
ABSTRACT
Objectives: Classically, immune thrombocytopenia (ITP) was thought to be caused by the destruction and insufficient production of platelets, as mediated by autoantibodies. More recently other immune mechanisms that contribute to the disease have been discovered. This review attempts to address the main unresolved questions in ITP.
Methods: We review the most current knowledge of the pathophysiology of ITP. Immunological effects of available therapies are also described.
Discussion: The trigger may be a loss of tolerance due to molecular mimicry with cross-reaction of antibodies arising from infectious agents or drugs, genetic factors, and/or platelet Toll receptors. This loss of tolerance activates autoreactive effector B and T lymphocytes, which in turn initiates platelet destruction, mediated by cytotoxic T lymphocytes and the release of pro-inflammatory cytokines (IL-2/IL-17) by T helper (Th) cells (Th1/Th17). Th2 (anti-inflammatory) and regulatory B (Breg) and Treg cells are also inhibited (with decrease in IL-10/TGF-β), which leads to the disease becoming chronic. Some isotypes of autoantibodies may increase the bleeding risk. Corticosteroids, rituximab, and thrombopoietin receptor agonists (A-TPOs) all increase levels of Tregs and TGF-β. The A-TPOs also increase Breg levels, which could explain why complete remission has been seen in some cases.
Conclusion: A better understanding of the immunomodulatory effects of each ITP therapy is needed to best manage the disease.
Introduction
Primary immune thrombocytopenia (primary ITP, formerly known as idiopathic thrombocytopenic purpura) is an organ-specific autoimmune disease in which the platelets and their precursors, megakaryocytes, are the targets of a disrupted immune system [Citation1]. It is estimated that approximately 4.5% of the Caucasian population suffers from an autoimmune disease [Citation2]. Primary ITP affects both sexes and occurs in both paediatric and adult populations, with an annual incidence rate of between 16 and 27 new cases per million [Citation3,Citation4]. The incidence is higher in women aged 30–59 years and in patients of both sexes older than 60 years [Citation3,Citation4]. The prevalence ranges from 4.5 to 10.5 per 100 000 in adults [Citation5,Citation6] and 4.6 per 100 000 in children [Citation7].
Traditionally, the pathophysiological model was based on platelet destruction mediated by autoantibodies [Citation8]. In 1951, Harrington and Hollingsworth [Citation9] demonstrated that ITP was caused by a circulating plasma factor, and in 1965 Shulman et al. [Citation10] identified this factor as platelet-specific immunoglobulin type G (IgG).
In 1994, thrombopoietin (TPO) was discovered [Citation11–Citation14]; the cytokine that stimulates the proliferation of megakaryocytes and platelet release via the TPO receptor (TPO-R, also known as c-Mpl) found in haematopoietic progenitor cells and megakaryocytes [Citation15]. The liver continuously synthesizes endogenous TPO (eTPO)[Citation16] which is subsequently cleared by being attached to circulating platelets [Citation17]. In patients with ITP, rather than a compensatory increase in eTPO, as seen in a megakaryocytic thrombocytopenia [Citation18], a reduced or normal level of eTPO is observed in approximately two-thirds of cases [Citation19]. Therefore, there is a functional deficit of eTPO [Citation20–Citation22]. Furthermore, although high levels of megakaryocytes are commonly found, most of them are immature or have been damaged by autoantibodies.
In parallel to these findings, several studies reported the presence of activated platelet-autoreactive T cells in ITP patients, with a cytokine imbalance towards interleukin (IL)-2 and interferon gamma (IFN-γ) [Citation23–Citation26], indicating its role in modulating the anti-platelet antibody response by B cells in ITP. More recently, the existence of other immunological mechanisms mediated by cytotoxic T lymphocytes has been discovered [Citation27,Citation28], providing the first evidence that the cell-mediated toxicity also participates in the pathogenesis of ITP. These lymphocyte subtypes cause both direct destruction of platelets and inhibition of their formation by inducing megakaryocyte maturation defects in the liver, kidney, spleen, and bone marrow [Citation29].
All this evidence supports the rationale for pharmacological therapy based on first-line corticosteroids with or without intravenous immunoglobulin (IVIG) [Citation30,Citation31]. Second-line therapies include splenectomy and thrombopoietin receptor agonists (A-TPOs) (romiplostim, eltrombopag) and, in refractory patients, different immunosuppressants such as azathioprine, rituximab, cyclosporin, cyclophosphamide, or danazol are used [Citation30]. Although in many cases the disease is controlled satisfactorily, there is still a significant percentage of patients who fail to achieve a complete and/or long-lasting remission [Citation32,Citation33].
