
ABSTRACT
Foot-and-mouth disease (FMD) is a contagious disease affecting cloven-hoofed animals, which has been well-documented as one of the major animal diseases that causes severe economic loss in livestock sectors. The disease is endemic in many countries, particularly in Asia and Africa. Indonesia has been declared a disease-free country since 1986, and the World Organization of Animal Health (WOAH/OIE) recognized Indonesia as an FMD-free country without vaccination in 1990. However, the FMD virus was detected in many disease outbreaks in cattle and goats in Indonesia in May 2022. This study reports the detection and identification of FMD serotype O viruses in Indonesia. Although these viruses appeared to belong to the ME-SA/Ind-2001e lineage, they formed a unique cluster with 95.3% average nucleotide sequence similarity of the FMD VP1 gene to Ind-2001e viruses from other Asia countries. The illegal trade of live animals from endemic areas in Southeast Asia is one of the possible routes regarding the incursion of FMD in Indonesia, however, it requires further investigation.
Introduction
Foot-and-mouth disease (FMD) is a contagious animal disease caused by an RNA virus from Picornaviridae, genus Aphthovirus, species foot-and-mouth disease virus. The virus has seven serotypes, including O, A, C, Asia 1, SAT 1, SAT 2 and SAT 3, and is further divided genetically into topotypes and lineages (Grubman and Baxt Citation2004). The disease affects cloven-hoofed animals causing a severe economic loss in livestock sectors and is endemic in many countries, particularly in Asia and Africa (Brito et al. Citation2015). To date, there are 10 topotypes of serotype O identified, including Europe-South America (Euro-SA), Middle East South Asia (ME-SA), South East Asia (SEA), Cathay (CHY), West Africa (WA), East Africa 1 (EA-1), East Africa 2 (EA2), East Africa 3 (EA-3), Indonesia-1 (ISA-1) and Indonesia-2 (ISA-2) (Brito et al. Citation2015; Blacksell et al. Citation2019).
In Southeast Asian (SEA) countries, serotype O has been recognized as the predominant serotype to cause more FMD outbreaks than serotype A and Asia 1 (Brito et al. Citation2015; Blacksell et al. Citation2019). Within the serotype O, South East Asia (SEA) and Middle East-South Asia (ME-SA) topotypes were frequently detected in FMD outbreaks in SEA countries (Blacksell et al. Citation2019). In particular ME-SA topotype, several virus lineages have been identified as strains that temporarily close related or belong to unique outbreaks occurring in certain regions including PanAsia, Ind2001 and Iran2001 based on phylogenetic relationships and a nucleotide difference of <5% based on VP1 protein (Hemadri et al. Citation2002; Knowles and Samuel Citation2003). The O/ME-SA/PanAsia lineage emerged from the Indian continent (Bangladesh, Bhutan, India and Nepal). It became the major serotype-O virus lineage that spread globally into the Middle East, Southeast Asia, North and South Africa, and Europe between 1990 and 2000 (Hemadri et al. Citation2002; Knowles et al. Citation2005; Bachanek-Bankowska et al. Citation2018). The Ind-2001 lineage of O/ME-SA topotype (O/ME-SA/Ind-2001), again, was initially identified in the Indian subcontinent in 2001 as a distinct group from the predominant PanAsia lineage (Hemadri et al. Citation2002; Knowles et al. Citation2005; Bachanek-Bankowska et al. Citation2018). Since then, the Ind-2001 lineage has diverged into five sub-lineages (Ind-2001a to Ind-2001e) and become significant in the Middle East, Asia and Africa (Bachanek-Bankowska et al. Citation2018; Blacksell et al. Citation2019).
Within the Ind-2001 lineage, a large proportion of FMD outbreaks were due to infection of the Ind-2001d lineage in South Asia, North Africa and Southeast Asia for 2–3 years from 2012 to 2015. However, viruses belonging to the Ind-2001e lineage have become predominant since 2016 (Bachanek-Bankowska et al. Citation2018). Moreover, since 2020 there has been an increasing dominance of O/ME-SA/Ind-2001e lineage in SEA countries, including Cambodia, Laos, Myanmar, Vietnam and Thailand (King Citation2021).
