
ABSTRACT
To investigate the genotype of Bovine herpesvirus-1 (BHV-1) isolated in Indonesia (n = 10), the DNA sequences of fragments of two genes, including the middle third of the gD gene and the downstream end of UL36, were determined using nested PCR. All the samples were classified as BHV-1.2 according to the deduced amino acid sequence of gD. On the other hand, analysis of the nucleotide sequence of UL36 indicated the presence of insertions or deletions (indels) compared to reference sequences and each other, which indels made more diverse than in gD sequence, and the classification of BHV-1 subtype based on the deduced amino acid sequence of UL36 differed from that obtained using the gD protein sequence. These results suggested that while the gD sequence analysis was suited for rough classification of BHV-1 subtype, the UL36 sequence permitted detection of BHV-1 subtype polymorphisms.
1. Introduction
Since the first outbreaks of infectious bovine rhinotracheitis (IBR) in 1950 in Colorado and in 1953 in California (Schroeder and Moys Citation1954; Miller Citation1955; Yates Citation1982), this disease has continued to spread to worldwide. The first clinical case of IBR disease in Indonesia was reported in the Lampung region (Marfiatiningsih Citation1982) in 1981, the disease has persisted in this country since then.
The classification of BHV-1 into various subtypes has changed as studies of the virus have provided further details on the viral genome. BHV-1 subtypes 1.1 and 1.2 are very closely related and are classified based on specific symptoms in infected cattle (Muylkens et al. Citation2007). Molecular methods, including enzymatic restriction fragment length polymorphism (RFLP) (Metzler, Schudel, et al. Citation1985; Rocha et al. Citation1998; Takiuchi et al. Citation2005; Kamiyoshi et al. Citation2008) subtype-specific monoclonal antibodies (Metzler, Matile, et al. Citation1985), restriction endonuclease mapping (Brake and Studdert Citation1985; Engels et al. Citation1986), and characterization using single nucleotide polymorphism (SNPs) (Fulton et al. Citation2016; Chase et al. Citation2017) has been used to differentiate types and subtypes.
The most extensively characterized gene is that encoding glycoprotein D (gD located in US, also known as US6); gD has functions related to viral immunity and antigenicity (Collins et al. Citation1993). UL36 gene also well-studied recently. UL36 gene was located in UL and forming tegument structure. The protein encoded by UL36 exhibits strong divergence among the various BHV-1 subtypes (d’Offay et al. Citation2016). In different subtypes of BHV-1, the upstream regions of the UL36 gene harbours insertions/deletions (indels) relative to its counterparts, and these differences are responsible for diversity among BHV-1.2b isolates (d’Offay et al. Citation2016). Herpesviruses may evolve faster than the host (Thiry et al. Citation2006), the herpesviral evolutionary rate estimated of 3 × 109 per amino acid per year (McGeoch et al. Citation2000).
The research described here had two related aims, both performed using Indonesian field isolates of BHV-1. The first goal was to characterize the molecular basis of the obvious subtype-specific differences in the sequences of middle third of the gD gene (a region known to contains SNPs) and in the downstream end of the UL36 gene (a region known to contain indels). The second goal was to compare the evolutionary process based on the sequence variations detected in gD and UL36.
2. Material and methods
2.1. Source of DNA samples and ethical approval
The samples consisted of nasal swabs and tracheal sections, and were obtained from The Animal Disease Investigation Centre (ADIC) in Lampung and Bukittinggi, Indonesia based on a letter of approval for material transfer No. 05010/PD.650/F5.H/06/2015 and No. 22022/PK.310/F4/02/2016. This study was approved by The Committee for Safe Handling of Living Modified Organism in Ehime University (Permission number: H28-05) and carried out according to the guidelines of the committee.
2.2. Preparation and amplification of DNA
The samples from nasal swabs were prepared per OIE instructions (OIE Citation2010). The samples from tracheal organ were prepared as described previously (Hidayati et al. Citation2018). DNA purification using a QIAamp DSP DNA Mini Kit (Qiagen, Hilden, Germany); the resulting nucleic acid was recovered in 200 µL elution buffer consisting of 10 mM Tris-Cl and 0.5 mM EDTA, pH 9.0. DNA fragments were amplified by nested PCR (Vilcek et al. Citation1994). The sequences of the gD primers were gD-1F 5′ACCGAGTGCGAGCCCAGGA3′; gD-1R 5′GGTTTCGGCTCGCCCTCG3′; gD-2F 5′ACCGCACACCCCCGTTTTG3′; gD-2R 5′TCGGGTCCTGGGGGGCC3′; gUL36-1F 5′TAGCGGCGACCCGCTATACG3′; gUL36-1R 5′TCTGGACGGCACGAAAGA; AGG3′; gUL36-2F 5′TAGACGACGAGATTGGCGA3′; gUL36-2R 5′GGCATGGAC; TTCCGTGGT3′.
