
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In this study, we used high-throughput sequencing technology to analyze the diversity and predict gene function of intestinal bacteria from captive black-capped capuchin in two wildlife parks. The results showed that the composition of intestinal gut bacterial diversity was significantly higher in black-capped capuchins monkeys from ES than HT. Moreover, high similarity of intestinal bacteria of black-capped capuchins at the same site was found by sample stratum clustering. Black-capped capuchins intestinal bacteria can be clustered into 2296 ASVs, belonging to 16 phyla, 99 families and 210 genera, respectively. The dominant phyla were Bacteroidota and Firmicutes in the intestinal bacteria of black-capped capuchins, but there were differences in the dominant bacterial families and dominant bacterial genera between ES and HT, and the bacterial families with significant differences had correspondence with bacterial genera. The results provide a basis for the study of intestinal bacteria in black-capped capuchins and the isolation and purification of dominant bacteria. The results of this study fill the gap in the study of intestinal bacterial diversity and its gene functions in black-capped capuchins, and provide a reference for the isolation of dominant bacteria in black-capped capuchins’ intestinal microbes and the discovery and study of novel functional genes.
1. Introduction
The black-capped capuchin (Sapajus apella), belonging to the capuchin family (Cebidae) of primates, is distributed in Central and South America, From Costa Rica TO the north of Paraguay. They inhabit the trees of tropical forests and live in familial clusters, but their survival is threatened by deforestation and destruction of the ecosystem (Taal et al. Citation2013). In CITES, the black-capped capuchin is listed in Appendix II (species that are not necessarily threatened with extinction, but whose trade must be controlled to avoid exploitation incompatible with their survival). As with other animals, the gastrointestinal tract of primates contains a complex microbial community, and the health of the host is closely related to the community structure of the intestinal microbes (Fehlberg et al. Citation2020). Intestinal microorganisms are able to retain genes that are beneficial to them and continuously improve their adaptive capacity during the process of long-term natural selection (Gaulke et al. Citation2018). In this process, there is always a relative balance among different microorganisms, between microorganisms and the host, and between the host and the internal environment, ultimately forming an interdependent and mutually restrictive relationship (Song et al. Citation2021). If the number, type and proportion of intestinal microorganisms change dramatically in a short period of time, the dynamic balance of host-microbial-internal environment will be disrupted, and the disrupted intestinal microecology will not be able to maintain normal physiological functions, which will affect the health of the host and cause diseases (Han et al. Citation2017, Liu et al. Citation2020). The composition of normal healthy intestinal microorganisms is complex and diverse (Beam et al. Citation2021), and intestinal microorganisms are important for the mechanisms of food digestion (Yao et al. Citation2021), nutrient absorption (Espirito Santo et al. Citation2021), intestinal diseases caused by disruption of the microecology of the intestinal flora (Srivastava et al. Citation2021) and the interactions between different flora (Bartek et al. Citation2021). In contrast, information regarding the gut microbiota of non-human primates is relatively limited. The results of applying non-human primate models to gut microbiota research may elucidate the role of the gut microbiota in human health and disease(Firrman et al. Citation2019). It has been reported that the relative abundance of Lachnospiraceae and Ruminococcaceae is higher under wild conditions in snub-nosed monkeys (Rhinopithecus), and microorganisms in these families can digest complex plant matter and produce butyric acid. However, the relative abundance of Prevotella and Bacteroides, which degrade monosaccharides and carbohydrates, is higher in captive conditions in snub-nosed monkeys (Hale et al. Citation2019). Furthermore, Mollicutes, Tenericutes, Coprobacillus and Faecalitale were found to be possible biomarkers of spontaneous osteoarthritis in rhesus monkeys (Macaca mulatta), while Prevotella and Desulfobacterales were higher in normal rhesus monkeys than in spontaneously osteoarthritic rhesus monkeys (Yan et al. Citation2021). The relative abundance of Succinivibrio increased significantly in winter, and Succinivibrio efficiently fermented glucose and produced acetate and succinate, and contributed to the metabolism of different types of fatty acids in Tibetan macaques (Macaca thibetana) in winter and spring (Sun et al. Citation2016). Therefore, intestinal flora composition is closely related to habitat, health, and seasonal factors.
High-throughput sequencing technology can elucidate the relationship between intestinal flora and host at the molecular level, providing the feasibility of improving the intestinal microenvironment through dietary interventions, preventing and treating diseases, and formulating appropriate feeding programmes (Zhao et al. Citation2018). The 16S rRNA gene is a highly conserved and specific DNA sequence that encodes the corresponding bacterial rRNA, which is present in the genome of all bacteria (Miyoshi and Chang Citation2017). Moreover, the rise of 16S rRNA gene detection technology and PICRUSt analysis methods will facilitate further exploration of the relationship between intestinal flora and host (Langille et al. Citation2013). Therefore, the study of the composition, genes, and functions of the gut microbiota in animals has became a research hotspot.