FMD is endemic in most Southeast Asia countries except Philippines, Singapore and Brunei. In Indonesia, the first FMD case occurred in East Java in 1887. Nevertheless, after implementing strict animal movement, strong quarantine measures and mass vaccination programs, no FMD has reported since December 1983, and Indonesia declared a disease-free country in 1986 (Blacksell et al. Citation2019). However, after 35 years free from FMD, at the beginning of May 2022, several disease outbreaks with FMD-like symptoms were reported in Aceh and East Java provinces, followed by several FMD cases in other areas in Java, Sumatera, Kalimantan and West Nusa Tenggara. This study reports the detection and identification of FMD-serotype O virus that belongs to the ME-SA/Ind-2001e lineage in Indonesia. In this study, we collected clinical samples from FMD outbreaks in Indonesia in the first 2 weeks of May 2022 and named the group of the isolates as ISA-22. Since this was the first case in Indonesia after 35 years free from FMD, we aimed to identify the genetic lineage of the viruses that cause the FMD outbreaks. This study also intended to determine the source of the virus introduction and to infer the molecular pattern of virus transmission amongst areas in Indonesia for a better understanding of disease control.
Material and methods
Clinical samples and diagnostics
During the initial outbreaks of disease in Java, Sumatera, Kalimantan and West Nusa Tenggara in May 2022 (), a total of 24 swabs and two blood samples were collected from cattle and goats (). These animals showed clinical symptoms of pyrexia, anorexia and vesicles either in buccal mucous membranes or between claws and coronary band; and hypersalivation in cattle. The presence of the FMD virus was detected directly from clinical samples (swabs and blood samples) through routine viral RNA extraction procedures followed by real-time reverse transcription polymerase chain reaction (rRT-PCR) technique using primers and probe targeting the 3D gene of FMD virus (WOAH Citation2021). Using these primers and probes, we could detect, on average, 92% of FMD cases positive from the total tested animals showing clinical signs (data not shown). This study is a collaborative work supported by Disease Investigation Centres (DICs) in Medan, Wates, Banjarbaru, and Denpasar and the Indonesian FMD Reference Laboratory, National Centre for Veterinary Biologics (NCVB) in Surabaya and National Veterinary Drug Assay Laboratory (NVDAL), Bogor, Indonesia.
Figure 1. Distribution of samples collected from Java, Sumatera, Kalimantan and West Nusa Tenggara for foot and mouth virus characterization. Numbers represent the sequence of collection dates that are in the same order as ID number in . Dots represent village-based centroid where samples were collected from FMD cases in livestock. A zoom-in of FMD case locations in Aceh and North Sumatera provinces and in Java is indicated in the bottom box.
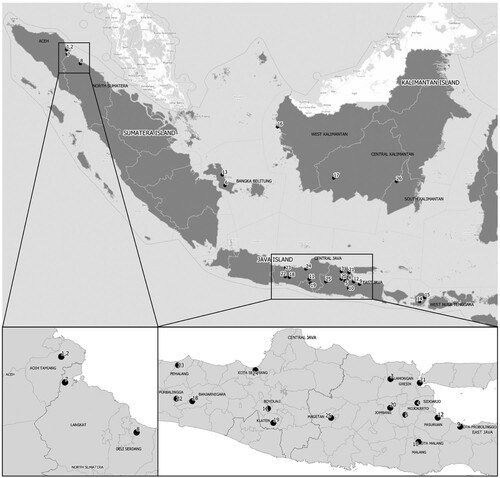
Table 1. Metadata of samples collected from initial FMD outbreaks in Indonesia, May 2022.
