
Abstract
Objective and importance: To verify the presence of β-thalassemia in subjects showing hematologic phenotype of α-thalassemia, conduct normal molecular sequence analysis of the α-globin genes, and detect the absence of the most frequent α-thalassemia deletions.
Clinical presentation: A patient from Apulia (Southern Italy) was referred to our institution for the occasional founding of hypochromic polyglobulia and microcytic red blood cells associated with normal levels of Hb A2 and Hb F and normal iron parameters.
Intervention and technique: The patient has been investigated using Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), quantitative real-time PCR, restriction analysis, and gap-PCR. A novel deletion, the Italian (ϵγδβ)0-thalassemia, has been identified. The 5′ breakpoint was within a LINE element of 80 kb 3′ of the ε-globin gene, and the 3′ breakpoint was within a 160-bp palindrome of about 30 kb 5′ of the β-globin gene. The breakpoint region was characterized by the presence of a microhomology (5′-TCT-3′) and of an insertion of 43 bp owing to the duplication of the 160-bp palindrome. Comparison of the Hb and Hb A2 values of (ϵγδβ)0-thalassemia from the literature with those of (molecularly known) thalassemia carriers indicated a higher level of Hb A2 with respect to α-thalassemia and a lower level of Hb with respect to β0-thalassemia carriers.
Conclusion: In this study, we report the first (ϵγδβ)0-thalassemia case identified in Italy. To avoid misdiagnosis of β-thalassemia, we suggest verifying the presence of large deletions of the β-globin gene cluster in subjects showing a higher border line level of Hb A2 and a lower level of Hb.
Introduction
Beta-thalassemia is one of the most frequent hereditary diseases in the Mediterranean area. The quantification of Hb A2 (the minor adult hemoglobin) is essential for the routine diagnosis of beta-thalassemia trait because its concentration is raised in most carriers of beta-thalassemia; moreover, the correct diagnosis is important to prevent the Cooley's anemia.Citation1
The (ϵγδβ)0-thalassemias are rare deletions identified only in heterozygous form. They are caused by long deletions in the beta-globin cluster and molecularly are classified into two groups: group I removes all, or a greater part of the beta-globin cluster, including the beta-globin gene; group II removes extensive upstream regions, leaving the beta-globin gene itself intact although its expression is silenced because of inactivation of the upstream beta-locus control region (β-LCR).Citation2,Citation3 Up to now, 32 (ϵγδβ)0-thalassemia deletions have been described, but the complete characterization has been achieved in only 14 of them.Citation3–Citation6
Adult heterozygotes show a phenotype similar to that of hematological carriers of beta-thalassemia, but with normal levels of Hb A2 due to the loss of one delta locus and normal or minimally increased Hb F; the red cells are relatively more hypochromic and small. The normal levels of Hb A2 make the hematologic phenotype similar to that of carriers of alpha-thalassemia. As already described, these deletions are characterized by a moderately severe neonatal hemolytic anemia and they are distinguished from beta-thalassemic patients because they may need red blood cell (RBC) transfusion for the first 6 months of life, but this remits spontaneously with the increasing production of beta-globin in the first year of life.Citation4
Despite the extreme heterogeneity of the molecular basis of the beta-thalassemia in Italy, the (ϵγδβ)0-thalassemia deletions have never been identified.Citation7,Citation8 We carried out an epidemiological project on the molecular basis of alpha-thalassemia in Southern Italy and we identified a patient with hematologic phenotype of alpha-thalassemia (microcytosis, polycythemia, and normal Hb A2), which showed normal molecular sequence analysis of the alpha- and beta-globin genes and absence of the most frequent alpha- or beta-thalassemia deletions. The analysis of the alpha- and beta-globin gene clusters by multiplex ligation probe amplification (MLPA) (MRC Holland) did not reveal the presence of deletions in the alpha-globin genes, but showed the presence of a novel deletion that removes all the beta-globin clusters.Citation9
Patients and methods
Clinical report
The patient was an Italian man, 30 years old, referred to our institution from a local hematological service for the occasional founding of hypochromic polyglobulia and microcytic RBCs. In the proband, normal levels of Hb A2 (3.1%) and Hb F (1.1%) were associated with normal iron parameters. Other members of the family were not available for the study. The proband was selected by the Thalassemia Center collaborating in this study, among those referred to it for the hematological diagnosis. This study adhered to the tenets of the Declaration of Helsinki. We obtained written informed consent from participants for the use of blood samples. This study was also approved by the institutional ethics committee.