In summary, the evaluation of gut microbial diversity and composition helps to provide a greater understanding of the resilience of wild primates and facilitates captive population management (Grassotti et al. Citation2021). Therefore, we conducted a study on the gut microbiota of black-capped capuchins using fecal samples collected from two wildlife parks, namely, the Erdos Wildlife Zoo and the Daqing Mountain Wildlife Zoo in Hohhot in Inner Mongolia. The purpose of this study is to reveal information about the types, abundance, and predicted gene functions of the gut bacteria in black-capped capuchins. Additionally, a comparative analysis will be conducted on the gut microbiota of black-capped capuchins raised in two different regions. This study aims to provide a foundation for a more in-depth investigation into the gut microbiota of black-capped capuchins.
2. Materials and methods
2.1. Fecal sample collection and transportation
Fecal samples of black-capped capuchins for this study were collected from November 11, 2021 to November 25, 2021 at Erdos Wildlife Park (ES) and Hohhot Daqingshan Wildlife Park (HT), Inner Mongolia Autonomous Region, China (). Specific samples were collected and transported as follows: the feces of black-capped capuchins were broken open with a sterile spoon, the central part of the feces was clamped and immediately loaded into sterile 50 mL centrifuge tubes. The tube was then sealed with sealing film, labelled, and stored in a cooler with dry ice. The samples were transported to the laboratory and stored in a −80°C freezer for further use.
Figure 1. Schematic diagram of fecal sample collection sites of black-capped capuchins; The triangle marker is the fecal sample collection point.
2.2. DNA extraction and PCR amplification
Total DNA extraction from microbial communities was performed according to the instructions of the E.Z.N. A.® soil DNA kit (Omega Bio-tek, Norcross, GA, U.S.). The quality of DNA extraction was detected using 1% agarose gel electrophoresis, and DNA concentration and purity were determined using NanoDrop2000; The 16S rRNA gene V3-V4 variable region was PCR-amplified using 338F (5'-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5'-GGACTACHVGGGTWTCT AAT-3’). Amplification was performed at (Chen, Zhang, et al. Citation2021) with the following amplification procedure: pre-denaturation at 95°C for 3 min, 27 cycles (denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 30 s), followed by stable extension at 72°C for 10 min, and finally storage at 4°C. The PCR reaction system was: 5× TransStart FastPfu buffer 4 μL, 2.5 mM dNTPs 2 μL, and Upstream primer (5 uM) 0.8 μL, downstream primer (5 uM) 0.8 μL, TransStart FastPfu DNA polymerase 0.4 μL, template DNA 10 ng, ddH2O to make up to 20 μL. 3 replicates of each sample were used.
2.3. Illumina Miseq sequencing
PCR products from the same sample were mixed and recovered using a 2% agarose gel, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), detected by 2% agarose gel electrophoresis and quantified by Quantus™ Fluorometer (Promega, USA). Fluorometer (Promega, USA) for quantification of recovered products (Xue et al. Citation2017). Library construction was performed using NEXTflexTM Rapid DNA-Seq Kit (Bioo Scientific, USA) with the following steps: (1) splice linkage; (2) removal of splice self-linked fragments using magnetic bead screening; (3) enrichment of library templates using PCR amplification; (4) recovery of PCR products from magnetic beads to obtain the final library. Sequencing was performed using Illumina’s Miseq PE300/NovaSeq PE250 platform (Shanghai Meiji Biomedical Technology Co., Ltd.). Raw data were uploaded to the NCBI SRA database (sequence number: PRJNA818119).
2.4. Data quality control
The raw sequencing data were subjected to quality control using the fastp software (https://github.com/OpenGene/fastp, version 0.20.0) (Chen et al. Citation2018) and the assembly was performed using the FLASH software (http://www.cbcb.umd.edu/software/flash, version 1.2.7) (Luo et al. Citation2021). Based on default parameters, denoising of the optimized sequences after quality control and merging was performed using the DADA2 (Callahan et al. Citation2016) plugin within the Qiime2 workflow (Bolyen et al. Citation2019).
2.5. Data analysis
Using the Sliva 16S rRNA gene database (v138), taxonomic classification analysis of Amplicon Sequence Variants (ASVs) was conducted using the Naive Bayes classifier within the Qiime2 framework. The Venn data table was analyzed using samples at a similarity level of 100% for ASVs or other taxonomic levels. The statistical analysis was conducted using the R language (version 3.3.1) tools. Using Mothur software (v.1.30.2) to calculate α diversity indices, including Chao, Shannon, and Shannon even indices, at different taxonomic levels for rarefied samples. Student’s t-test and False Rate Discovery (FRD) multiple testing correction were employed to assess the significance of inter-group differences in a two-tailed test. A test was conducted based on the Hierarchical clustering analysis was performed based on the diversity distance matrix, and a tree structure was constructed using the Bray–Curtis distance algorithm to visualize the degree of similarity or difference in community composition across environmental samples. Qiime software was used to calculate
the diversity distance matrix was used to draw the tree using the R language (version 3.3.1). Circos plots were calculated using Circos-0.67-7 software (http://circos.ca/) at different taxonomic levels for the sampled flats, and visual circle plots reflecting the proportion of dominant species distributed in each sample (or group), and the proportion of each dominant species distributed in different samples (or groups). Differential analysis of species abundance at the top 10 taxa between groups using the ‘stats’ package in R software (version 3.3.1). The analysis employed Student’s t-test with False Rate Discovery (FRD) correction for multiple testing, assessing the significance of interspecies differences across different taxonomic levels. Additionally, utilizing PICRUSt2 software and leveraging information from the COG database, we parsed descriptive and gene metabolic function information for each COG from the eggNOG database (http://eggnog.embl.de/). This process allowed us to generate a gene metabolic function abundance spectrum. Statistical analysis of inter-group differences in gene metabolic functions was performed using Student’s t-test with FRD correction in a two-tailed test.