DNA sequencing and phylogenetic analysis
This study conducted the molecular genetic assay mainly in the VP1 region to identify the serotype and lineage of FMD virus. The Sanger dideoxy sequencing targeting the full length of VP1 coding region [639 nucleotides (nt) total length] was performed in the National Centre of Veterinary Biologics (NCVB), Surabaya and National Veterinary Drug Assay Laboratory (NVDAL), Bogor Indonesia, by using the Big Dye Terminator v3.1 Cycle sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA) on the ABI 3130 Genetic Analyser (Applied Biosystems, Foster City, CA, USA). DNA sequencing was done using clinical samples without prior isolation in cell culture. It was performed using a previously developed protocol (Le et al. Citation2012) with modified primers for amplification and sequencing reactions (primer sequences are available upon request). Genome assembly and analysis were performed using the AB1 files extension in UGENE v.40 software (https://ugene.net/). The VP1 sequence data were submitted to GenBank under accession numbers, as shown in . The maximum likelihood (ML) method with 1000 bootstrap replicates using the fittest and best substitution model of HKY+G (based on the lowest Akaike information criterion/AIK) was used to construct a phylogenetic tree based on 639 nt length of complete VP1 coding sequence from 26 FMD viruses of this study and 60 FMD virus sequences that available in GenBank (3 viruses of ISA-1, 2 of ISA-2, and 55 of ME-SA topotype viruses). Evolutionary distances were computed using average pairwise distance within and between sequence groups in MEGA X (Kumar et al. Citation2018).
Results
To determine genetic relationships and molecular epidemiology of the FMD virus, the viral capsid protein VP1 (encoded by 1D gene) has been widely used for FMD phylogenetic analysis (Knowles et al. Citation2016). Phylogenetic analysis based on the VP1 coding sequence showed that all FMD viruses detected during the disease outbreaks in Indonesia in May 2022 (named ISA-22) belonged to the O/ME-SA/Ind-2001e sequence (). Although all the ISA-22 viruses were clustered under the Ind-2001e lineage, they formed a distinct cluster separated from those of Ind-2001e from other Asian countries with 95.3% average nucleotide sequence similarity (). Homology analysis using the BLAST search tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) found that the nearest nucleotide sequence identity of VP1-FMD that was publicly available in GenBank was an FMD virus sample from China, XJ/CHA/2017 (Ind-2001e virus lineage, Accession Number MF461724), with VP1 nucleotide similarity ranging from 95.6% to 96.2% to ISA-22 viruses. Sequence analysis using the between-group average pairwise distance indicated low nucleotide identity of the VP1 gene of ISA-22 to that of other virus lineages of O/ME-SA topotype, including the earlier Indonesian FMD viruses that are now extinct (ISA-1 and ISA-2). By contrast, high nucleotide identity of VP1 sequence (99.6%) was detected within ISA-22 viruses, as indicated by a low replication pairwise distance within this group (0.004) ().
Figure 2. Phylogenetic analysis of Foot-and-Mouth Disease (FMD) Subtype O virus collected from disease outbreaks in Indonesia in May 2022. The evolutionary history was inferred by using the Maximum Likelihood method and the best-fit substitution model for the dataset (HKY+G; Hasegawa-Kishino-Yano with a discrete Gamma distribution) with 1000 bootstrap replication. The percentage of bootstrap statistical value in which the associated taxa clustered together is shown next to the branches with values equal or more than 50% are only shown. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (0.05) shown in the left. This analysis involved 96 nucleotide sequences with a total of 639 positions of the VP1 gene of the FMD virus in the final dataset. Evolutionary analyses were conducted in MEGA X. Three viruses from Cambodia isolated in 2019 (CAM-22) and the ISA-22 viruses (bold-italic taxa name) formed two distinct clusters within O/ME-SA/Ind-2001e lineage. Three viruses were collected from goats including ISA/Jombang/PVTW-J11/2022, ISA/Mempawah/BBR11/2022 and ISA/HSU/A0522099-5/2022, while the other twenty-three viruses collected from cattle.
