
Abstract
Background and objectives: The clinical manifestation in sickle cell disease (SCD) patients varies from one individual to another due to factors like the presence of alpha-thalassaemia mutation, foetal haemoglobin, and β-globin gene haplotype. The present study enumerates the clinical profile of sickle cell anaemia patients from Central India.
Methods: Seven hundred seventy-six SCD patients from Jabalpur and surrounding districts (Madhya Pradesh) in central India were registered with the sickle cell clinic of NIRTH, Jabalpur. The present study reveals recorded signs and symptoms of genetically confirmed sickle cell anaemia (404) and sickle beta thalassaemia (92) patients.
Results: Majority of the patients were from scheduled caste communities (47.9%) and Gond tribal community (13.8%). Splenomegaly was the most common clinical manifestation observed (71.4%). Overall, 63.5% patients had a history of blood transfusion. The most frequent signs and symptoms observed were Pallor, Icterus, Joint pain, Fever, and Fatigue. Majority of the patients revealed onset of disease prior to attaining the age of 3 years (sickle cell anaemia 44.3% and sickle beta thalassaemia 35.9%). Mean haemoglobin levels among SCA individuals were marginally higher than SBT patients. On the other hand, mean foetal haemoglobin levels among SBT individuals showed the reverse trend. Notably, the present study reports the first incidence of priapism recorded in Central India.
Conclusions: The study revealed a high prevalence of SCD among scheduled caste, backward caste, and tribal communities. Dissemination of study findings, screening, pre-marriage counselling, and pre-natal diagnosis are fundamental to preventing or lowering of birth of sickle cell anaemia children in the affected populations.
Keywords:
Introduction
Sickle cell disease (SCD) is a structural disorder of haemoglobin causing anaemia among affected individuals. In India, it is reported mainly among tribal populations of Central and Southern parts of India.Citation1–Citation5 SCD is a monogenic disorder caused by a point mutation in the beta globin gene on chromosome 11 and is responsible for the production of sickle haemoglobin (HbS). However, the clinical phenotype among SCD patients varies from one individual to another due to factors like β-globin gene haplotype, alpha-thalassaemia mutation(s), foetal haemoglobin etc. The sickle haemoglobin gene haplotype observed among various tribal populations in Central and Southern India is similar to that of the Asian-Indian haplotypeCitation6,Citation7 and is associated with high HbF levels. The current study provides a detailed clinical profile of SCD patients attending the sickle cell clinic of the National Institute for Research in Tribal Health (NIRTH) situated at the Netaji Subhash Chandra Bose (NSCB) Medical College, Jabalpur.
Material and methods
This is an ongoing follow-up study cum service provided by NIRTH beginning from year 2000 till date at the Department of Pediatrics, NSCB, Medical College and Hospital Jabalpur. Subjects are sickle cell patients registered with the NIRTH SCD clinic. Patients suspected to be suffering from haemolytic anaemia were referred to the genetics laboratory of the NIRTH, Jabalpur, by the Government Medical College and hospitals in and around Jabalpur district for diagnosis. Patients diagnosed with SCA (sickle cell anaemia) or SBT (sickle beta thalassaemia) were encouraged to register for detailed clinical assessment and follow-up. The clinical history and haematological investigations were recorded in a structured proforma after obtaining informed written consent. Data presented here correspond to the year 2000–March 2014. Data are mainly presented in univariate and bivariate tables and graphs. Appropriate statistical tests like t-test for comparing means, z-test for comparing proportion and χ2 (chi square test) for assessing association of age at onset of symptoms between SCA and SBT were used.
Procedures
About 2–3 ml of peripheral blood was drawn in EDTA vials. Complete blood count and red cell indices were measured by automated analyzer. The presence of sickle haemoglobin was identified by sickling test with 2% sodium metabisulphite and confirmed by membrane electrophoresis.Citation8 Estimation of HbF was done by alkaline denaturation method.Citation9 Haemoglobin electrophoresis was also done on both parents of registered patients to ascertain their genetic zygosity. They were categorized as SCA, if both parents were heterozygous for sickle haemoglobin. If one parent was heterozygous for sickle haemoglobin and the other showed a normal haemoglobin pattern, HbA2 quantization was carried out. If the level of HbA2 was greater than 4.0%, then that individual was classified as having Beta thalassaemia trait and their child was designated as having SBT. The clinical data presented represent 496 patients whose genetic makeup was confirmed. Genetic makeup of 99 patients could not be ascertained and hence not included in the analysis. Further 81 patients died during the study period.