Hematological data
Blood counts and Hb analysis were done using standard methods. The hematological data and the serum iron values were determined in the hospitals, by using standard methods. Hb analysis was carried out by cation exchange high-performance liquid chromatography, using the HA-8160 ADAMS (Menarini, Firenze, Italy). The proband was selected on the basis of the normal iron parameters and normal Hb A2 ≥ 2.0–2.2% and ≤3.2–3.4%.Citation10
Molecular analysis
For the identification of alpha- or beta-thalassemia defects, molecular analyses have been conducted as already reportedCitation10–Citation13 (see Supplementary Data).
MLPA and qRT-PCR analysis
Two MLPA kits (MRC-Holland, Amsterdam, the Netherlands), for the identification of rearrangement in the alpha-globin gene cluster (SALSA MLPA KIT P140 HBA) and in the beta-globin gene cluster (SALSA MLPA KIT P102 HBB), were used according to the manufacturer's recommendations and as previously reported.Citation14MLPA products were separated by ABI-3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), quantified with the Coffalyser software (MRC-Holland), and compared with a pool of normal subjects (Fig. A). qRT-PCR was performed with ABI 7900HT System and the Power SYBR-Green PCR-Master mix (Applied Biosystems) with primers chosen outside repeated sequences (Table and Supplementary Data). The beta-2-microglobulin was used as the reference gene.
Figure 1 Molecular characterization of the Italian (ϵγδβ)0-thalassemia and comparison with others’ deletions. (A) MLPA analysis of the new deletion. Ratio between the values obtained from the proband and normal subjects: the position of the different probes on the beta-globin gene cluster is indicated on the x-axis. Shown in gray are the probes with half dosage and in black, the probe with full dosage. (B) Quantitative real-time PCR analysis (primers in Table ) probing the regions not tested by MLPA: 12 fragments (gray diamonds) show half dosage and seven fragments (black diamonds) show full dosage. The localization of MLPA fragments 28, 27, and 1 is shown. (C) Scheme of the beta-globin gene cluster with location of the MLPA probes (indicated with numbers) and of the quantitative real-time PCR fragments (indicated with small letter in italic); the s and a fragments are at 5′ and 3′ arm of depicted region. Genes are indicated by filled rectangles; pseudo gene is indicated by an empty rectangle; DNA hypersensitive sites (HS) are indicated by empty triangles. The positions of the LINE element and of the 160-bp palindrome are reported in the section repeat elements. The position of the Italian (ϵγδβ)0-thalassemia deletion (gray bar), of the Chilean (ϵγδβ)0-thalassemia deletion (most similar), and of three deletions with 5′ breakpoints localized in the LINE element are indicated by thick lines.