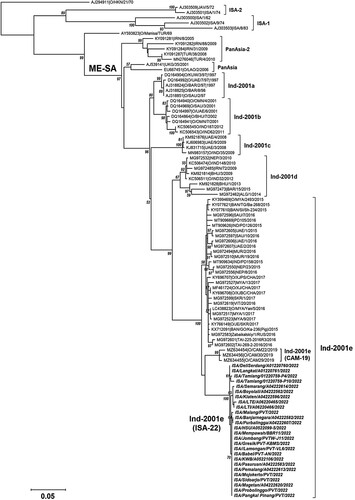
Table 2. Average pairwise distance, number of base differences and homology of ISA-22 viruses to the other virus lineages belonging to O Serotype circulating in Asia.
The most recent common ancestral node of the ISA-22 virus lineage could not yet be determined using the 26 sequences used for phylogenetic analysis. At first glance from the phylogenetic tree, three Ind-2001e viruses from Cambodia isolated in 2019 (CAM-19) seemed to have a potential relationship as virus ancestors to the ISA-22 group (). However, this was not supported by strong statistical evidence as the bootstrap value in the common node of these two groups was less than 50% although the best substitution model (HKY+G) was used for generating the tree. The more appropriate conclusion to define this connection is that they are genetically close related. This topology inference was found to be reversed with the results of molecular analysis of VP1 gene of CAM-19 which revealed only 94.5% nucleotide sequence identity to ISA-22 (), however, this may be due to the inclusion of calculated ORFs and ORF coding.
Phylogenetic analysis inferred that ISA-22 viruses from North Sumatera (ISA/Langkat/A01220761/2022 and ISA/Deli Serdang/A01220762/22) have likely emerged preceding ISA-22 viruses from Aceh (ISA/Tamiang/01220759-P4/2022 and ISA/Tamiang/01220759-P10/2022). Furthermore, genetically similar viruses were detected in East Java, Central Java, Bangka Belitung, South Kalimantan, Central Kalimantan and West Kalimantan. Interestingly, ISA-22 viruses from West Nusa Tenggara (ISA/LTE/A06220465/2022 and ISA/LTI/A06220466/2022) were clustered with three ISA-22 viruses from Central Java (ISA/Boyolali/A04222562/2022, ISA/Klaten/A04222596/2022 and ISA/Semarang/A04222614/2022).
Sequence analysis on the VP1 protein of ISA-22 viruses revealed that amino acid variations were found at position 13 (A/T), 129 (V/A), 142 (T/A), 158 (A/G) and 201 (Y/H) (). Amino acid changes in VP1 protein of three virus samples collected from goat (ISA/Jombang/PVTW-J11/2022, ISA/Mempawah/BBR11/2022 and ISA/HSU/A0522099-5/2022) were also found in most of virus samples collected from cattle, and no changes were found either at the critical amino acid sites at position 144 (V), 148 (L), 154 (K) and 208 (P), or in the position 145–147 (retained RGD sequence motif).
Table 3. Amino acid changes detected in VP-1 of ISA-22 viruses and critical amino acids found at the VP1 epitope and sites that involved in the adsorption of virus to host cell.
Discussion
This study aimed to identify the genetic lineage of viruses responsible for FMD outbreaks, to infer the virus transmission across regions in Indonesia in the initial phase of the outbreaks. In combination with recent epidemiological data of FMD outbreaks in Asia, this study could be used as a preliminary study to obtain possible consideration of the virus introduction into Indonesia. FMD viruses of O/ME-SA/Ind-2001e were detected and identified from disease outbreaks with FMD symptoms in cattle and goats in Indonesia in May 2022. Our study showed that the nucleotide difference of this FMD virus was >4.7% to the existing O/ME-SA/Ind-2001e viruses that circulating in ASIA; hence, additional study is necessary to determine the phylogenetic relationships of this Indonesian virus lineage.
We found that there was no significant difference in both nucleotide and amino acid sequences of VP1 protein amongst virus samples collected from goats and most of those collected from cattle. In addition, no amino acid substitutions were found either at the critical amino acid sites within the VP1 epitope at position 144 (V), 148 (L), 154 (K) and 208 (P), or in the position 145–147 of VP1 protein (retained RGD sequence motif) that involved in the adsorption of FMD virus to host cell where their alteration more or less affecting antigenic loci and corresponding monoclonal antibody reactivity (Jinding et al. Citation2006).