Results
A total of 776 patients were registered in the sickle cell clinic between 2000 and March 2014. In general, 67.9% patients were males and 32.1% patients were females. The male:female ratio was 2.1:1. The highest numbers of females attending the clinic were in the age group of 20–25 (44.3%). Similarly highest numbers of male patients (73.1%) were in the age group of 5–10. Overall majority of the patients (30.2%) were in the age group of 5–10 years followed by the 10–15-year group (21%) and below 5 years (19.3%). Caste-wise distribution revealed that 47.9% patients were from scheduled caste communities (Jharia, Mehra, Deharia etc.) followed by Other Backward Classes (OBC) [28.4%] comprising Lodhi, Kurmi, and Yadav. The Gonds (scheduled tribe) constituted 13.8% of the patients and the rest comprised Muslims and Brahmins.
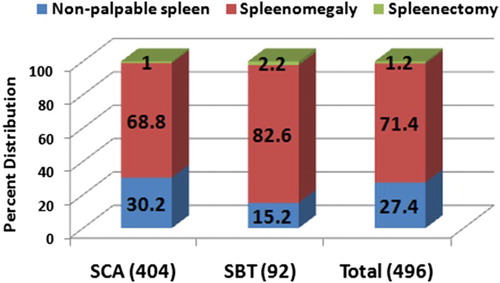
A comparison of the splenic status among SCA and SBT patients is given in Fig. . Splenomegaly was observed in the majority of the patients (71.4%) and it was significantly higher (P < 0.05) among SBT patients (82.6%) as compared to the SCA patients (68.8%). No palpable spleen was observed in SCA individuals (30.2%). SBT Individuals showed 2-fold higher splenectomy (2.2%) as compared to SCA (1%) individuals. Ultra-sonography of 18 patients revealed auto-splenectomy in two individuals.
Figure 1 Percent distribution of splenic status in SCD individuals representing non palpable splenomegaly and splenectomy in SCA, SBT, and total SCD groups
Requirement of blood transfusion was evaluated; 36.5% patients did not require any blood transfusion and 35.5% patients required only a single transfusion from enrolment up to their last follow-up visit. On the other hand, 16.3% required three or more transfusions [16.1% of SCA and 17.4% of SBT] and 11.7% patients received two blood transfusions (Fig. ).
Figure 2 Percent distribution of blood transfusions among three different groups (SCA, SBT, and Total SCD)
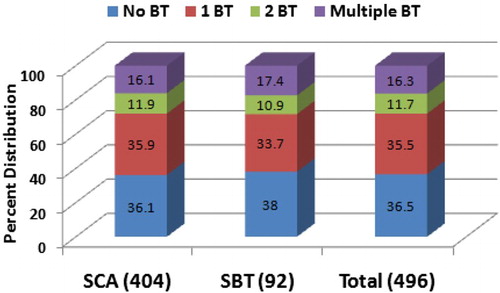
Table shows the clinical symptoms observed among SCD patients in the study population. Pallor was noted in nearly all patients irrespective of their genetic status (SCA-95.5% and SBT-94.6%). Icterus (86.1% in SCA and 81.5% in SBT) and joint pain (81.2% in SCA and 80.4% in SBT) were also routinely noted. Swelling of the joints was significantly higher in SCA patients (P < 0.05). The bilirubin levels were evaluated in 64 participants and 87.5% of these patients showed 2 mg/dl or higher levels. Incidence of dactilytis was more common in SCA (4.2%) children of the age group 0–5 years as compared to SBT children (1%). Among the clinical manifestations, avascular necrosis of the femur head was more pronounced among SBT (2.2%) subjects as compared to SCA (0.5%) ones and was established by X-ray radiography also. Non-healing leg ulcers were noted only in 1.0% of SCA group individuals. Notably, only a single incidence of priapism was observed; thus far no report of priapism has been reported from this region. Occurrence of gall stones was nearly similar in the two groups. However, exhaustion was more common among SBT (83.7%) individuals as compared to SCA individuals (75.2%).