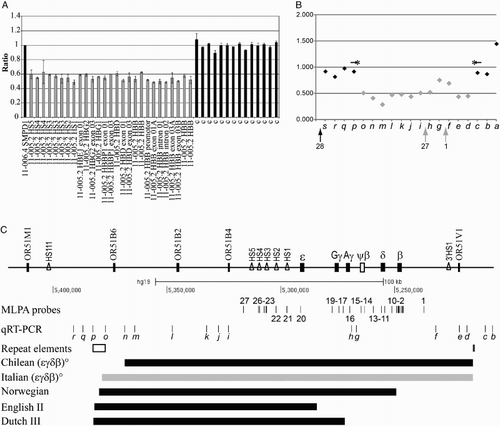
Table 1 Primers and position of amplicons for quantitative real-time PCR
Breakpoint characterization
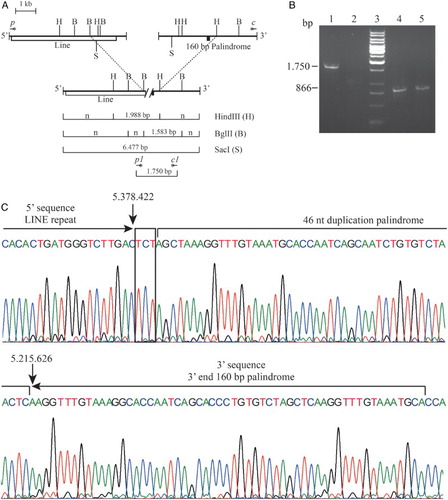
The genomic DNA encompassing the deletion breakpoint was amplified by long-range gap-PCR using the TripleMasterPCR System (Eppendorf, Hamburg, Germany) with the forward primers p 5′-GCATCTAGGTTAGGGAATTAGCCA-3′ and the reverse primer c 5′-TCAATGTGATCAAGATTTGGGAGTT-3′, giving rise to a fragment of about 6.5 kb. The region of the breakpoint was reduced by restriction mapping analysis using the enzymes BglII, HindIII, and SacI; new primers were designed (forward primer p1 5′-AGATGTCTATTAGGTCCGCTTGGT-3′ and reverse primer c1 5′-CAACTGCTGTGTTCTTTTGTTCAGA-3′) and a new fragment of about 1.7 kb was amplified by long-range gap-PCR and then sequenced (Figs. B and 2A).Citation14
A gap-PCR for the screening of carriers for the Italian (ϵγδβ)0-thalassemia was set up. The specific deletion amplicon was 1.750 bp (primers c1 and p1); the internal control amplicon was 866 bp (forward primers control A 5′-TCAAATGTATCATGCCTCTTTGCACC-3′ and reverse primer control B 5′-TGGAGTCAAGGCTGAGAAGATGCAGGA-3′) (Fig. B).
Figure 2. Molecular characterization of the Italian (ϵγδβ)0-thalassemia. (A) Long-range PCR fragment obtained using the qRT-PCR primers p and c. In the upper part of the normal cluster in the lower line the recombinant cluster. Restriction mapping of the enzymes HindIII, BglII, and SacI are reported. The vertical dashed lines connect the same restriction sites. The length of the anomalous fragments is reported. The position of the primers are indicated with the symbol *; n indicated fragment of normal length. (B) Gap-PCR for the screening of carriers for the Italian (ϵγδβ)0-thalassemia. Lines 1–2: Gap-PCR for the Italian (ϵγδβ)0-thalassemia (1.750 bp); Lines 4–5: PCR for the internal control amplicon (867 bp). Lanes 1 and 4: proband; lanes 2 and 5: normal controls; lane 3: reagent control. (C) Electropherogram of the sequence of the Italian (ϵγδβ)0-thalassemia deletion breakpoint. The LINE sequence in the 5′ and the 160-bp palindrome in the 3′ breakpoint are reported; the microhomology found is in a box. Deletion breakpoints are highlighted with arrows. The nucleotide numbering of the normal sequences, according to reference sequence GRCh37/hg19, is shown.
Statistical analysis
Statistical analysis was conducted with the software Minitab v.16 using the one-way ANOVA method to compare the hematological data from the carriers of the (ϵγδβ)0-thalassemia with that of previously studied α0- and β0-thalassemia carriers. P < 0.05 was considered statistically different.
Database
All the experimental results (hematological, biochemical, and mutations) and the family relationships were collected in anonymous form in a database, developed on Microsoft Visual Fox platform and interfaced with external software such as Microsoft Excel, Microsoft Word, Progeny (Progeny software, LLC, Winsford, UK).