The course of the virus’s incursion into Indonesia remains unknown. We could not yet determine whether the Ind-2001e virus group from Cambodia collected in 2019 (CAM-19) is a direct precursor of the Ind-2001e virus group in Indonesia (ISA-22) since the statistical bootstrap value supporting the node that linking these two groups was low. However, the tree suggests that CAM-19 strains are closely related to those circulated in Indonesia or strains circulated in Cambodia in 2019 and in Indonesia in 2022 belonging to the same lineage (Ind-2001e) and probably share a close common ancestor, but emerged at different outbreak events. A recent report from The World Reference Laboratory for Foot-and-Mouth Disease (WRLFMD) showed that some virus sequences from Thailand and Malaysia in 2021 were genetically more closely related to ISA-22 (WRLFMD Citation2022). These highlight that sharing genetic data is important for better understanding virus evolution to assist FMD control strategies between affected countries.
Based on the current FMD situation in neighbouring countries of Indonesia where O/ME-SA/Ind-2001e virus is endemic (Bachanek-Bankowska et al. Citation2018; Blacksell et al. Citation2019; King Citation2021) and the role of animal movement in the spread of FMD in Asia and other regions (Rweyemamu et al. Citation2008; Di Nardo et al. Citation2011; Smith et al. Citation2015), the introduction of virus into Indonesia could be predisposed by a transboundary ‘illegal’ trade of live animals or FMD-contaminated materials from endemic areas in Southeast Asia. A study by Bachanek-Bankowska et al. (Citation2018) indicated that although many outbreaks of the Ind-2001d and Ind-2001e lineages were due to single independent introductions of the virus into new regions, some became established within different geographic areas, providing additional sources for onward transmission. This study also indicated that the predominant disease occurrences of the Ind-2001e lineage in diverse and distant geographic regions since 2016 might be linked to increased or altered trading routes as well as movement of people, and such virus-specific factors for example replication and/or transmission fitness across susceptible host species have played an important role in the establishment of this lineage (Bachanek-Bankowska et al. Citation2018).
As shown in , Aceh and North Sumatera provinces are in the same Sumatera Island where they share land borders. Phylogenetic analysis inferred that there might have been an incidence of FMD virus transmission and circulation on the border areas between North Sumatera and Aceh before the transmission event occurred in Java causing outbreaks in domestic livestock and then spread to other areas in Indonesia. Previous studies identified critical points of disease amplification and transmission, such as holding facilities and livestock markets that provide opportunities for extensive mixing of livestock from different origins that are destined for different locations (Di Nardo et al. Citation2011; Smith et al. Citation2015). Furthermore, the spread of disease can also be affected by shared border areas, unofficial cross-border movement, lack of effective biosecurity and low levels of compliance with animal health regulations (Smith et al. Citation2015). However, the lack of extensive data hamper the understanding of how the virus was transmitted in the early stage of FMD cases in Indonesia. One possibility is that the virus might be transmitted over longer distances by infected small ruminants (e.g. sheep and goats) that show inapparent or mild clinical signs of FMD (Barnett and Cox Citation1999) before the disease was reported and spread in cattle. This hypothesis should be investigated further since FMD viruses seemed able to persist in a sheep population with the reproduction ratio only slightly larger than one (Orsel et al. Citation2007), yet in a mixed population of sheep and cattle, sheep play a more limited role in the transmission of FMDV than cattle (Bravo de Rueda et al. Citation2014).