Table 1 Common signs and symptoms observed in SCD patients
The comparison of mean haematological parameters noted among the SCA and SBT patients has been given in Table . In general, mean haemoglobin levels of SCA individuals were marginally higher than in the SBT patients. On the other hand, the mean foetal haemoglobin (Hb F) levels showed a reverse trend. In the SCA population, the mean haemoglobin level of adult male, adult female, male child, and female children was 8.5 ± 2.3 g/dl, 7.8 ± 2.0 g/dl, 7.1 ± 1.9 and 7.5 ± 2, respectively. In contrast, in the SBT subjects, the corresponding mean haemoglobin levels were 7.8 ± 1.8 g/dl, 7.5 ± 2.5 g/dl, 6.9. ± 1.7, and 7.4 ± 1.5, respectively. Mean corpuscular volume and mean corpuscular haemoglobin were significantly higher (P < 0.05) among SCA subjects in all four age-sex categories (men, women, male children, and female children). Analysis of the mean foetal haemoglobin levels showed significantly higher levels in SBT males (16.5 ± 7.1) as compared to SCA males (13.3 ± 5.6). No appreciable difference was detected in the mean HbF levels of the adult females or in children. Clinically, the presence of foetal haemoglobin in SCD patients diminished the frequency and severity of disease symptoms such as joint pain, swelling, bony pain, abdominal pain, chest pain, and fatigue (Table ). Moreover, bony pain, abdominal pain, chest pain, and fatigue showed a linear decrease with increase in foetal haemoglobin among SCA patients. In contrast, blood transfusion requirement was significantly reduced in SBT patients with increase in HbF.
Table 2 Haematological profile of SCD patients
Table 3 Comparison of some clinical signs with foetal haemoglobin levels
The age at initial or first appearance of any of the common signs or symptoms of the disease was arrived at through discussion with the parents of the patients and their medical records wherever available. The presenting signs and symptoms at the time of early clinical manifestation were often fever, joint pain, musculo-skeletal pain, anaemia, jaundice, and chest infection. It was observed that 44.3% of SCA and 35.9% of SBT patients (Table ) had first appearance of signs or symptoms of the disease prior to attaining the age of 3 followed by 3–6 years (30%). In the case of SBT, 21.7% individuals exhibited first symptoms of the disease only after 9 years while in comparison to 12.9% SCA individuals who developed disease symptoms after the age of 9 years. Clinically, 12% of both categories exhibited the first sign/symptom of the disease between 6 and 9 years. Table also summarizes the onset of other clinical signs in these patients. Majority of the patients showed onset of clinical symptoms prior to attaining the age of 3 years. Notably, the incidence of joint swelling and chest infection were comparatively lower among those over the age of 6 as compared to individuals less than 3 years.
Table 4 Age at first onset of clinical symptoms in SCD patients
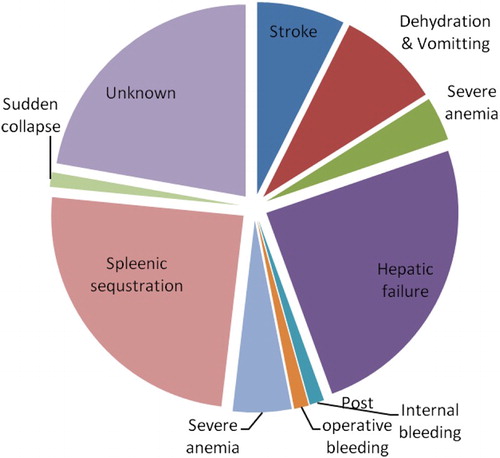
In Central India, particularly in Madhya Pradesh, no diagnostic facilities or dedicated SCD clinics exist in public health establishments. The present study is an effort to provide diagnostic as well as supportive care for patients in the outpatient mode. The registered patients are requested to present themselves for routine follow-up. Barring 110 patients, majority reported for quarterly follow-up. Efforts were made to visit these 110 patients to ascertain the cause of their absence. Many of the registered patients indicated distance as a major factor in addition to wage loss. Further, it was identified that 81 patients among non-respondents had died during the period. Owing to economic conditions and low educational status, families failed to save or collect clinical record sheets. Fig. depicts the probable cause of death discerned based on the information provided by the family members. Hepatic failure and splenic sequestration were predominant causes of death followed by dehydration and stroke. In one-fifth of the cases, the causes of death in affected children could not be identified.