The PCR reaction mixtures in each round were generated using KAPA HiFi HotStart ReadyMix (KAPABiosystem, Wilmington, MA) and consisted of 2.5 mM MgCl2., 0.3 mM each of dNTPs, 1 U of DNA polymerase, 10 pmol each of the forward and reverse primers, and less than 100 ng template DNA per reaction, with the balance of the volume consisting of PCR-grade water. First-round amplification of the gD fragment was performed: initial denaturation at 95°C for 3 min; 35 cycles of denaturation at 98°C/20 sec, annealing at 55°C/15 sec, extension at 72°C/15 sec; and a final extension at 72°C/5 min. Second-round amplification of the gD fragment was performed according to the same programme but with annealing at 59°C. First-round amplification of the UL36 fragment was performed: initial denaturation at 95°C for 3 min; 35 cycles of denaturation at 98°C /20 sec, annealing at 60.7°C/15 sec, extension at 72°C/5 sec; and a final extension for 72°C/5. Second-round amplification of the UL36 fragment was performed according to the same programme but with annealing at 57.6°C for 5 sec. PCR reactions were run using a Thermal Cycler Dice Gradient TP600 apparatus (TAKARA, Otsu, Shiga, Japan). The PCR products were examined by agarose gel electrophoresis in Tris acetate-EDTA (TAE) buffer; the resulting gels were stained with ethidium bromide (0.5 µg /mL) and bands were visualized by UV transillumination.
2.3. DNA cloning and sequencing
DNA fragments from each second-round PCR were incubated with the enzyme portion of the 10× A-attachment Mix (TOYOBO, Osaka, Japan) to generate overhanging adenines at the 3′-ends. The resulting products were purified using a FastGene Gel/PCR Extraction Kit (Nippon Genetics, Tokyo, Japan) and the DNA fragments were ligated into a plasmid using the pGEM®-T vector system (Promega Corp., Madison, WI); the ligation products were recovered by transformation into E. coli using NEB® stable, high-efficiency cells (New England BioLabs, Ipswich, MA) according to the manufacturer’s instructions. Transformants were selected on Luria–Bertani (LB) agar plate containing ampicillin (100 µg/mL).
Nucleotide sequence determination was performed using an Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. The purified plasmids were used as templates. When sequencing using native plasmid failed, sequencing was performed using the linearized plasmids as templates. Specifically, plasmids were linearized by digestion with the restriction enzyme ScaI (TAKARA), which cuts at a single site in the plasmid. When sequencing using the plasmid as template failed, the insert fragment was amplified using the same conditions as the 2nd-round PCR reactions described above, but using the cloned plasmid as the template. The resulting amplicon was purified using a FastGene Gel/PCR Extraction Kit prior to use as the template in the sequencing reaction.
2.4. In silico alignment and computational analysis
Alignment was analyzed using Clustal W (MegAlign DNAStar Lasergene, version 7; Madison. WI, USA). The prediction of AluI and TaqI restriction sites was performed using SeqBuilder (DNAStar Lasergene, version 7). The prediction of recombination sites was performed using DNA Sequence Polymorphism (version 6; Universitat de Barcelona).
3. Results and discussion
3.1. Amplification of gD and UL36 fragments
The sequences were deposited in DDBJ (DNA Data Bank of Japan), and the accession numbers are LC318523-LC318532 and LC368822-LC368831. The gD primers were designed to yield a 511-bp amplicon; the UL36 primers were designed to yield a 211-, 184-, or 157-bp amplicon ().
Figure 1. DNA sequence alignment of the UL36 partial open reading frame. The indel region is indicated by a 27-nt insertion (at a copy number of 2–5 presented as boxes outlined with black or red, and the background of blue, yellow, purple, and grey). The differentiation of the BHV-1.2 allele vs. BHV-1.1 allele is indicated by some degeneracy due to point mutation of C to T in the indel (red block letter, in sample B/1; B/31; B/32, and L/33). The dashes indicate spaces introduced to provide alignment among fragments of different lengths (due to the presence/absence of the indel).