In summary, after 36 years of maintaining the FMD-free country, Indonesia reported FMD outbreaks in livestock to World Organization for Animal Health in May 2022 (WOAH Citation2022). Our study reports the detection and identification of the FMD O/ME-SA/Ind-2001e virus in Indonesia. This finding provided immediate direction for the government of Indonesia for FMD control, particularly for vaccination strategy since stamping-out policy was not fully implemented as the disease has been spread in multiple areas across islands within the country. How the virus was introduced into Indonesia remains undetermined; nonetheless, unauthorized movement of live animals from areas where FMD is currently endemic is a possible route that requires more investigation. More advanced studies using whole genome sequencing data of the FMD virus will provide better insight to infer the source of virus incursion and to determine the molecular pattern of virus transmission between areas in Indonesia.
Ethic statement
Sample collection was performed as part of routine field outbreak investigations from which all cases described herein occurred spontaneously in domestic livestock with no experimental inoculation or treatment of live animals and no animals were euthanized for this study. Therefore, no ethical approval was required for this study as the sample collection was performed using standard diagnostic procedures with no harm to the animals.
Author’s contributions
Study conception and design: E.B. Susila, H. Wibawa. Drafting the manuscript: E.B. Susila, H. Wibawa, Y. Yupiana. Revised the manuscript: D.N. Hidayati, H. Wibawa. Virus identification through sequencing and phylogenetic analysis: R.S.D. Daulay, E. Andesfha, H. Wibawa. Performed virus detection through laboratory diagnostics and provided outbreak data: S.R.B. Prasetyowati, Wriningati, D.N. Hidayati, S.H. Irianingsih, I.N. Dibia, Faisal, A. Supriyadi. Provided epidemiological data and analysis: Y. Yupiana, M.M. Muharram. Facilitated the study and provided final approval of the manuscript to be published: N. Zainuddin.
Acknowledgements
The authors acknowledge the Director General of Livestock and Animal Health Services (DGLAHS), Ministry of Agriculture of Indonesia in facilitating epidemiological investigations and laboratory confirmation conducted by DGLAHS’ veterinary laboratories. We thank the veterinary laboratory directors including from DICs, NCVB Surabaya, and NVDAL Bogor and their staff for their support on field sampling and laboratory diagnostics for the detection and identification of FMD agents. We also acknowledge the United Nations Food and Agriculture Organization Emergency Centre for Transboundary Animal Diseases, Jakarta, Indonesia, for supporting a diagnostic algorithm and reagents for molecular identification through rRT-PCR and DNA sequencing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All sequence data of the FMD-VP1 gene are available in GenBank under accession numbers: ON783873, ON783874, and ON854934 to ON854957.
References
- Bachanek-Bankowska K, Di Nardo A, Wadsworth J, Mioulet V, Pezzoni G, Grazioli S, Brocchi E, Kafle SC, Hettiarachchi R, Kumarawadu PL, et al. 2018. Reconstructing the evolutionary history of pandemic foot-and-mouth disease viruses: the impact of recombination within the emerging O/ME-SA/Ind-2001 lineage. Sci Rep. 8(1):14693. doi:10.1038/s41598-018-32693-8.
- Barnett PV, Cox SJ. 1999. The role of small ruminants in the epidemiology and transmission of foot-and-mouth disease. Vet J. 158(1):6–13. doi:10.1053/tvjl.1998.0338.
- Blacksell SD, Siengsanan-Lamont J, Kamolsiripichaiporn S, Gleeson LJ, Windsor PA. 2019. A history of FMD research and control programmes in Southeast Asia: lessons from the past informing the future. Epidemiol Infect. 147(e171):1–13. doi:10.1017/S0950268819000578.
- Bravo de Rueda C, de Jong MC, Eble PL, Dekker A. 2014. Estimation of the transmission of foot-and-mouth disease virus from infected sheep to cattle. Vet Res. 45(1):45. doi:10.1186/1297-9716-45-58.
- Brito BP, Rodriguez LL, Hammond JM, Pinto J, Perez AM. 2015. Review of the global distribution of foot-and-mouth disease virus from 2007 to 2014. Transbound Emerg Dis. 64(2):316–332. doi:10.1111/tbed.12373.
- Di Nardo A, Knowles NJ, Paton DJ. 2011. Combining livestock trade patterns with phylogenetics to help understand the spread of foot and mouth disease in sub-Saharan Africa, the Middle East and Southeast Asia. Rev Sci Tech. 30(1):63–85. doi:10.20506/rst.30.1.2022.