Results and discussion
The proband, showing an alpha-thalassemia phenotype, was enrolled in an epidemiological project on the molecular basis of alpha-thalassemia in Southern Italy (Table ). The molecular screening for alpha-thalassemia gave negative result: the alpha-globin gene sequencing analysis revealed that the patient was heterozygote for the SNP α2 + 4 C > G, leading to exclude the presence of deletions involving the alpha-globin genes. We hypothesized the presence of double heterozygosis for the alleles of delta- and beta-globin, but the beta-globin gene sequencing showed absence of mutation and homozygosis for the beta-globin gene framework 3.Citation15
Table 2 Hematological data of the proband and of the current findings in heterozygotes for the ϵγδβ-deletion
The MLPA analysis, performed to verify the presence of deletions or rearrangements, showed a normal pattern of the alpha-globin cluster while the analysis of the beta-globin cluster revealed the presence of a long deletion, removing all the beta-globin clusters, including the LCR regions (Fig. A). The analysis by qRT-PCR indicated that the deletion starts in a region located about 80 kb 3′ of the epsilon-globin gene, extends for at least 150 kb, and ends about 30 kb 5′ of the beta-globin gene (Fig. ). At a first analysis, the deletions looked very similar to the Chilean (ϵγδβ)0-thalassemia.Citation16
By qRT-PCR with primers probing the regions not tested by MLPA, we restricted the possible breakpoint region: the 5′ breakpoint was localized between the qRT-PCR amplicons p and o, and the 3′ breakpoint was localized between the qRT-PCR amplicons d and c. The long-range PCR using the amplicons’ primers c and p (Fig. B and Table ) gave rise to an anomalous fragment of about 6.500 bp; the breakpoint region was further reduced by restriction mapping analysis with the enzymes BglII, HindIII, and SacI. A new fragment of about 1.700 bp was amplified. The Sanger sequence showed that the deletion ranged from position 5.215.626 to position 5.378.422 for a total length of 162.796 bp (Fig. B). The analysis of breakpoints showed that both are within repeated sequencing. In particular, the 5′ breakpoint was in a 5.247-bp LINE element containing the breakpoint of three different deletions: the Norwegian (ϵγδβ)0-thalassemia, the Dutch III (ϵγδβ)0-thalassemia, and the English II (ϵγδβ)0-thalassemia deletions (Fig. C).Citation17–Citation19 However, the 3′ breakpoint was located within a perfect 160-bp palindrome region that contains the deletion breakpoints of five other deletions: the Chilean (ϵγδβ)0-thalassemia; the Italian Gγ(Aγδβ)0-thalassemia; the Belgian Gγ(Aγδβ)0-thalassemia; the Pakistani I (ϵγδβ)0-thalassemia; and the Indian HPFH3.Citation14,Citation16,Citation20,Citation21 All these five deletion breakpoints are very close to the mid-point of the palindrome and localized in a region of 18 bp, as reported in the Italian Gγ(Aγδβ)0-thalassemia.Citation14 The breakpoint of the Italian (ϵγδβ)0-thalassemia was localized in the 3′ end of the 160 bp palindrome at position +103 and characterized by the presence of an insertion of 46 nucleotide sequences created by the duplication of the part of the palindrome from position +98 to position +143. A short homology TCT was present in the breakpoint. Palindrome sequences can form stem-loop structures that could favor unequal crossing-over. No direct homology was detected between the 5′ and 3′ normal sequences surrounding the deletion. The exact mechanism causing the breakage event remains speculative, but the cluster of deletion breakpoints suggests that the two regions (LINE and 160-bp palindrome) are highly prone to recombination. In addition, the presence of a short homology (5′-TCT-3′) could favor the non-homologous recombination.Citation22,Citation23
Complete characterization of deletion is important for genetic counseling and may lead to delineation of novel regulative regions. In the present case, we showed that the definition of the breakpoints of deletion is possible also in the presence of repeats although technically challenging.