3. Analysis of results
3.1. Analysis of the species composition of intestinal bacteria
The analysis was conducted at different taxonomic levels, including phylum, family, and genus, using 100% ASV clustering to obtain the composition of bacterial species at each taxonomic level. The results showed () that the intestinal bacteria of captive black-capped capuchins in the two wildlife parks could be clustered into 2296 ASVs, belonging to 16 phyla, 99 families and 210 genera, respectively. At the ASV level, the shared number of ASVs was 36 for both groups, but the number of unique ASVs was 1345 and 915 for ES and HT, respectively. At the genus level, the number of genera shared by the two groups was 102, but the number of genera unique to ES and HT was 50 and 58, respectively. At the family level, the number of families shared by both groups was 56, but the number of families unique to ES and HT was 24 and 19, respectively. At the phylum level, the number of shared phyla in both groups is 11, but the number of phyla unique to ES and HT was 2 and 3. Thus, the intestinal bacteria of black-capped capuchins in captivity in these two wildlife parks differed in species composition, and the number of intestinal bacterial species at different taxonomic levels of ES was greater than that of HT. Therefore, there are differences in the species composition of the gut bacteria in captive black-capped capuchins between these two wildlife parks. Apart from the phylum level, ES has a greater number of gut bacterial species compared to HT.
Table 1. Species composition of bacteria at different taxonomic levels.
3.2. Analysis of the α-diversity of intestinal bacteria
The intestinal bacterial α-diversity of black-capped capuchins was evaluated by three indices, including richness (Chao), diversity (Shannon) and evenness (Shannon even). The results showed () that there were no significant differences (P > 0.05) in the richness of bacterial species composition (Chao index) between ES and HT at the ASV, genus, family, and phylum taxonomic levels. However, ES exhibited significantly higher bacterial diversity (Shannon index) compared to HT at the ASV, genus, family, and phylum levels (P < 0.05). Similarly, the bacterial diversity in ES, as measured by the Shannon even index, was significantly higher than HT at the ASV, genus, family, and phylum levels (P < 0.05). These findings indicate that there is a difference in the diversity of gut bacteria in captive black-capped capuchins between the two wildlife parks, with ES consistently showing significantly higher evenness in bacterial species composition than HT.
Table 2. Analysis summary of α diversity index at different classification levels.
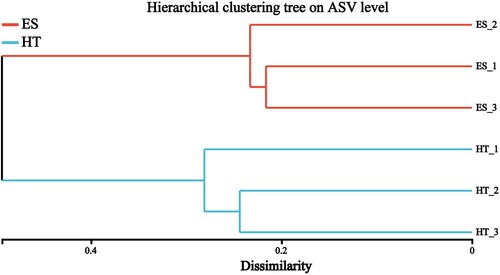
3.3. Cluster analysis of stratified sets of intestinal bacteria samples
According to Bray_curtis difference analysis showed (), the intestinal bacteria of black-capped capuchins at the same location have high flora similarity.
Figure 2. Cluster analysis of intestinal bacterial flora of black-capped capuchins in different regions.
3.4. Analysis of the composition of the intestinal bacterial community
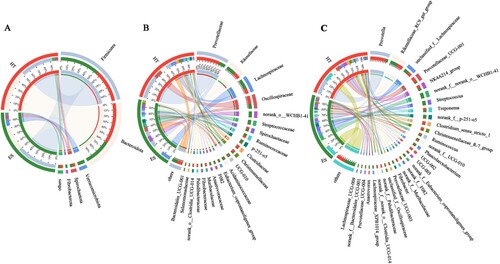
Circos plots were used to reflect the proportion of dominant species composition in each group and also the relative abundance distribution of each dominant species in different groups (). At the phylum level (A), the dominant bacterial phylum for ES and HT was Bacteroidota (33%, 51%) and Firmicutes (51%, 41%). Moreover, the sum of the relative abundance of these two phyla was greater than 80% in both ES and HT. Moreover, Verrucomicrobiota and Fibrobacterota were clearly higher in ES than HT, but Bacteroidota was clearly lower in ES than HT. At the family level (B), the dominant bacterial families in ES and HT were Prevotellaceae (9%, 35%), Rikenellaceae (13%, 9%), Lachnospiraceae (15%, 8%) and Oscillospiraceae (7%, 12%). Moreover, p-251-o5, Streptococcaceae, norank_o__WCHB1-41 were clearly higher in ES than HT, but Prevotellaceae were clearly lower in ES than HT. At the genus level (C), ES and HT shared the dominant bacterial genera Prevotella (4%, 24%), Rikenellaceae_RC9_gut_group (12%, 9%) and Prevotellaceae_UCG-001 (2%, 8%). Moreover, norank_f__norank_o__WCHB1-41 and Streptococcus were clearly higher in ES than HT, but Prevotella and Prevotellaceae_UCG-001 were clearly lower in ES than HT. In addition, we found many unclassified genera in the intestinal bacteria of black-capped capuchins, such as norank_f_norank_o_WCHB1-41 (9%, 0%), norank_f_p-251-05 (5%, 1%) and unclassified_f_ Lachnospiraceae (8%, 6%), etc., to be studied in depth.