Figure 3 Probable cause of death among SCD patients who died during the study period
Discussion
The present study provides an overview on the wide prevalence of SCD and its clinical manifestation in patients attending the SCD clinic of NIRTH, Jabalpur. The data presented here do not indicate the prevalence rate of the sickle cell gene in any particular community, but only indicate wide disease occurrence in the study area. It is often considered that the prevalence of the Arab-India beta globin haplotype results only in milder phenotypes.Citation10,Citation11
In the present study it was observed that the majority of the patients attending the clinic were males. Even though the disease is not sex-linked, the high proportion of the male patients may be due to commonly prevalent male preferential treatment in the Indian society.Citation12,Citation13 Number of patients seeking medical intervention decreased with age more than 15 years; this may be due to lower morbidity load or increased fatality above 14 years (unpublished data). It was earlier reported in Gujarat, India, that 30% tribal SCD children die prior to adulthood.Citation14
The number of females registered with SCD clinics was comparatively higher in the age group of 20–25, exhibiting morbidities such as anaemia, weakness, breathlessness, pneumonia, transfusion, postpartum infection, sepsis, gestational hypertension/preeclampsia, preterm labour, and foetal growth restriction and painful crisis during pregnancy. This corresponds to the detection of the underlying sickle disease status during their visits to gynaecologists. The high rate of complications in pregnant patients with SCD is well known.Citation15,Citation16
In general, disease prevalence was higher among the scheduled caste patients belonging to Jharia, Deharia, and Mehra communities followed by tribals, suggesting that these endogamous populations are at a greater risk of acquiring SCD. These findings are in agreement with earlier studies.Citation17 Furthermore, the study revealed a majority of these patients (74.3% of SCA and 66.3% of SBT) indicated onset of disease prior to attaining the age of 6 years. It has been shown that in Odisha, majority of SCD patients seeking hospital care were younger than 10 years of age.Citation18
Sickle mutation in homozygous condition is responsible for the commonly observed severe anaemia, whose severity depends on the co-inherited beta thalassaemia mutation.Citation19 When the β0-thalassaemia gene is co-inherited with the sickle gene, resulting in Sβ0-thalassaemia its severity is comparable to homozygous sickle genotype.Citation20 However, when the β+-thalassaemia gene is co-inherited with the sickle gene, resulting in Sβ+-thalassaemia it results in a wide range of clinical severity.Citation21 However, it may be noted that in the present studies no molecular characterizations of beta thalassaemia mutations were carried out.
Impaired splenic function and auto-splenectomy are commonly encountered in SCD patients. However, splenomegaly is known to persist beyond the first decade of life and into adult life.Citation22 In the present studies, 71.4% patients showed splenomegaly. Among them significantly higher number of SBT individuals (82.6%) showed splenomegaly as compared to SCA patients (68.8%). About 1.2% patients underwent surgical splenectomy due to massive spleen reaching up to the umbilicus and frequent pain in the splenic region. Post splenectomy, these patients were advised penicillin therapy to avoid any complications. We also report here the first case detection of priapism in Central India. So far no case of priapism has been reported from the region. About 64% patients had a history of blood transfusion with at least one blood transfusion from enrolment up to their last follow-up visit. Blood transfusion was only prescribed to patients with haemoglobin lower than 6.5 g/dl as it decreases the proportion of sickle red cells in the circulation, thereby improving the microvascular perfusion and the oxygen-carrying capacity.