3.2. Sequence analysis of the gD and UL36 fragments
The conclusion of the sequence variability of gD and gUL36 fragments summarized in Table S1.
The resulting alignment of the gD fragments among BHV-1.1, BHV-1.2, and the study samples revealed strong sequence similarity, with mean ± SD identities of 98.04 ± 0.32% (BHV-1.1 vs. samples), 98.13 ± 0.42% (BHV-1.2 vs. samples), and 98.34 ± 0.42% (among samples). Specifically, 4 separate point mutations were detected in this region (observed at nucleotide residues 462, 631, 666, and 912). Two of the SNPs (nt 631 and 666) were predicted to result in changes in the amino acid sequence (aa 211 and 222) of the gD glycoprotein when comparing BHV-1.1 and BHV-1.2 ((a)). The reassortment of the SNPs, yielding the reassortment of amino acids at the corresponding positions in the gD protein, suggests that homologous recombination has occurred within the gD sequence. The calculated synonymous and nonsynonymous ratio (dn/ds) was 0.22 (dn/ds < 1). This value indicates negative selection (H0 = dn < ds), meaning that these sequences have been subjected to purifying selection (Traesel et al. Citation2014). A previous paper (Saepulloh and Adjid Citation2010) using AluI and TaqI restriction enzymes confirming that the Indonesian isolates grouped with BHV-1.1. In this study, this region contains variations that included AluI and TaqI sites, as well as palindromic and inverted sequences (TCGAAGCTCGACGCAGCT), indicating that the samples should be assigned to subtype BHV-1.2. Notably, other work has revealed that the US region (within which gD is located) exhibits flipping with respect to the direction of replication (Chowdhury and Sharma Citation2012), although the possible function of this inversion remains unknown (Hammerschmidt et al. Citation1988). Several laboratories have hypothesized that this inversion plays a role in viral recombination, yielding different isomeric forms of the virus (Sheldrick and Berthelot Citation1975; Dutch et al. Citation1992; Schynts et al. Citation2003).
Figure 2. (a) Sequence alignment of deduced amino acid (aa) sequence of the gD protein, extending from aa 137– 306 of the full-length protein. Arrows indicate the positions of aa residues corresponding to the SNPs. Blue and red backgrounds indicate amino acids that differ between BHV-1.1 and BHV-1.2 (respectively). (b) Sequence alignment of the deduced aa sequence of the UL36 protein extending from aa 2428–2496 of the full-length protein. The arrow indicates the position of the aa residue that differs between BHV-1.1 and BHV-1.2. The red-outlined box indicates the sequence region corresponding to the indel. The black box indicates the sequences that are unique to BHV-1.2 as a result of the indel and the changed amino acid residue. Green, blue, yellow, and purple backgrounds indicate the positions of the repeated 9-aa peptide sequence encoded by the indel.
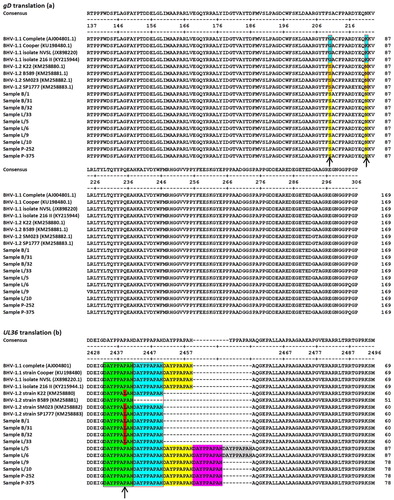
An alignment of the UL36 fragments from the Indonesian samples (n = 10) with the corresponding sequences from the reference genomes (n = 8) suggested a shift in the position of the deleted region without changing the length of the missing nucleotides. The experimental samples showed mean ± SD identities of 95.28 ± 0.05% (BHV-1.1 vs. samples), 95.39 ± 0.09% (BHV-1.2 vs. samples) and 93.24 ± 0.12% (among samples). The indel in the Indonesian samples (B/1, B/31, B/32 and L/33) corresponded to a loss of 27 nt and exhibiting occasional degeneracy due to point mutations (C to T of nt 7610 and 7662 relative to accession no. AJ004801), while samples (L/5, L/6, L/9, L/10, P-252 and P-357) present of 27 nt (with a copy number 2–5) relative to accession no. AJ004801 (nt 7625–7651) (). The 27-nt indel repeat in the BHV-1.1 samples is predicted to encode the peptide DAYPPAPAH ((b)). In the BHV-1.2 samples (B/1, B/31, B/32, and L/33), the first of the aforementioned nucleotide substitutions (at nt 20 of the first repeat) results in an amino acid substitution of Pro to Leu, yielding the peptide DAYPPALAH (where the underlined L indicates the altered aa sequence compared to that encoded by BHV-1.1). In contrast, the second substitution (at nt 18 of the second repeat) converts a GCC codon to GCT; because of the degeneracy of the code, both codons correspond to Ala, and the deduced peptide is unaltered compared to those encoded by the BHV-1.1 strains.