- Grubman MJ, Baxt B. 2004. Foot-and-mouth disease. Clin Microbiol Rev. 17(2):465–493. doi:10.1128/CMR.17.2.465-493.2004.
- Hemadri D, Tosh C, Sanyal A, Venkataramanan R. 2002. Emergence of a new strain of type O foot-and-mouth disease virus: its phylogenetic and evolutionary relationship with the PanAsia pandemic strain. Virus Genes. 25:23–34. doi:10.1023/A:1020165923805.
- Jinding C, Mingqiu Z, Hui KH, Leung FC. 2006. Molecular characterization of foot-and-mouth disease virus in Hong Kong during 2001–2002. Virus Genes. 32:139–143. doi:10.1007/s11262-005-6869-1.
- King D. 2021. Global foot-and-mouth disease situation: risk and new development. The 24th SEACFMD National Coordinators Meeting-OIE-Asia. https://rr-asia.woah.org/wp-content/uploads/2021/07/01_donald_king_seacfmd-july-2021.pdf.
- Knowles NJ, Samuel AR. 2003. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 91(1):65–80. doi:10.1016/S0168-1702(02)00260-5.
- Knowles NJ, Samuel AR, Davies PR, Midgley RJ, Valarche JF. 2005. Pandemic strain of foot-and-mouth disease virus serotype O. Emerg Infect Dis. 11(12):1887–1893. doi:10.3201/eid1112.050908.
- Knowles NJ, Wadsworth J, Bachanek-Bankowska K, King DP. 2016. VP1 sequencing protocol for foot and mouth disease virus molecular epidemiology. Rev Sci Tech. 35(3):741–755. doi:10.20506/rst.35.3.2565.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549. doi:10.1093/molbev/msy096.
- Le VP, Lee KN, Nguyen T, Kim SM, Cho IS, Khang DD, Hien Nguyen B, Van Quyen D, Park JH. 2012. A rapid molecular strategy for early detection and characterization of Vietnamese foot-and-mouth disease virus serotypes O, A, and Asia 1. J Virol Methods. 180(1–2):1–6. doi:10.1016/j.jviromet.2011.11.028.
- Orsel K, Dekker A, Bouma A, Stegeman JA, de Jong MCM. 2007. Quantification of foot and mouth disease virus excretion and transmission within groups of lambs with and without vaccination. Vaccine. 25(14):2673–2679. doi:10.1016/j.vaccine.2006.11.048.
- Rweyemamu M, Roeder P, Mackay K, Sumption K, Brownlie Y, Leforban J, Valarcher J-F, Knowles NJ, Saraiva V. 2008. Epidemiological patterns of foot-and-mouth disease worldwide. Transbound Emerg Dis. 55(1):57–72. doi:10.1111/j.1865-1682.2007.01013.x.
- Smith P, Lüthi NB, Huachun L, Oo KN, Phonvisay A, Premashthira S, Abila R, Widders P, Kukreja K, Miller C. 2015. Movement pathways and market chains of large ruminants in the Greater Mekong Sub-region. OIE Sub-Regional Representation for South-East Asia; [accessed 2022 June 6]. https://rr-asia.woah.org/wp-content/uploads/2019/10/livestock_movement_pathways_and_markets_in_the_gms__final_.pdf.
- WOAH. 2021. Chapter 3.1.8: foot and mouth disease (infection with foot and mouth disease virus). OIE Terrestrial Manual 2021. https://rr-asia.woah.org/en/projects/foot-and-mouth-disease-fmd/.
- WOAH. 2022. Immediate notification of foot-and-mouth disease status from Indonesia (Reported on 9 May 2022). https://wahis.woah.org/#/report-info?reportId=53545.
- WRLFMD. 2022. WHO-FAO FMD reference laboratory report, July-September 2022. Quarterly Report. https://www.wrlfmd.org/ref-lab-reports.