Figure 3. Circos plot of the composition of the intestinal bacterial community at different taxonomic levels; A, Phylum; B, Family; C, Genus.
3.5. Analysis of differences between groups of intestinal bacterial species
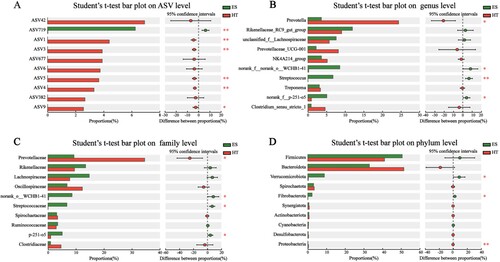
Analysis of between-group differences in relative abundance among the top 10 species of intestinal bacteria using Student’s t-test showed that at the ASV level (A), ASV719 was significantly higher in ES than in HT (P < 0.01). On the other hand, ASV1, ASV3, ASV4, and ASV5 were significantly higher in HT than in ES (P < 0.01), and ASV9 was significantly higher in HT than in ES (P < 0.05). The remaining four ASVs, although showing differences, were not statistically significant (P > 0.05). At the genus level (B), norank_f_p-251-o5 and norank_f_norank_o_WCHB1-41 were significantly higher in ES than in HT (P < 0.05), while Streptococcus was significantly higher in HT than in ES (P < 0.01). Prevotella was significantly higher in HT than in ES (P < 0.05), and the remaining seven genera, although showing differences, were not statistically significant (P > 0.05). At the family level (C), Prevotellaceae, norank_o_WCHB1-41, and p-251-o5 were significantly higher in ES than in HT (P < 0.05). In HT, Prevotellaceae was significantly higher than in ES (P < 0.05), and the remaining six families, although showing differences, were not statistically significant (P > 0.05). At the phylum level (D), Verrucomicrobiota and Fibrobacterota were significantly higher in ES than in HT (P < 0.05). In HT, Proteobacteria was significantly higher than in ES (P < 0.01), and the remaining seven phyla, although showing differences, were not statistically significant (P > 0.05). This shows that there are significant differences in the composition of the intestinal bacterial communities of captive black-capped capuchins in the two wildlife parks, and that the bacterial families with significant differences (p-251-o5 and Streptococcaceae) correspond to the bacterial genera (norank_f__p-251-o5 and Streptococcus) with corresponding relationships. The results provide a basis for the study of intestinal bacteria of black-capped capuchins and the isolation and purification of dominant bacteria.
Figure 4. Analysis of differences and significance tests between species groups at different taxonomic levels; *P < 0.05 and **P < 0.01.
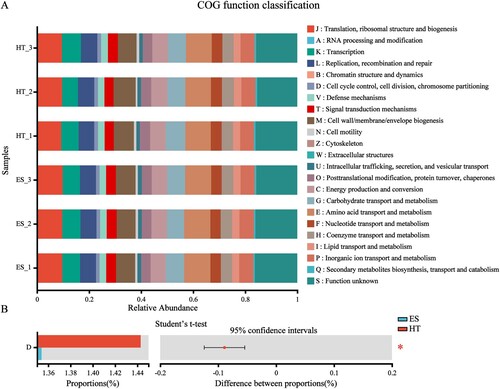
3.6. Prediction of gene function in intestinal bacterial metabolism
PICRUSt2 analysis using 16S rRNA gene data from each sample to predict intestinal bacterial metabolic gene function in black-capped capuchins is shown in . A total of 23 metabolic gene functions were predicted in the intestinal bacteria of black-capped capuchins (A), among which the main gene functions were amino acid transport and metabolism, translation/ribosomal structure and biogenesis, cell wall/membrane/envelope biogenesis, etc. As indicated by Student’s t-test (B), only 1 metabolic gene function was significantly different between study sites, i.e. Cell cycle control, cell division, and chromosome partitioning were significantly higher in HT than ES (P < 0.05). In addition, the mean relative abundance of function unknown in ES and HT was 16.0% and 15.7%, respectively. thus we found that a large proportion of gene functions in the intestinal bacteria of black-capped capuchins have not been elucidated and studied, which provides us with good research directions.
Figure 5. Functional classification statistics and difference test of metabolic genes in intestinal bacteria; A, COG functional abundance statistics; B, COG functional abundance Student’s t-test; *P < 0.05 and **P < 0.01.