3.3. Phylogram variability of gD and UL36 fragments
Phylogenetic trees for the protein sequences are shown in . The tree derived from the gD amino acid sequences (predicted from amplified gD fragments) ((a)) suggested that all 10 of the novel Indonesian samples formed a clade separate from the BHV-1.1 reference strain. Specifically, the gD peptides encoded by the gD fragments from these samples were most similar to the BHV-1.2 reference strains, i.e. BHV-1.2 strain B589 from Australia (KM258881.1) and BHV-1.2 strain K-22 from the USA (KM258880.1). The gD peptide encoded by sample L/33 was identical to those encoded by BHV-1.2 reference strains SM023 (an American isolate) and SP1777 (a European isolate). Karlin et al. (Citation1994) suggested that divergence at the genomic and protein levels can differ, given that the DNA sequence emphasizes sequence specificity. However, most studies employing phylogenetic tree construction are based on protein sequence comparisons (Karlin et al. Citation1994).
Figure 3. (a) Phylogenetic tree based on analysis of amino acid sequences encoded by the gD ORF fragment. Red indicates the BHV-1.1 group. Green indicates BHV-1.2, and blue indicates samples characterized in the present study. Red stars indicate the sample isolates that move from one group to another between the trees based on DNA vs. protein sequences. (b) Phylogenetic tree based on analysis of amino acid sequences encoded by the UL36 ORF fragment.
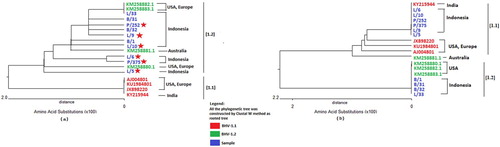
In contrast, the tree derived from the of UL36 amino acid sequences (predicted from amplified UL36 fragments) ((b)) exhibited a branching pattern distinct from that obtained with the gD peptides. Specifically, some samples that were affiliated with the BHV-1.2 group in the gD peptide-based tree (marked in (a) with red stars) were instead affiliated with the BHV-1.1 group in the UL36 peptide-derived tree ((b)). Notably, the UL36-based tree showed that samples L/5, L/6, L/9, L10, P-252, and P-375 sorted with BHV-1.1 isolate 216 II (accession no. KY215944; a strain from India) rather than with isolate NVSL, Cooper strain and complete strain (accession no. JX898220, KU1984801, and AJ004801 respectively; from the USA and Europe). Genetic divergence at gD (US6) and UL36 appeared to differ. The indel in UL36 permitted differentiation of the novel isolates into two groups, while the SNPs in gD suggested that the new viruses formed a single group (based on protein sequence). These findings are supported by those of Karlin et al. (Citation1994), who highlighted the evolutionary divergence of US compared to that of UL. In particular, the tegument protein (UL36) appears to have evolved more rapidly (as inferred from aa sequences) than proteins encoded by the US region (e.g. gD) (Karlin et al. Citation1994). In the present study, we observed a mixture of point mutations affecting palindromic and inverted sequences in a fragment of the gD ORF, which is located in the US region. However, the fragment of the UL36 ORF examined in this study contains a transposon (Robinson et al. Citation2008) that may affect the structure and/or function of the UL36 tegument protein (Möhl et al. Citation2010), with resulting effects on cellular phenotypes (Casacuberta and González Citation2013).
4. Conclusions
All 10 novel Indonesian field isolates of BHV-1 were classified as members of the BHV-1.2 subtype based on the deduced amino acid sequence of the protein encoded by an amplified fragment of the gD ORF. In contrast, the sequence of an amplified fragment of the UL36 ORF suggested that the novel strains be grouped differently than indicated by the gD sequence analysis. Given that the variation in UL36 was higher than that in gD, analysis of the UL36 sequence may be suited for detecting diversity among BHV-1 isolates. It concludes that the gD sequence analysis was suited for rough classification of BHV-1 subtype, the UL36 sequence permitted detection of BHV-1 subtype polymorphisms.