The new deletion removes all the genes of the beta-globin cluster and it was for the first time identified in an Italian patient and therefore has been called Italian (ϵγδβ)0-thalassemia. The Hugo nomenclature was NC_000011.10:g.[5194397_5357192del;5194356_5194401insAGCTAAAGGTTTTGTAAATGCACCAATCAGCAATCTGTGTCTAACTC]. Although these kinds of deletions affected a moderately severe neonatal hemolytic anemia, our patient did not show any such occurrence.
Several deletions (β)0-thalassemia, (δβ)0-thalassemia (Sicilian, Spanish, Turkish/Macedonian), and (γδβ)0-thalassemia deletions (Turkish, Italian) have been described in Italy and in the Mediterranean area. All of them are characterized by microcytosis, normal Hb A2, and increased Hb F level, with the last marker indicating the possible presence of a deletion of the beta-globin gene cluster.Citation24–Citation27 Conversely, the carrier of the Italian (ϵγδβ)0-thalassemia shows a beta-thalassemia phenotype with normal Hb A2 and Hb F level.
Analyzing the phenotype of the new deletion, we were intrigued by the Hb A2 (3.1%) that is border line and by the low hemoglobin level (11.8 g/dL). To test the significance of these values, we looked for the phenotype of the (ϵγδβ)0-thalassemia carriers from literature (Table ).Citation3–Citation6,Citation16–Citation19,Citation28–Citation30 We compared (i) the Hb A2 values of the 19 (ϵγδβ)0-thalassemia carriers with those of 92 males and females, selected from our database, heterozygotes for an α0-thalassemia deletion [-(α)20.5, - -MED, - -CAL] and showing normal iron parameters;Citation7 (ii) the Hb levels of the (ϵγδβ)0-thalassemia carriers with those of the 92 α0-thalassemia and 291 β0-thalassemia carriers, with known genotype, selected from our database. The Hb A2 level of the (ϵγδβ)0-thalassemia carriers was of 2.9 ± 0.34 (range 2.4–3.8), while the Hb A2 level of the α0-thalassemia carriers was of 2.39 ± 0.25 (range 2.0–2.9), with Student's t-test indicating a significance at >0.01. The Hb level of the (ϵγδβ)0-thalassemia showed a mean of 10.18 ± 1.56 (range 6.8–13.2) significantly lower than that of the β0-thalassemia carriers 12.16 ± 1.22 (range 9.5–16.0) or α0-thalassemia carriers 12.88 ± 1.19 (range 10.4–16.4), with Student's t-test indicating a significance at >0.01. These Hb values are similar to those observed in thalassemia intermediate patients (Fig. ).
Figure 3 Comparison of the HbA2 and Hb values between (ϵγδβ)0-thalassemia and α0- and β0-thalassemia carriers. (A) Histogram of the Hb A2 value of 92 heterozygotes for α0-thalassemia and of the 19 heterozygotes for (ϵγδβ)0-thalassemia from literature. The ranges, averages, and SD of the Hb A2 of the two groups of carriers were reported in a comparative scheme with horizontal overlapping lines. On the left, the number of subjects. (B) Interval plot of the Hb levels of α0-, β0- and (ϵγδβ)0-thalassemia carriers. The Hb level of the (ϵγδβ)0-thalassemia was significantly lower than the others two class of thalassemia carriers.
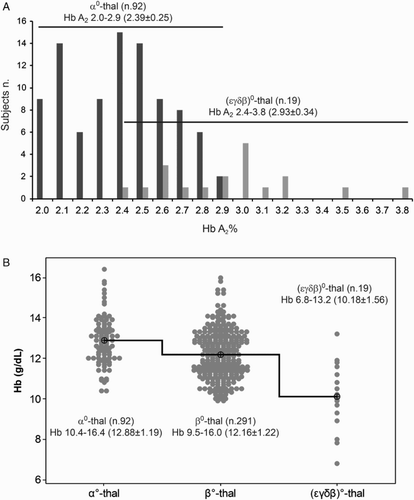
In conclusion, the beta-thalassemia with normal Hb A2 could be due to the presence of double heterozygosis for the alleles of delta- and beta-globin genes, as we already reported in 18 unrelated families, or due to the presence of long deletions involving all the beta-globin gene clusters.Citation31 In the first case, the segregation patterns and the sequencing analysis of the beta- and delta-globin genes are of great help. In case of (ϵγδβ)0-thalassemia heterozygous, we showed that the Hb A2 value is expected to be a little bit higher than in case of α0-thalassemia carriers and the Hb level is expected to be lower than β0-thalassemia carriers. These two data could favor a differential diagnosis of (ϵγδβ)0-thalassemia (Table and Fig. ).