4. Discussion
The intestinal flora is a complex dynamic ecosystem, involved in host digestion, metabolism, regulation of immunity and inhibition of pathogen colonization, among other roles (Khurana et al. Citation2021). There are many factors affecting the structure of the intestinal flora of monkeys, including host factors (species type, health, developmental stage), environmental factors (food intake, habitat type), microbial factors (biotin, enzymes, metabolic capacity), etc (Kohl et al. Citation2013, Long et al. Citation2020, Tong et al. Citation2020). Animals raised in different regions may have variations in the composition of their intestinal microbiota due to differences in temperature, range of activity, and types of food (Stumpf et al. Citation2016). In this paper, we analyzed the structure of the intestinal flora of captive black-capped capuchins in two wildlife parks based on high-throughput sequencing of 16S rRNA gene, in order to provide a basis for the isolation and screening of dominant bacteria in the intestinal flora. Bacterial alpha diversity analysis indicated that there were no significant differences (P > 0.05) in richness (Chao index) at the ASV, genus, family, and phylum levels between ES and HT. However, the bacterial diversity (Shannon index) and evenness (Shannon even index) in ES were significantly higher than in HT at the ASV, genus, family, and phylum levels (P < 0.05). Thus, there were significant differences in the intestinal bacterial diversity of captive black-capped capuchins in the two wildlife parks, and the bacterial species composition was higher in ES than HT, due to the effect of rearing conditions on the intestinal bacterial diversity and richness of black-capped capuchins. This was further validated by the bacterial community Bray_curtis difference analysis, i.e. the intestinal bacteria of black-capped capuchins in the same region had high flora similarity. Therefore, we determined that there is a correlation between the intestinal bacterial diversity of black-capped capuchins and the rearing environment and rearing conditions. The dominant phyla of intestinal bacteria in black-capped capuchins are both Firmicutes and Bacteroidota, which are similar to the dominant phyla of non-human primates such as Sichuan snub-nosed monkeys (rhinopithecus roxellana) (Liu et al. Citation2018, Wang et al. Citation2021), and rhesus monkeys (Rendina et al. Citation2019), etc. Therefore, we can determine that Firmicutes and Bacteroidota are also intrinsic flora in the intestine of black-capped capuchins. Moreover, the former mainly promotes the degradation of fibre in food and converts cellulose into volatile fatty acids to promote food digestion and growth and development, while the latter mainly promotes the digestion and absorption of protein and carbohydrate in food, thus promoting the development of the gastrointestinal immune system (Jiang et al. Citation2021). As shown in Circos plot, the ratio of Firmicutes/Bacteroidota is much higher in ES (1.54) than HT (0.80), and we know from field observations and assessments that black-capped capuchins in ES are more obese than in HT, a result that it is consistent with previous findings (Grigor’eva. Citation2020) who reported that obesity leads to elevated results of Firmicutes/Bacteroidota ratio. Furthermore, two phyla with relatively high abundance in ES, Fibrobacterota (2%) and Verrucomicrobiota (9%), were also present. Among them, Fibrobacterota is one of the main contributors to the digestion of lipopolysaccharides (LPS) (Chen, Wang, et al. Citation2021), While Verrucomicrobiota plays an important role in degrading polysaccharides and is a key degrading phylum of complex sulfate polysaccharides (e.g. polysaccharides containing rockulose and rhamnose) (Orellana et al. Citation2022). Thus, we can hypothesize that the type (e.g. fresh vegetables and fresh fruits) and amount of polysaccharides in the diet of black-capped capuchins in ES is superior to that in HT. Moreover, this result further indicates that the level of feeding of black-capped capuchins in ES is higher than that in HT and is consistent with the finding that the ratio of Firmicutes/Bacteroidota is much higher in ES than HT.
The dominant families in the family level ES and HT were Lachnospiraceae (15%, 8%), Rikenellaceae (13%, 9%), Prevotellaceae (9%, 35%) and Oscillospiraceae (7%, 12%). It has been reported that Lachnospiraceae can produce large amounts of short-chain fatty acids and that short-chain fatty acids are important in regulating the immune and inflammatory responses of the body (Guo et al. Citation2020); while Rikenellaceae are considered to be the main cause of degradation of plant polysaccharides such as cellulose, pectin and starch (Su et al. Citation2014). Prevotellaceae were significantly lower in ES than HT (P < 0.05), and Prevotellaceae had an important role in the breakdown and utilization of hemicellulose, pectin, starch, carbohydrates and monosaccharides derived from fruits, cereals and young leaves(Tremaroli and Backhed. Citation2012). In addition, Streptococcaceae were significantly higher in ES (7.65%) than in HT (<1%), and Streptococcaceae were frequently recovered from intestinal microorganisms of domesticated animals, unpasteurized dairy products or spoiled food. It can be assumed that the black-capped capuchins at ES may have been fed dairy products (e.g. fresh milk, yogurt) or there were cases of feeding by visitors. Therefore, the main roles of the dominant bacterial families in the gut bacteria of black-capped capuchins are related to the maintenance of body immunity and the enhancement of digestion and absorption (polysaccharides, fibre, etc.). Moreover, the bacterial families with significant differences corresponded to bacterial genera. The results provide a basis for the study of intestinal bacteria of black-capped capuchins and the isolation and purification of dominant bacteria. Moreover, we found that there are many unclassified genera in the gut bacteria of black-capped capuchins, such as norank_f_norank_o_WCHB1-41, norank_f_p-251-05 and unclassified_f_Lachnospiraceae, which need to be studied in depth.