TAAR_1600521_Supplementary_TableS1
Download MS Word (57 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Brake F , Studdert MI. 1985. Molecular epidemiology and pathogenesis of ruminant herpesviruses including bovine, buffalo and caprine herpesviruses 1 and bovine encephalitis herpesvirus. Aust Vet J. 62:331–334. DOI:10.1007/BF01315300 doi: 10.1111/j.1751-0813.1985.tb07652.x
- Casacuberta E , González J. 2013. The impact of transposable elements in environmental adaptation. Mol Ecol. 22(6):1503–1517. doi: 10.1111/mec.12170
- Chase CC , Fulton RW , O’Toole D , Gillette B , Daly RF , Perry G , Clement T. 2017. Bovine herpesvirus 1 modified live virus vaccines for cattle reproduction: balancing protection with undesired effects. Vet Microbiol. 206:69–77. doi: 10.1016/j.vetmic.2017.03.016
- Chowdhury S , Sharma B. 2012. Transcription of gD and gI genes in BHV1-infected cells. J Biosci. 37:971–977. DOI:10.1007/s12038-012-9258-7.
- Collins JK , Ayers VK , Whetstone CA , van Drunen Littel-van den Hurk S. 1993. Antigenic differences between the major glycoproteins of bovine herpesvirus type 1.1 and bovine encephalitis herpesvirus type 1.3. J Gen Virol. 74:1509–1517. DOI:10.1099/0022-1317-74-8-1509; PMID 7688409.
- d’Offay JM , Eberle R , Fulton RW , Kirkland PD. 2016. Complete genomic sequence and comparative analysis of four genital and respiratory isolates of bovine herpesvirus subtype 1.2 b (BoHV-1.2 b), including the prototype virus strain K22. Arch Virol. 161:3269–3274. DOI:10.1007/s00705-016-3026-1.
- Dutch RE , Bruckner RC , Mocarski ES , Lehman IR. 1992. Herpes simplex virus type 1 recombination: role of DNA replication and viral a sequences. J Virol. 66:277–285.
- Engels M , Giuliani C , Wild P , Beck TM , Loepfe E , Wyler R. 1986. The genome of bovine herpesvirus 1 (BHV-1) strains exhibiting a neuropathogenic potential compared to known BHV-1 strains by restriction site mapping and cross-hybridization. Virus Res. 6:57–73. DOI:10.1016/0168-1702(86)90057-2.
- Fulton RW , d’Offay JM , Dubovi EJ , Eberle R. 2016. Bovine herpesvirus-1: genetic diversity of field strains from cattle with respiratory disease, genital, fetal disease and systemic neonatal disease and their relationship to vaccine strains. Virus Res. 223:115–121. DOI:10.1016/j.virusres.2016.06.017.
- Hammerschmidt W , Ludwig H , Buhk HJ. 1988. Specificity of cleavage in replicative-form DNA of bovine herpesvirus 1. J Virol. 62:1355–1363.
- Hidayati DN , Untari T , Wibowo MH , Akiyama K , Asmara W. 2018. Cloning and sequencing gB, gD, and gM genes to perform the genetic variability of bovine herpesvirus-1 from Indonesia. Vet World. 11:1255–1261. doi: 10.14202/vetworld.2018.1255-1261
- Kamiyoshi T , Murakami K , Konishi M , Izumi Y , Sentsui H. 2008. The presence of a deletion sequence in the BHV-1 UL49 homolog in a live attenuated vaccine for infectious bovine rhinotracheitis (IBR). Vaccine. 26:477–485. DOI:10.1016/j.vaccine.2007.11.037; PMID 18164789.
- Karlin S , Mocarski ES , Schachtel GA. 1994. Molecular evolution of herpesviruses: genomic and protein sequence comparisons. J. Virol. 68:1886–1902.
- Marfiatiningsih S. 1982. Diagnosa infectious bovine rhinotracheitis-like disease pada sapi bali di lampung tengah. Annual report of animal disease investigation in Indonesia in 1976–1981. Directorate general of animal health and animal husbandary, Ministry of agriculture of the republic of Indonesia, Jakarta. p. 122–124 (in Indonesia).