For providing genetic counseling and to prevent misdiagnosis of beta-thalassemia, further to the first identification of an Italian (ϵγδβ)0-thalassemia, we suggest that MLPA analysis (for the identification of beta-globin gene cluster deletions/duplication) or screening for the Italian (ϵγδβ)0-thalassemia by gap-PCR should be carried out in patients showing alpha-thalassemia phenotype, but detection of the absence of common or rare alpha-thalassemia deletions and perhaps normal beta-globin gene sequence, in order to ascertain the presence of deletions causing (ϵγδβ)0-thalassemia. In particular, the search for deletions should be tested in carriers analyzed in the past showing a biosynthetic ratio of beta-thalassemia but the absence of point mutations or deletion, in subjects showing thalassemia, low level of Hb, and border line level of Hb A2 but the absence of the frequent mutations/deletions in the beta- or alpha-globin gene cluster. In our experience, the first identification of deletions and the screening could allow subsequent identification, as reported in the case of the Italian Gγ(Aγδβ)0-thal deletion and of the (α)α 5.3.Citation14,Citation32
Disclaimer statements
Contributors Conceived and designed the experiments: G.L. Performed the experiments: G.C., F.D.B., G.L. Analyzed the data: G.L. Contributed reagents/materials/analysis tools: F.D.B., C.S., R.P., G.L. Wrote the paper: G.L. Revised the article critically: C.S. Selected the patients and contributed clinical data: S.D., C.S.
Conflict of interest The authors declare that they have no competing interest.
Funding This research was supported by a grant from Ministero Istruzione, Università e Ricerca (MIUR), Legge 488/92 (Cluster C02, Project 2).
Ethics approval This study adhered to the tenets of the Declaration of Helsinki. We obtained written informed consent from participant for the use of blood sample. This study was also approved by the institutional ethical committee.
supplementary_data_February.docx
Download MS Word (126.1 KB)Acknowledgments
We would like to thank the patients for their participation and cooperation.
ORCID
Giuseppina Lacerra http://orcid.org/0000-0003-1038-8363
Related Research Data
References
- Stephens AD, Angastiniotis M, Baysal E, Chan V, Fucharoen S, Giordano PC, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1–13. doi: 10.1111/j.1751-553X.2011.01368.x
- Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4th ed. Oxford: Blackwell Science Press; 2001. p. 846.
- Rooks H, Clark B, Best S, Rushton P, Oakley M, Thein OS, et al. A novel 506kb deletion causing epsilongammadeltabeta thalassemia. Blood Cells Mol Dis. 2012;49(3–4):121–7. doi: 10.1016/j.bcmd.2012.05.010
- Shalev H, Landau D, Pissard S, Krasnov T, Kapelushnik J, Gilad O, et al. A novel epsilon gamma delta beta thalassemia presenting with pregnancy complications and severe neonatal anemia. European J Haematol. 2013;90(2):127–33. doi: 10.1111/ejh.12047
- Zebisch A, Schulz E, Grosso M, Lombardo B, Acierno G, Sill H, et al. Identification of a novel variant of epsilon-gamma-delta-beta thalassemia highlights limitations of next generation sequencing. Am J Hematol. 2015;90(3):E52–4. doi: 10.1002/ajh.23913
- Shooter C, Rooks H, Thein SL, Clark B. Next generation sequencing identifies a novel rearrangement in the HBB cluster permitting to-the-base characterization. Hum Mutat. 2015;36(1):142–50. doi: 10.1002/humu.22707
- Hardison RC, Chui DH, Riemer C, Giardine B, Lehväslaiho H, Wajcman H, et al. Databases of human hemoglobin variants and other resources at the globin gene server. Hemoglobin. 2001;25(2):183–93. doi: 10.1081/HEM-100104027
- Rosatelli MC, Tuveri T, Scalas MT, Leoni GB, Sardu R, Faa V, et al. Molecular screening and fetal diagnosis of beta-thalassemia in the Italian population. Hum Genet. 1992;89(6):585–9.