The PICRUSt2 analysis revealed that a total of 23 metabolic gene functions were predicted in the intestinal bacteria of black-capped capuchins, among which amino acid transport and metabolism, carbohydrate transport and metabolism and energy production and conversion were relatively abundant, which may be related to the dietary habits of black-capped capuchins, as their main food items are fruits, vegetables, plants, cereals, and a small amount of meat food. In addition, the relative abundance of translation/ribosomal structure and biogenesis and cell wall/membrane/envelope biogenesis was higher in this study, which may be closely related to bacterial growth and reproduction and host resistance (Lian et al. Citation2021). Moreover, the relative abundance of gene families without known functions in the gut bacteria of black-capped capuchins surpassed that of gene families with known functions, so we found that a large proportion of gene functions in the gut flora of black-capped capuchins had not been elucidated and studied, which provided us with good research directions. In conclusion, the results of this study fill the gap in the study of intestinal bacterial diversity and its gene function in black-capped capuchins, and provide a reference for the isolation and screening of the dominant flora in the intestine of black-capped capuchins, as well as a good research direction.
5. Conclusion
This study is the first analysis of intestinal bacterial diversity and gene function prediction in black-capped capuchins by high-throughput sequencing of 16S rRNA genes. The results of the study revealed significant differences in intestinal bacterial diversity and community composition between captive black-capped capuchins from two wildlife parks. Moreover, the intestinal bacteria of black-capped capuchin at the same site had high flora similarity. Black-capped capuchins intestinal bacteria can be clustered into 2296 ASVs, belonging to 16 phyla, 99 families and 210 genera, respectively. However, there were differences in the dominant bacterial families and dominant bacterial genera between ES and HT, and the bacterial families with significant differences corresponded to the bacterial genera. In addition, we found many unclassified genera in the intestinal bacteria of black-capped capuchins, which need to be studied in depth. A total of 23 metabolic gene functions were predicted by PICRUSt2 analysis in the intestinal bacteria of black-capped capuchins, among which the main known gene functions were amino acid transport and metabolism, translation/ribosomal structure and biogenesis, cell wall/membrane/envelope biogenesis, etc. Moreover, we found that a large proportion of gene functions in the intestinal bacteria of black-capped capuchins have not been elucidated and studied, which provides us with good research directions. In summary, the results of this study fill the gap in the study of intestinal bacterial diversity and its gene functions in black-capped capuchins, and provide a reference for exploring the isolation and screening of dominant bacteria and novel functional genes in the intestinal microbes of black-capped capuchins.
Author contributions statement
Conceptualization: W.B.; Methodology: W.B., J.Y., G.B., Y.H., M.L., X.Y.; Formal; Analysis: W.B., J.Y; Investigation: J.L., Y.S., F.L.; Data Curation: J.Y., Y.H.; Writing: W.B., J.Y.; Original Draft Preparation: W.B., J.Y.; Writing – Review and Editing: W.B.; All authors agree to be accountable for all aspects of the work.
Ethics Statement
This project is led by Professor Wuyundalai at Inner Mongolia Agricultural University and is a major project under the Science and Technology Plan of Inner Mongolia Autonomous Region. The project is titled ‘Research on Probiotic Technology and Its Application in Agriculture and Animal Husbandry.’ It involves animal experiments, and after review by the Experimental Animal Welfare and Ethics Committee of Inner Mongolia Agricultural University, it has been deemed to comply with ethical principles, and the implementation of the project has been approved. Additionally, Erdos Wildlife Park and Daqingshan Wildlife Park have agreed to allow Professor Wuyundalai’s team to collect animal feces in the wildlife parks for research on the microbial diversity associated with animal feces.
Appendix_II_1
Download PDF (1.5 MB)Acknowledgments
We acknowledge the support and assistance from Ordos Wildlife Park and Hohhot Daqingshan Wildlife Park in collecting fecal samples of black-capped capuchins.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
Genetic sequencing data (BioSample SAMN26816508, SAMN26816509, SAMN26816510, SAMN26816511, SAMN26816512, SAMN26816513) of six fecal samples from black-capped capuchin completed by Shanghai Majorbio Bio-Pharm Technology Co., Ltd, and this study is available in GenBank under BioProject number PRJNA818119.
Additional information
Funding
References
- Bartek N, Bj B, Gil B, Milena J. 2021. Gut microbiota-informed precision nutrition in the generally healthy individual: Are We There Yet. Current Developments in Nutrition. 5(9):nzab107. doi:10.1093/cdn/nzab107.
- Beam A, Clinger E, Hao L. 2021. Effect of diet and dietary components on the composition of the gut microbiota. Nutrients. 13(8):2795–2795. doi:10.3390/nu13082795.
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 37(8):852–857. doi:10.1038/s41587-019-0209-9.
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 13(7):581–583. doi:10.1038/nmeth.3869.
- Chen H, Wang C, Huasai S, Chen A. 2021. Effects of dietary forage to concentrate ratio on nutrient digestibility, ruminal fermentation and rumen bacterial composition in Angus cows. Sci Rep. 11(1):17023. doi:10.1038/s41598-021-96580-5.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890. doi:10.1093/bioinformatics/bty560.
