA, De Nucci G, Fortes ZB. 1990. Comparison of the effect of endothelin on microvessels and macrovessels in Goldblatt II and deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 15:I68–I68. doi: 10.1161/01.HYP.15.2_Suppl.I68
- Dupuis J, Jazmin JF, Prie S, Cernacek P. 2000. Importance of local production of endothelin-1 and the of the ETB receptor in the regulation of pulmonary vascular tone. Pulm Pharmacol Ther. 13:135–140. doi: 10.1006/pupt.2000.0242
- Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. 1993. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 328:1732–1739. doi: 10.1056/NEJM199306173282402
- Godfraind T. 1983. Actions of nifedipine on calcium fluxes and contraction in isolated rat arteries. JPET. 224:443–450.
- Gómez AP, Moreno MJ, Iglesias A, Coral PX, Hernández A. 2007. Endothelin 1, its endothelin type A receptor, connective tissue growth factor, platelet-derived growth factor, and adrenomedullin expresión in lungs of pulmonary hypertensive and nonhypertensive chickens. Poult Sci. 86:909–916. doi: 10.1093/ps/86.5.909
- Gosling M, Poll C, Li S. 2005. TRP channels in airway smooth muscle as therapeutic targets. Naunyn-Schmiedebergs Arch Pharmacol. 371:277–284. doi: 10.1007/s00210-005-1058-2
- Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, Kimura S, Yanagisawa M, Masaki T. 1989. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci USA. 86:3915–3918. doi: 10.1073/pnas.86.10.3915
- Gray GA, Loffler BM, Clozel M. 1994. Characterization of endothelin receptors mediating contraction of rabbit saphenous vein. Am J Physiol. 266:H959–966.
- Hall J, Jones TH, Channer KS, Jones RD. 2009. Mechanisms of agonist-induced constriction in isolated human pulmonary arteries. Vasc Pharmacol. 51:8–12. doi: 10.1016/j.vph.2009.01.007
- Inagami T, Mitsuhide N, Hoover R. 1995. Endothelium as an endocrine organ. Annu Rev Physiol 57:171–189. doi: 10.1146/annurev.ph.57.030195.001131
- Itoh H, Yokochi A, Yamauchi-Kohno R, Maruyama K. 1999. Effects of the endothelin ETA receptor antagonist, TA-0201, on pulmonary arteries isolated from hypoxic rats. EJP. 376:233–238.
- Jannsen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee, S. 2004. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 287:L852–L858. doi: 10.1152/ajplung.00130.2004
- Katz AM. 1997. Molecular Biology of Calcium Channels in the Cardiovascular System. Am J Cardiol. 80:17I–22I. doi: 10.1016/S0002-9149(97)00792-3
- Kawanabe Y, Hashimoto N, Masaki, T. 2002. Characterization of Ca2+ channels involved in endothelin-1-induced contraction of rabbit basilar artery. J Cardiovasc Pharmacol. 40:438–447. doi: 10.1097/00005344-200209000-00013
- Kawanabe Y, Nauli SM. 2005. Involvement of extracellular Ca2+ influx through voltage-independent Ca2+ channels in endothelin-1 function. Cellular Signaling. 17:911–916. doi: 10.1016/j.cellsig.2005.01.001
- Komuro T, Miwa S, Zhang X-F, Minowa T, Enoki T, Kobayashi S, Okamoto Y, Ninomiya H, Sawamura T, Kikuta K, et al. 1997. Physiological role of Ca2+ permeable nonselective cation channel in endothelin-1-induced contraction of rabbit aorta. J Cardiovasc Pharmacol. 30:504–509. doi: 10.1097/00005344-199710000-00015
- Laurant P, Berthelot A. 1996. Endothelin-1-induced contraction in isolated aortae from normotensive and DOCA-salt hypertensive rats: effect of magnesium. Brit J Pharmacol. 119:1367–1374. doi: 10.1111/j.1476-5381.1996.tb16048.x
- Lin SY, Cai H, Gong QY, Yang ZC, Zhou JH, Zhang D, Zhang ZK. 1990. Role of endothelin-1 in the pathogenesis of hypertension in spontaneously hypertensive and 2 kidneys 1 clip rats. Chin Med J (Engl). 103:748–753.
- Lin M-J, Leung GPH, Zhang W-M, Yang X-R, Yip K-P, Tse C-M, Sham JSK. 2004. Chronic hypoxia-induced upregulation os store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells. Circ Res. 95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad
- Linder AE, Bendhack LM. 2002. Endothelin-1 induced contraction is impaired in the tail artery of renal hypertensive rats. Vasc Pharmacol. 39:77–82. doi: 10.1016/S1537-1891(02)00282-3
- Lipscombe D, Helton TD, Xu W. 2004. L-type calcium channels: the low down. J Neurophysiol. 92:2633–2641. doi: 10.1152/jn.00486.2004
- Mam V, Tanbe AF, Vitali SH, Arons E, Christou HA, Khalil RA. 2010. Impaired vasoconstriction and nitric oxide-mediated relaxation in pulmonary arteries of hypoxia- and monocrotaline-induced pulmonary hypertensive rats. JPET. 332:455–462. doi: 10.1124/jpet.109.160119
- McCulloch KM, Docherty CH, Maclean MR. 1998. Endothelin receptors mediating contraction of rat and human pulmonary resistance arteries: effect of chronic hypoxia in the rat. Brit J Pharmacol. 123:1621–1630. doi: 10.1038/sj.bjp.0701785
- Michel RP, Langleben D, Dupuis J. 2003. The endothelin system in pulmonary hypertension. Can J Physiol Pharmacol. 81:542–554. doi: 10.1139/y03-008
- Minowa T, Miwa S, Kobayashi S, Enoki T, Zhang X-F, Komuro T, Iwamuro Y, Masaki T. 1997. Inhibitory effect of nitrovasodilators and cyclic GMP on ET-1 – activated Ca2+-permeable nonselective cation channel in rat aortic smooth muscle cells. Brit J Pharmacol. 120:1536–1544. doi: 10.1038/sj.bjp.0701059
- Miwa S, Kawanabe Y, Okamoto Y, Masaki T. 2005. Ca2+ entry channels involved in endothelin-1-induced contractions of vascular smooth muscle cells. J Smooth Muscle Res. 41:61–75. doi: 10.1540/jsmr.41.61
- Nelson MT, Quayle JB. 1995. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 268:799–822.