- Musollino G, Prezioso R, Scarano C, Piluso G, Del Vecchio Blanco F, Nigro V, et al., editors. Caratterizzazione molecolare mediante MLPA e RT-PCR di una nuova delezione beta0 talassemica con Hb A2 nella norma: la Italian (epsilongammadeltabeta)0-thalassemia, XV Congresso Nazionale SIGU; 2012; Hilton Sorrento Palace, Sorrento. P392
- Lacerra G, Scarano C, Lagona LF, Testa R, Caruso DG, Medulla E, et al. Genotype–phenotype relationship of the delta-thalassemia and Hb A(2) variants: observation of 52 genotypes. Hemoglobin. 2010;34(5):407–23. doi: 10.3109/03630269.2010.511586
- Lacerra G, Musollino G, Di Noce F, Prezioso R, Carestia C. Genotyping for known Mediterranean alpha-thalassemia point mutations using a multiplex amplification refractory mutation system. Haematologica. 2007;92(2):254–5. doi: 10.3324/haematol.10736
- Lacerra G, Fiorito M, Musollino G, Di Noce F, Esposito M, Nigro V, et al. Sequence variations of the alpha-globin genes: scanning of high CG content genes with DHPLC and DG-DGGE. Hum Mutat. 2004;24(4):338–49. doi: 10.1002/humu.20088
- Lacerra G, Testa R, De Angioletti M, Schilirò G, Carestia C. Hb Bronte or alpha93(FG5)Val-->Gly: a new unstable variant of the alpha2-globin gene, associated with a mild alpha(+)-thalassemia phenotype. Hemoglobin. 2003;27(3):149–59. doi: 10.1081/HEM-120023378
- Lacerra G, Prezioso R, Musollino G, Piluso G, Mastrullo L, De Angioletti M. Identification and molecular characterization of a novel 55-kb deletion recurrent in southern Italy: the Italian (G) gamma((A) gammadeltabeta)0-thalassemia. Eur J Haematol. 2013;90(3):214–9. doi: 10.1111/ejh.12066
- Carestia C, Pagano L, Lacerra G, Fioretti G, De Angioletti M, de Bonis C, et al. Beta-globin gene framework variant: an African type? Nucleic Acids Res. 1991;19(24):6968. doi: 10.1093/nar/19.24.6968-a
- Game L, Bergounioux J, Close JP, Marzouka BE, Thein SL. A novel deletion causing (epsilon gamma delta beta)0 thalassaemia in a Chilean family. Br J Haematol. 2003;123(1):154–9. doi: 10.1046/j.1365-2141.2003.04564.x
- Brantberg A, Eik-Nes SH, Roberts N, Fisher C, Wood WG. Severe intrauterine anemia: a new form of epsilongammagammadeltabeta thalassemia presenting in utero in a Norwegian family. Haematologica. 2009;94(8):1157–9. doi: 10.3324/haematol.2009.007534
- Rooks H, Bergounioux J, Game L, Close JP, Osborne C, Best S, et al. Heterogeneity of the epsilon gamma delta beta-thalassaemias: characterization of three novel English deletions. Br J Haematol. 2005;128(5):722–9. doi: 10.1111/j.1365-2141.2005.05368.x
- Harteveld CL, Osborne CS, Peters M, van der Werf S, Plug R, Fraser P, et al. Novel 112 kb (epsilonGgammaAgamma) deltabeta-thalassaemia deletion in a Dutch family. Br J Haematol. 2003;122(5):855–8. doi: 10.1046/j.1365-2141.2003.04505.x
- Fodde R, Losekoot M, Casula L, Bernini LF. Nucleotide sequence of the Belgian G gamma+(A gamma delta beta)0-thalassemia deletion breakpoint suggests a common mechanism for a number of such recombination events. Genomics. 1990;8(4):732–5. doi: 10.1016/0888-7543(90)90263-T