- Chen T, Zhang YQ, Zhang YY, Shan CJ, Zhang YY, Fang K, Xia YK, Shi ZH. 2021. Relationships between gut microbiota, plasma glucose and gestational diabetes mellitus. J Diabetes Investig. 12(4):641–650. doi:10.1111/jdi.13373.
- Espirito Santo C, Caseiro C, Martins MJ, Monteiro R, Brandao I. 2021. Gut microbiota, in the halfway between nutrition and lung function. Nutrients. 13(5):1716. doi:10.3390/nu13051716.
- Fehlberg LK, Lattimer JM, Vahl CI, Drouillard JS, Douthit TL. 2020. Digestibility of diets containing calcium salts of fatty acids or soybean oil in horses1. Transl Anim Sci. 4(2):txaa001. doi:10.1093/tas/txaa001.
- Firrman J, Liu L, Tanes C, Bittinger K, Mahalak K, Rinaldi W. 2019. Metagenomic assessment of the Cebus apella gut microbiota. Am J Primatol. 81(10-11):e23023. doi:10.1002/ajp.23023.
- Gaulke CA, Arnold HK, Humphreys IR, Kembel SW, O’Dwyer JP, Sharpton TJ. 2018. Ecophylogenetics clarifies the evolutionary association between mammals and their gut microbiota. mBio. 9(5):e01348–e01318. doi:10.1128/mBio.01348-18.
- Grassotti TT, Kothe CI, Prichula J, Mohellibi N, Mann MB, Wagner PGC, Campos FS, Campos AAS, Frazzon J, Frazzon APG. 2021. Fecal bacterial communities of wild black capuchin monkeys (Sapajus nigritus) from the Atlantic Forest biome in Southern Brazil are divergent from those of other non-human primates. Curr Res Microb Sci. 2:100048. doi:10.1016/j.crmicr.2021.100048.
- Grigor’eva IN. 2020. Gallstone disease, obesity and the firmicutes/bacteroidetes ratio as a possible biomarker of gut dysbiosis. J Pers Med. 11(1):13. doi:10.3390/jpm11010013.
- Guo H, Chou WC, Lai Y, Liang K, Tam JW, Brickey WJ, Chen L, Montgomery ND, Li X, Bohannon LM, et al. 2020. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science. 370(6516):eaay9097. doi:10.1126/science.aay9097.
- Hale VL, Tan CL, Niu K, Yang Y, Zhang Q, Knight R, Amato KR. 2019. Gut microbiota in wild and captive Guizhou snub-nosed monkeys, Rhinopithecus brelichi. Am J Primatol. 81(10-11):e22989. doi:10.1002/ajp.22989.
- Han Z, Li K, Shahzad M, Zhang H, Luo H, Qiu G, Lan Y, Wang X, Mehmood K, Li J. 2017. Analysis of the intestinal microbial community in healthy and diarrheal perinatal yaks by high-throughput sequencing. Microb Pathog. 111:60–70. doi:10.1016/j.micpath.2017.08.025.
- Jiang F, Gao H, Qin W, Song P, Wang H, Zhang J, Liu D, Wang D, Zhang T. 2021. Marked seasonal variation in structure and function of gut microbiota in forest and alpine musk deer. Front Microbiol. 12:699797. doi:10.3389/fmicb.2021.699797.
- Khurana H, Sharma M, Bharti M, Singh DN, Negi RK. 2021. Gut milieu shapes the bacterial communities of invasive silver carp. Genomics. 113(2):815–826. doi:10.1016/j.ygeno.2021.01.013.
- Kohl KD, Cary TL, Karasov WH, Dearing MD. 2013. Restructuring of the amphibian gut microbiota through metamorphosis. Environ Microbiol Rep. 5(6):899–903. doi:10.1111/1758-2229.12092.
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 31(9):814–821. doi:10.1038/nbt.2676.
- Levi T, Silvius KM, Oliveira LF, Cummings AR, Fragoso JM. 2013. Competition and facilitation in the capuchin–squirrel monkey relationship. Biotropica. 45(5):636–643. doi:10.1111/btp.12046.
- Lian C, Shuxian L, Qi X, Ying L, Xuexia L, Yanfu Q, Guogan W, Hong L. 2021. Composition and diversity of gut microbiota in pomacea canaliculata in sexes and between developmental stages. BMC Microbiol. 21(1):200. doi:10.1186/s12866-021-02259-2.
- Liu L, Wang Q, Wu X, Qi H, Das R, Lin H, Shi J, Wang S, Yang J, Xue Y, et al. 2020. Vancomycin exposure caused opportunistic pathogens bloom in intestinal microbiome by simulator of the human intestinal microbial ecosystem (SHIME). Environ Pollut. 265(Pt B):114399. doi:10.1016/j.envpol.2020.114399.
- Liu X, Fan P, Che R, Li H, Yi L, Zhao N, Garber PA, Li F, Jiang Z. 2018. Fecal bacterial diversity of wild Sichuan snub-nosed monkeys (Rhinopithecus roxellana). Am J Primatol. 80(4):e22753. doi:10.1002/ajp.22753.