- Ohlstein EH, Elliott JD, Feuerstein GZ, Ruffolo Jr RR. 1996. Endothelin receptors classification, novel receptor antagonist, and potential therapeutic targets. Med Res Rev. 16:365–390. doi: 10.1002/(SICI)1098-1128(199607)16:4<365::AID-MED4>3.0.CO;2-V
- Okatani Y, Taniguchi K, Sagara Y. 1995. Amplifying effect of endothelin-1 on serotonin-induced vasoconstriction of human umbilical artery. Am J Obstet Gynecol. 172:1240–1245. doi: 10.1016/0002-9378(95)91486-2
- Opitz CF, Ewert R. 2006. Dual ETA/ETB vs. selective ETA endothelin receptor antagonism in patients with pulmonary hypertension. Eur J Clin Invest. 36:1–9. doi: 10.1111/j.1365-2362.2006.01691.x
- Patel, A., Sharif-Naeini R, Folgering JRH, Bichet D, Duprat F, Honoré E. 2010. Canonical TPR channels and mechanotransduction: from physiology to disease states. Pflugers Arch–Eur J Physiol. 460:571–581. doi: 10.1007/s00424-010-0847-8
- Peng W, Hoidal JR, Karwande SV, Farrukh IM. 1997. Effect of chronic hypoxia on K+ channels: regulation in human pulmonary vascular smooth muscle cells. Am J Physiol. 272:C1271–C1278.
- Pollock DM. 1998. Endothelin: molecular biology, physiology, and pathology. In Endothelin receptor subtypes and tissue distribution. Totowa: Humana Press. p. 8.
- Ramírez JH, Palacios M, Gutiérrez O. 2007. Implementation of the isolated vascular organ technique as a tool for evaluation of medicinal plants: a study of the vasodilatation effect of Salvia scutellarioides. Colombia Médica. 38:28–33.
- Shimoda LA, Sham JSK, Sylvester, JT. 2000. Altered pulmonary vasoconstriction in the chronically hypoxic lung. Physiol Res. 49:549–560.
- Shimoda LA, Sham JSK, Shimoda TH, Sylvester JT. 2000. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocites. Am J Physiol Lung Cell Mol Physiol. 279:L884–894.
- Shimoda LA, Sylvester JT, Sham JSK. 1998. Inhibition of voltage-gated K1 current in rat intrapulmonary arterial myocytes by endothelin. Am J Physiol. 274:L842–L853.
- Sudjarwo SA, Hori M, Tanaka T, Matsuda Y, Okada T, Karaki H. 1994. Subtypes of endothelin ETA and ETB receptors mediating venous smooth muscle contraction. Biochem Biophys Res Commun. 200:627–633. doi: 10.1006/bbrc.1994.1494
- Takuwa Y, Kasuya Y, Takuwa N, Kudo M, Yanagisawa M, Goto K, Masaki T, Yamashita K. 1990. Endothelin receptor is coupled to phospholipasa C via pertussis toxin insensitive guanine nucleotide binding, regulatory protein in vascular smooth muscle cells. J Clin Invest. 85:653–658. doi: 10.1172/JCI114488
- Vásquez IC, Hernández A. 2011. Pulmonary hypertension in chickens, lapse in the exposure to hypobaric exposure and relation to pulmonary weight: body weight under temperature-controlled conditions. Rev Col Cienc Pec. 25:85–89.
- Wang J, Juhaszova M, Rubin LJ, Yuan XJ. 1997. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary arterial smooth muscle cells. J Clin Invest. 100:2347–2353. doi: 10.1172/JCI119774
- Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. 2006. Hypoxia inducible factor 1 mediates hypoxia-induced TPRC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res. 98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98
- Warner TD, Battistini B, Allcock GH, Vane JR. 1993. Endothelin ETA and ETB receptors mediate vasoconstriction and prostanoid release in the isolated kidney of the rat. Eur J Pharmacol. 250:447–453. doi: 10.1016/0014-2999(93)90032-D
- Weigand L, Sylvester JT, Shimoda LA. 2006. Mechanisms of endothelin-1 induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 290:L284–L290. doi: 10.1152/ajplung.00449.2004
- Yanagisawa M, Kurihara H, Kimura S, Tomote Y, Kobayashi Y. 1988. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 332:411–415. doi: 10.1038/332411a0
- Zhang X-F, Komuro T, Miwa S, Minowa T, Iwamuro Y, Okamoto Y, Ninomiya H, Sawamura T, Masaki T. 1998. Role of nonselective cation channels as Ca2+ entry pathway in endothelin-1 induced contraction and their suppression by nitric oxide. EJP. 352:237–245.