- McGeoch DJ , Dolan A , Ralph AC. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol. 74:10401–6. doi: 10.1128/JVI.74.22.10401-10406.2000
- Metzler AE , Matile H , Gassmann U , Engels M , Wyler R. 1985. European isolates of bovine herpesvirus 1: a comparison of restriction endonuclease sites, polypeptides, and reactivity with monoclonal antibodies. Archi Virol. 85:57–69. doi: 10.1007/BF01317006
- Metzler AE , Schudel AA , Engels M. 1985. Bovine herpesvirus 1: molecular and antigenic characteristics of variant viruses isolated from calves with neurological disease. Archi Virol. 87:205–217. DOI:10.1007/BF01315300.
- Miller NJ. 1955. Infectious necrotic rhinotracheitis of cattle. J Am Vet Med Assoc. 126:463–467.
- Möhl BS , Böttcher S , Granzow H , Fuchs W , Klupp BG , Mettenleiter TC. 2010. Random transposon-mediated mutagenesis of the essential large tegument protein pUL36 of pseudorabies virus. J Virol. 84(16):8153–8162. doi: 10.1128/JVI.00953-10
- Muylkens B , Thiry J , Kirten P , Schynts F , Thiry E. 2007. Bovine herpesvirus 1 infection and infectious bovine rhinotracheitis. Vet res. 38:181–209. DOI:10.1051/vetres:2006059; PMID 17257569.
- OIE Resources page . 2010. [accessed 2017 April 5]. http://www.oie.int/animal-health-in-the-world/oie-listed-diseases-2017 .
- Robinson KE , Meers J , Gravel JL , McCarthy FM , Mahony TJ. 2008. The essential and non-essential genes of Bovine herpesvirus 1. J Gen Virol. 89:2851–2863. doi: 10.1099/vir.0.2008/002501-0
- Rocha MA , Barbosa EF , Guimaraes SE , Neto ED , Gouveia AM. 1998. A high sensitivity-nested PCR assay for BHV-1 detection in semen of naturally infected bulls. Vet Microbiol. 63:1–11. DOI:10.1016/S0378-1135(98)00213-2; PMID 9810617.
- Saepulloh M , Adjid R. 2010. Isolasi dan Identifikasi Bovine herpesvirus-1 pada Sapi Perah dan Potong di Indonesia. JITV. 19:376–387.
- Schroeder RJ , Moys MD. 1954. An acute upper respiratory infection of dairy cattle. J Am Vet Med Assoc. 125:471.
- Schynts F , McVoy MA , Meurens F , Detry B , Epstein AL , Thiry E. 2003. The structures of bovine herpesvirus 1 virion and concatemeric DNA: implications for cleavage and packaging of herpesvirus genomes. Virology. 314:326–335. DOI:10.1016/S0042-6822(03)00437-9.
- Sheldrick P , Berthelot N. 1975. Inverted repetitions in the chromosome of herpes simplex virus. Cold Spring Harbor Symp. Quant. Biol. 39:667–678. doi: 10.1101/SQB.1974.039.01.080
- Takiuchi E , Médici KC , Alfieri AF , Alfieri AA. 2005. Bovine herpesvirus type 1 abortions detected by a semi-nested PCR in Brazilian cattle herds. Res Vet Sci. 79:85–88. DOI:10.1016/j.rvsc.2004.11.005; PMID 15894030.
- Thiry E , Muylkens B , Meurens F , Gogev S , Thiry J , Vanderplasschen A , Schynts F. 2006. Recombination in the alphaherpesvirus bovine herpesvirus 1. Vet Microbiol. 113:171–177. doi: 10.1016/j.vetmic.2005.11.012
- Traesel CK , e Silva MS , Weiss M , Spilki FR , Weiblen R , Flores EF. 2014. Genetic diversity of 3′ region of glycoprotein D gene of bovine herpesvirus 1 and 5. Virus Genes. 48:438–447. DOI:10.1007/s11262-014-1040-5; PMID 24482291.
- Vilcek S , Nettleton PF , Herring JA , Herring AJ. 1994. Rapid detection of bovine herpesvirus 1 (BHV 1) using the polymerase chain reaction. Vet Microbiol. 42:53–64. DOI:10.1016/0378-1135(94)90077-9.
- Yates WD. 1982. A review of infectious bovine rhinotracheitis, shipping fever pneumonia and viral-bacterial synergism in respiratory disease of cattle. Can J Comp Med Vet Sci. 46:225. http://www.ncbi.nlm.nih.gov/pubmed/6290011 .