- Long J, Xiang J, He T, Zhang N, Pan W. 2020. Gut microbiota differences during metamorphosis in sick and healthy giant spiny frogs (Paa spinosa) tadpoles. Lett Appl Microbiol. 70(2):109–117. doi:10.1111/lam.13251.
- Luo J, Zhang HF, Lu JC, Ma CL, Chen TT. 2021. Antidiabetic effect of an engineered bacterium Lactobacillus plantarum-pMG36e-GLP-1 in monkey model. Syn Syst Biotechno. 6(4):272–282. doi:10.1016/j.synbio.2021.09.009.
- Miyoshi J, Chang EB. 2017. The gut microbiota and inflammatory bowel diseases. Transl Res. 179:38–48. doi:10.1016/j.trsl.2016.06.002.
- Nogal B, Blumberg JB, Blander G, Jorge M. 2021. Gut microbiota–informed precision nutrition in the generally healthy individual: are we there yet? Curr Dev Nutr. 5(9):nzab107. doi:10.1093/cdn/nzab107.
- Orellana LH, Francis TB, Ferraro M, Hehemann JH, Fuchs BM, Amann RI. 2022. Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms. ISME J. 16(3):630–641. doi:10.1038/s41396-021-01105-7.
- Rendina DN, Lubach GR, Phillips GJ, Lyte M, Coe CL. 2019. Maternal and breast milk influences on the infant gut microbiome, enteric health and growth outcomes of rhesus monkeys. J Pediatr Gastroenterol Nutr. 69(3):363–369. doi:10.1097/MPG.0000000000002394.
- Song Q, Wang Y, Huang L, Shen M, Yu Y, Yu Q, Chen Y, Xie J. 2021. Review of the relationships among polysaccharides, gut microbiota, and human health. Food Res Int. 140:109858. doi:10.1016/j.foodres.2020.109858.
- Srivastava A, Prabhakar MR, Mohanty A, Meena SS. 2021. Influence of gut microbiome on the human physiology. Syst Microbiol Biomanuf. 2(2):1–15. doi:10.1007/S43393-021-00052-W.
- Stumpf RM, Gomez A, Amato KR, Yeoman CJ, Polk JD, Wilson BA, Nelson KE, White BA, Leigh SR. 2016. Microbiomes, metagenomics, and primate conservation: new strategies, tools, and applications. Biol Conserv. 199:56–66. doi:10.1016/j.biocon.2016.03.035.
- Su XL, Tian Q, Zhang J, Yuan XZ, Shi XS, Guo RB, Qiu YL. 2014. Acetobacteroides hydrogenigenes gen. nov., sp. nov., an anaerobic hydrogen-producing bacterium in the family Rikenellaceae isolated from a reed swamp. Int J Syst Evol Microbiol. 64(Pt 9):2986–2991. doi:10.1099/ijs.0.063917-0.
- Sun B, Wang X, Bernstein S, Huffman MA, Xia DP, Gu Z, Chen R, Sheeran LK, Wagner RS, Li J. 2016. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep. 6:26035. doi:10.1038/srep26035.
- Taal Levi, Kirsten M Silvius, Luiz FB Oliveira, Anthony R Cummings, Jose MV Fragoso. 2013. Competition and facilitation in the capuchin-squirrel monkey relationship. Biotropica. 45(5):636–643. doi:10.1111/btp.12046.
- Tong Q, Cui LY, Hu ZF, Du XP, Abid HM, Wang HB. 2020. Environmental and host factors shaping the gut microbiota diversity of brown frog Rana dybowskii. Sci Total Environ. 741:140142. doi:10.1016/j.scitotenv.2020.140142.
- Tremaroli V, Backhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature. 489(7415):242–249. doi:10.1038/nature11552.
- Wang X, Wang Z, Pan H, Qi J, Li D, Zhang L, Shen Y, Xiang Z, Li M. 2021. Captivity influences the gut microbiome of rhinopithecus roxellana. Front Microbiol. 12:763022. doi:10.3389/fmicb.2021.763022.
- Xue L, Ren H, Li S, Leng X, Yao X. 2017. Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front Microbiol. 8:2377. doi:10.3389/fmicb.2017.02377.
- Yan YP, Yi XY, Duan YC, Jiang B, Huang TZ, Inglis BM, Zheng BR, Si W. 2021. Alteration of the gut microbiota in rhesus monkey with spontaneous osteoarthritis. BMC Microbiol. 21(1):328. doi:10.1186/s12866-021-02390-0.
- Yao LJ, Li X, Zhou ZT, Shi DS, Li ZL, Li SF, Yao H, Yang JY, Yu HL, Xiao YC. 2021. Age-based variations in the gut microbiome of the shennongjia (Hubei) golden snub-nosed monkey (Rhinopithecus roxellana hubeiensis). Biomed Res Int. 2021:6667715.
- Zhao LL, Yin HC, Lu TF, Niu YJ, Zhang YY, Li SQ, Wang YP, Chen HY. 2018. Application of high-throughput sequencing for microbial diversity detection in feces of specific-pathogen-free ducks. Poult Sci. 97(7):2278–2286. doi:10.3382/ps/pex348.