Pallor, Icterus, joint pain, fever, fatigue, abdominal pain, joint swelling, and pain crisis are the main signs and symptoms observed in the present study. Relative incidences of these conditions were more often noted among SCA patients as compared to SBT patients. In addition, joint swelling and dactilytis were also higher in SCA patients. We observed that SCD patients with foetal haemoglobin (HbF) exhibited lower incidence of painful crisis as it acts as an important modulator of the clinical and haematological parameters. High levels of foetal haemoglobin are known to reduce the rate of acute painful episodes and leg ulcers.Citation23–Citation26
In the present study, patients were counselled to seek immediate medical help to avoid crisis and disease-precipitating factors like exposure to extreme climatic conditions, dehydration, and physical and mental stress. Patients were also administered folic acid (1 mg/day) and B-complex. Notably, these patients were treated in outdoor patient mode and referred to appropriate clinics in case of any emergency.
The presence of a large number of SCD patients in the area necessitates an urgent need for starting public health-based diagnostic facilities to lower morbidity. Furthermore, initiation of pre-marriage counselling and blood testing to control/avoid birth of an SCD child in the affected communities is warranted. The knowledge of the prevalence of the trait in this region is indispensable to control haemoglobinopathies in the affected communities who are often engaged in endogamous marriage practices.
One factor that might be considered as a bias in this study is that data of clinical events are based on the patients ‘medical records’ which at times may be deficient. On the other hand, the patients followed-up in the institution, not only as outpatients but also during emergencies and hospitalizations, reduce or discard bias caused by dependence on the memory of the patient's family.
Disclaimer statements
Contributors RY, ML, PG and SK are Clinicians involved in the study and care of subjects. MPSS is involved in data compilation, analysis and Manuscript preparation, RBG designed study, RKS is responsible for statistical analysis and RS designed, coordinated study and prepared manuscript and finalized.
Funding None.
Conflict of interest R. Yadav, M. Lazarus, P. Ghnaghoria, M.P.S.S. Singh, R.B. Gupta, S. Kumar, R.K. Sharma and S. Rajasubramaniam declare that they have no conflict of interest.
Ethics approval Institutional Ethics Committee of NIRTH, Jabalpur.
Acknowledgements
The authors are grateful to Dr Neeru Singh, Director, NIRTH (ICMR), Jabalpur, for providing facilities and necessary funds for the study. The authors are also grateful to staff of Genetics department, Mr Subhash Godbole, Mr C. P. Vishwakarma, Mr Ashok Gupta, Mr R. L. Neelkar and Mr Anil Gwal, for their help and support in diagnosis of these cases.
References
- Bhatia HM, Rao VR. Genetic atlas of the Indian Tribes: a report by Institute of Immunohaematology. Bombay, India: ICMR; 1986.
- Kar BC, Devi S, Dash KG, Das M. The sickle cell gene is widespread in India. Trans R Soc Trop Med Hyg 1987;81:275–78. doi: 10.1016/0035-9203(87)90239-2
- Kar BC, Devi S. Clinical profile of sickle cell disease in Orissa. Indian J Paediatr 1997;64:73–7. doi: 10.1007/BF02795780
- Balgir RS. Genetic epidemiology of the three predominant abnormal haemoglobins in India. JAPI. 1996;44:25–8.
- Nimgaonkar V, Krishnamurti L, Hari Prabhakar BA, Menon N. Comprehensive integrated care for patients with sickle cell disease in a remote aboriginal tribal population in Southern India. Pediatr Blood Cancer 2014;61:702–5. doi: 10.1002/pbc.24723
- Kuzolik AE, Wainscoat JS, Seargant GR, Kar BC, Al-Awamy B, Essan GJF, et al. Geographical survey of βS-globin gene haplotypes: evidence for an independent Asian origin of the sickle cell mutation. Am J Hum Genet 1986;39:239–44.