- Henthorn PS, Mager DL, Huisman TH, Smithies O. A gene deletion ending within a complex array of repeated sequences 3′ to the human beta-globin gene cluster. Proc Natl Acad Sci U S A. 1986;83(14):5194–8. doi: 10.1073/pnas.83.14.5194
- Kurahashi H, Inagaki H, Ohye T, Kogo H, Kato T, Emanuel BS. Palindrome-mediated chromosomal translocations in humans. DNA Repair. 2006;5(9–10):1136–45. doi: 10.1016/j.dnarep.2006.05.035
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131
- Camaschella C, Serra A, Bertero MT, Trento M, Dall'Acqua M, Gottardi E, et al. Molecular characterization and hematological phenotype of Sicilian delta beta-thalassemia. Haematologica. 1986;71(4):287–92.
- Baiget M, Gimferrer E, Fernandez I, Romero C, Mira Y, Perez ML, et al. Spanish delta-beta-thalassemia: hematological studies and composition of the gamma-chains in ten homozygous patients. Acta Haematol. 1983;70(5):341–4. doi: 10.1159/000206771
- Craig JE, Efremov GD, Fisher C, Thein SL. Macedonian (delta beta) zero thalassemia has the same molecular basis as Turkish inversion-deletion (delta beta) zero thalassemia. Blood. 1995;85(4):1146–7.
- Henthorn PS, Smithies O, Mager DL. Molecular analysis of deletions in the human beta-globin gene cluster: deletion junctions and locations of breakpoints. Genomics. 1990;6(2):226–37. doi: 10.1016/0888-7543(90)90561-8
- Rose C, Rossignol J, Lambilliotte A, Depret S, Le Metayer N, Pissard S. A novel (epsilongammadeltabeta)(0)-thalassemia deletion associated with an alpha globin gene triplication leading to a severe transfusion dependent fetal thalassemic syndrome. Haematologica. 2009;94(4):593–4. doi: 10.3324/haematol.2008.002675
- Von Kanel T, Rothlisberger B, Schanz U, Dutly F, Huber AR, Saller E. A Swiss (epsilongammadeltabeta)(0)-thalassemia patient with a novel 3-Mb deletion associated with mild mental impairment. Am J Hematol. 2013;88(2):158–9. doi: 10.1002/ajh.23364
- Joly P, Lacan P, Garcia C, Meley R, Pondarre C, Francina A. A novel deletion/insertion caused by a replication error in the beta-globin gene locus control region. Hemoglobin. 2011;35(4):316–22. doi: 10.3109/03630269.2011.571331
- Lacerra G, Scarano C, Musollino G, Testa R, Prezioso R, Caruso DG, et al. HbA2-Partinico or delta(A2)Pro-->Thr, a new genetic variation in the delta-globin gene in cis to the beta(+) thal IVS-I-110 G>A, and the heterogeneity of delta-globin alleles in double heterozygotes for beta- and delta-globin gene defects. Ann Hematol. 2010;89(2):127–34. doi: 10.1007/s00277-009-0784-9
- Lacerra G, Fioretti G, De Angioletti M, Pagano L, Guarino E, de Bonis C, et al. (Alpha)alpha 5.3: a novel alpha(+)-thalassemia deletion with the breakpoints in the alpha 2-globin gene and in close proximity to an Alu family repeat between the psi alpha 2- and psi alpha 1-globin genes. Blood. 1991;78(10):2740–6.