
Abstract
Objectives: In the last decade the identification of germline mutations in several genes such as EPOR, VHL, EGLN1, and EPAS1, helped the definition of several different subtypes of familial (congenital) erythrocytosis. Being rare disorders these entities often remain unrecognized or misdiagnosed, which necessitates the extensive reporting of newly identified cases.
Methods: We applied a genetic approach including whole exome sequencing and Sanger sequencing for the identification of the causative germline mutation in a Bulgarian family with congential erythrocytosis.
Results: We identified EPAS1 (HIF2A) p. M535T heterozygous mutation carried by four members of the family over three generations. We provide also an extensive description of the clinical features of the affected family members.
Discussion: EPAS1 p.M535T appears to be found in different populations as a causative variation in familial erythrocytosis. Our findings support the notion that the affected patients present with variable clinical features and disease course. Furthermore, close clinical follow-up with phlebotomies on demand and regular intake of low doses of anticoagulants seem to prevent from serious complications such as thrombembolic events and pulmonary hypertension.
Conclusion: This is the first description of an entire family with EPAS1 p. M535T mutation expanding our knowledge about the clinical features of the disease.
Introduction
Congenital (familial) erythrocytosis is characterized by hereditary elevation of the red cell mass (RCM). Mutations in several genes have been reported as associated with this disorder, including EPOR, VHL, EGLN1, EPAS1, HBB, HBA1, HBA2, BPGM, and PKLR genes. The identification of disease causing mutations in the oxygen sensing pathway genes VHL, EGLN1, and EPAS1 led to the definition of three different erythrocytosis entities (ECYT2, ECYT3, and ECYT4, respectively) and allowed for novel insights in the molecular mechanisms of hypoxia response.Citation1,Citation2 Here, we report a Bulgarian family of Caucasian origin with congenital erythrocytosis identified as harboring EPAS1 p.M535T mutation. Although this mutation has been reported previously,Citation3 this is the first report of an entire family and the extensive genetic and clinical evaluation of the affected individuals contributes to our understanding of this extremely rare disease.
Cases description
The index patient was a 44-years-old female (patient II-1, Fig. , Table ), who was referred to our out-patient department in 2015 with a 10-year history of elevated RCM. She had never been phlebotomized before and was chronically treated with low doses aspirin. At our department she was phlebotomized twice because of Hct level of 0.514 and complaints of whole body pruritus. Upon medical history taking she reported that her father and brother had known erythrocytosis and were undergoing occasional phlebotomies. She did not report any thrombembolic event. That prompted us to perform an extensive genealogy analysis as shown in Fig. . For the last one year of follow-up, the patient continued taking aspirin and her Hct level never reached above 0.49.
Fig. 1 Summary of the genetic analysis of the family. (a) A pedigree tree of the studied family. The arrow indicates the index patient. (b) WES analysis showing the heterozygous mutation in EPAS1 p. M535T in the index patient. Data were visualized with the Integrative Genomics Viewer (IGV). (c) Sanger sequencing of EPAS1 exon 12 of all members of the family. The arrows show the position of the heterozygous mutation
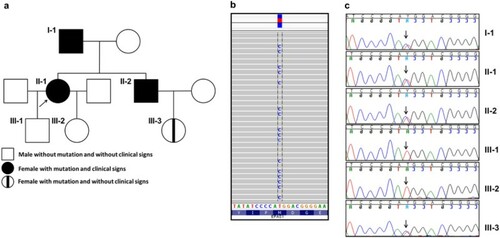
Table 1 Summary of the demographic, clinical and hematological parameters of the studied subjects.
The father (patient I-1) of the index patient reported that he had known erythrocytosis since 1987, when he was initially diagnosed with polycythemia vera. Over the last three decades he had had many phlebotomies and had taken low dose aspirin until 2006 and never been prescribed any cytoreductive treatment. He reported a single thrombembolic event in 2006 when he was diagnosed with pulmonary embolism. He had been taking acenocoumarol ever since then and his INR value was kept between 2.0 and 3.0.
The brother (patient II-2) of the index patient was diagnosed with elevated hemoglobin levels in 2007 when he had Hgb of 211 g/l and normal erythropoietin level (Table ). He had a single phlebotomy and then underwent alternative treatment with bloodletting using leeches. His Hct level was kept under 0.55 until he was referred to our department in 2015 with Hgb level of 205 g/l and Hct of 0.59. In 2015, he had a total of five phlebotomies to reach a Hct level below 0.53 during the 10 months follow-up afterwards.
The son (subject III-1) and the daughter (subject III-2) of the index patient as well as her niece (subject III-3) did not have any clinical and laboratory signs of elevated RCM as of January 2016.
The index patient was subjected to whole exome sequencing (WES) using Illumina HiSeq platform. WES revealed a heterozygous missense mutation in the Endothelial PAS domain protein 1 (EPAS1) (also known as hypoxia-inducible factor-2alpha (HIF2A)) gene at position c. 1604 T > C in exon 12, translating into p.M535T substitution at protein level (Fig. B). The presence of this mutation was validated by Sanger sequencing of the entire exon 12 of the EPAS1 gene (Fig. C). No mutations were found in the VHL, HBB, HBA1, HBA2, and EPOR genes, as well as mutations in JAK2 (exons 12 and 14), CALR (exon 9), MPL (exon 10) (data not shown). We then confirmed the presence of the same EPAS1 heterozygous mutation in the father and the brother of the index patient. The mutation was absent in the children of the index patient (subjects III-1, III-2) but was present in her niece (subject III-3).