- Gupta RB, Tiwary RS, Pande PL, Kultar F, Oner C, Oner R, et al. Haemoglobinopathies among the Gond tribal groups of central India. Interaction of alpha and beta thalassaemia with beta chain variants. Haemoglobin 1991;15:441–58. doi: 10.3109/03630269108998864
- Chanarin I, editor. Laboratory haematology: an account of laboratory techniques. 1st ed. London: Churchill Livingstone; 1989.
- Dacie JV, Lewis SM, editors. Practical haematology. 7th ed. London: Churchill Livingstone; 1991.
- Mukherjee MB, Lu CY, Ducrocq R, Gangakhedkar RR, Colah RB, Kadam MD, et al. Effect of alpha-thalassemia on sickle-cell anemia linked to the Arab-Indian haplotype in India. Am J Hematol 1997;55:104–9. doi: 10.1002/(SICI)1096-8652(199706)55:2<104::AID-AJH9>3.0.CO;2-X
- Nagel RL, Fleming AF. Genetic epidemiology of the βs gene. Bailliere's Clin Haematol 1992;5:331–65. doi: 10.1016/S0950-3536(11)80023-5
- Kar BC, Satapathy RK, Kulozik AE, Sirr S, Kulozik M, Serjeant BE, et al. Sickle cell disease in Orissa State, India. The Lancet 1986;328:1198–201. doi: 10.1016/S0140-6736(86)92205-1
- Kar BC. Sickle cell disease in India. JAPI. 1991;39(12):954–60.
- In Government of Gujarat. Sickle cell anaemia control project. Ahmedabad: Govt. of Gujarat; 2012.
- Patrícia RSC, Regina ALPeA, Marcos BV. Clinical complications in pregnant women with sickle cell disease: prospective study of factors predicting maternal death or near miss. Rev Bras Hematol Hemoter 2014;36(4):256–63. doi: 10.1016/j.bjhh.2014.05.007
- Villers MS, Jamison MG, De Castro LM, James AH. Morbidity associated with sickle cell disease in pregnancy. Am J Obstet Gynecol 2008;199:125.e1–125.e5. doi: 10.1016/j.ajog.2008.04.016
- Urade BP. Haemoglobin S and βThal: their distribution in Maharashtra, India. Int J Biomed Sci 2013;9(2):75–81.
- Dash BP, Das RK. Age, sex and seasonal variations of sickle cell disorder cases in Orissa. J Hum Ecol 1998;9:281–4.
- Paul SF, George FA. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest 2007;117(4):850–858. doi: 10.1172/JCI30920
- Serjeant GR, Sommereux AM, Stevenson M, Mason K, Serjeant BE. Comparison of sickle cell-β° thalassaemia with homozygous sickle cell disease. Br J Haematol 1979;41:83–93. doi: 10.1111/j.1365-2141.1979.tb03684.x
- Serjeant GR, Ashcroft MT, Serjeant BE, Milner PF. The clinical features of sickle-cell-thalassaemia in Jamaica. Br J Haematol 1973;24:19–30. doi: 10.1111/j.1365-2141.1973.tb05723.x
- Al-Awamy B, Wilson WA, Pearson HA. Splenic function in sickle cell disease in the Eastern Province of Saudi Arabia. J Pediatr 1984;104(5):714–7. doi: 10.1016/S0022-3476(84)80950-6
- Steinberg MH. Pathophysiolocally based drug treatment of sickle cell disease. Trends Pharmacol Sci 2006;27:204–10. doi: 10.1016/j.tips.2006.02.007
- Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol 2012;87(8):795–803. doi: 10.1002/ajh.23232
- Nadkarni A, Pooja D, Roshan BC, Ghosh K. Fetal haemoglobin in sickle cell anemia. Blood Cells Mol Dis 2014;52:175. doi: 10.1016/j.bcmd.2013.11.007
- Idowu AI, Alsultan A, Solovieff N, Duyen Ngo, Clinton TB, Paola Sebastiani P, et al. Fetal haemoglobin in sickle cell anemia. Blood 2011;118(1):19–27. doi: 10.1182/blood-2011-03-325258