Discussion
To the best of our knowledge there are only 22 patients with ECYT4 reported worldwide.Citation1 Of note all the mutations are scattered between I533 and F540 residues, suggesting that they all affect a specific molecular feature of the protein.Citation3Citation4Citation5Citation6Citation7–Citation8 The M535T mutation was reported recently in two unrelated erythrocytosis patients lacking extensive family history.Citation3 In addition to the original observation of this mutation our genealogy and genetic analyses confirmed its autosomal dominant type of inheritance. Notably, all our affected patients showed clinical presentation after the first two decades of life. Our report includes also the oldest reported person with EPAS1 mutation with almost 30 years of follow-up. In spite of the high Hgb levels he had experienced for long periods of his life he never had any severe health problems likely related to the erythrocytosis except for a single pulmonary embolism event. This observation suggests that either this type of mutation does not dramatically increase the thrombotic risk or that chronic low-dose aspirin intake reduces it significantly. It is reasonable to speculate that other yet unidentified genetic factors may also affect the disease severity and the cumulative risk for thrombembolic events. Furthermore, it has been previously shown in humansCitation9 and mouse modelsCitation10 that EPAS1 mutations can cause pulmonary hypertension. None of our patients with M535T mutation showed symptoms or signs of pulmonary hypertension further demonstrating the variable clinical phenotype of ECYT4.
Interestingly, the Epo levels in the affected patients varied from normal to very high levels. This is in accord with previous reports on EPAS1 mutated patients with familial erythrocytosis. Perrotta et al.Citation7 proposed two possible hypotheses that could explain this observation. These are modulation of the effect of EPAS1 mutation through different genetic background, and the other is the age of the patients. Notably, mutant HIF2A knock-in mice also do not have significantly elevated Epo levels, which could be explained by potential pleiotropic effects of these mutations.Citation10
Position M535 appears to be a mutational hotspot as along with the two already known cases of M535TCitation3 two other types of mutations were reported, namely M535V and M535I.Citation4,Citation5 Obviously in the latter two mutations the hydrophobic methionine is substituted with other hydrophobic residues, whereas in the case of M535T methionine is substituted for the hydrophilic threonine. It has been proposed that M535 residue participates in the contact with VHL.Citation5 It is possible that M535T because of the substitution with a hydrophilic amino acid affects more severely such an interaction than the M535V and M535I mutations but this hypothesis requires further experimental testing.
Conclusion
In conclusion, here we reported the first extensive genetic and clinical study of a family with four members carrying the EPAS1 p.M535T mutation. This study demonstrates the utility of WES as a tool for identification of mutations in congenital erythrocytosis as well as helps the definition of the clinical characteristics of the disease.
Disclaimer statement
Contributors TA provided clinical data, MI performed analyses, VS provided clinical data, performed analyses and wrote the paper. All authors approved the final version of the manuscript.
Funding None.
Conflicts of interest The authors have no conflict of interest to disclose.
Ethics approval The guidelines of the Declaration of Helsinki were followed strictly.
Related Research Data
References
- Bento C, Percy MJ, Gardie B, Maia TM, van Wijk R, Perrotta S, et al. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat. 2014;35:15–26. doi: 10.1002/humu.22448
- Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol. 2011;6:165–92. doi: 10.1146/annurev-pathol-011110-130321
- Percy MJ, Chung YJ, Harrison C, Mercieca J, Hoffbrand AV, Dinardo CL, et al. Two new mutations in the HIF2A gene associated with erythrocytosis. Am J Hematol. 2012;87:439–42. doi: 10.1002/ajh.23123
- Martini M, Teofili L, Cenci T, Giona F, Torti L, Rea M, et al. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica 2008;93:1068–71. doi: 10.3324/haematol.13210
- Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood 2008;111:5400–2. doi: 10.1182/blood-2008-02-137703
- Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008;112:919–21. doi: 10.1182/blood-2008-04-153718
- Perrotta S, Stiehl DP, Punzo F, Scianguetta S, Borriello A, Bencivenga D, et al. Congenital erythrocytosis associated with gain-of-function HIF2A gene mutations and erythropoietin levels in the normal range. Haematologica 2013;98:1624–32. doi: 10.3324/haematol.2013.088369
- van Wijk R, Sutherland S, Van Wesel AC, Huizinga EG, Percy MJ, Bierings M, et al. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica 2010;95:829–32. doi: 10.3324/haematol.2009.017582
- Formenti F, Beer PA, Croft QP, Dorrington KL, Gale DP, Lappin TR, et al. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel-Lindau disease and HIF-2alpha gain-of-function mutation. FASEB J 2011;25:2001–11. doi: 10.1096/fj.10-177378
- Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, et al. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem. 2013;288:17134–44. doi: 10.1074/jbc.M112.444059