This paper attempts to address the main unresolved questions in ITP: what is the alteration that initiates platelet destruction, which mechanisms perpetuate the disease, why do patients with the same platelet count have different clinical haemorrhagic manifestations, and why do some patients respond to some therapies while others do not?
We review the most current knowledge of the pathophysiology of ITP, including the alteration of B lymphocytes, the loss of immunological tolerance, and alterations in T cells. Furthermore, the immunological effects of available therapies are described in an attempt to contribute to the decision that clinicians make with regard to choosing the appropriate form of treatment.
Immune system physiology
Role of B and T lymphocytes
The B and T lymphocytes are the main cells of the specific immune response (Fig. ). B lymphocytes generate a humoral response [Citation34] when differentiating into plasma cells and memory B cells after recognition of the corresponding antigen. Plasma cells produce large amounts of IgG subtype antibodies, which neutralize bacterial toxins and viruses by agglutination, activation of the complement system, phagocytosis, and the activity of natural killer cells [Citation34]. There are also some regulatory B lymphocytes (Bregs) which inhibit activation of T cells and monocytes by secreting anti-inflammatory interleukins such as IL-10 [Citation35]. These cells regulate the differentiation of the different subtypes of T helper lymphocytes (Th), the pro-inflammatory differentiation of the antigen-presenting cells (APCs) and other autoimmune responses [Citation36].
Figure 1 Mechanisms of cellular and humoral immune response: the role of T and B lymphocytes.
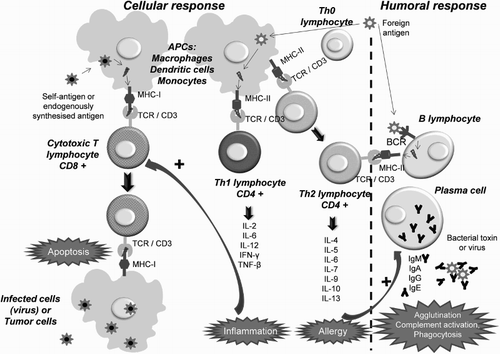
T lymphocytes, which mature in the thymus, generate a cellular response. Through their receptors (TCR) they recognize antigens bound to molecules of the major histocompatibility complex (MHC) on the surface of APCs (dendritic cells, monocytes, and macrophages) [Citation34]. T-cell precursors split and differentiate into effector T cells and memory T cells. The effector cells are classified into three main types cytotoxic (CTL) [CD8 positive (+)], which induce apoptosis; helper 1 (Th1) (CD4+), which activate CTLs and macrophages; and helper 2 (Th2) (CD4+), which activate B cells [Citation34]. The effector response of Th1 and Th2 consists of the secretion of various growth factors and ILs. The Th1 are mainly linked to inflammation [IL-2, IL-6, IL-12, IFN-γ, tumour necrosis factor beta (TNF-β)], and Th2 to the allergic response (IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-13) [Citation34].
There are also regulatory T cells (Treg) that monitor autoreactivity in the peripheral blood and inhibit the activity of both CTL and Th lymphocytes, and inhibit the production of antibodies through direct contact with B lymphocytes or by secreting cytokines such as IL-10 or TGF-β. Treg cells are CD4+CD25+ and can inhibit CD4+CD25− and CD8+ lymphocytes. They are therefore responsible for the ‘tolerance’ of the immune system [Citation34].
Cellular and humoral responses are interrelated (Fig. ). The Th effector cells stimulate both T and B cells and, in turn, the B cells require both antigen recognition and a signal by Th cells. In addition, Treg and Breg lymphocytes exert reciprocal control mechanisms between humoral and cellular responses [Citation34].
Natural tolerance mechanisms
The immune system has several mechanisms that inactivate the specific receptors that recognize self-antigens.
The first mechanism is central, and occurs in the thymus and bone marrow. Normally, in these organs, immature lymphocytes only meet with self-antigens presented by APCs as external antigens are presented in peripheral organs (lymph nodes and spleen). The TCR is capable of huge clonotypic variation that allows recognition of over 108 different antigens [Citation37]. After a process of positive and negative selection in the thymus, T cells with a TCR of low-or-intermediate affinity for the self-peptide/MHC complex are positively selected and allowed to mature. However, if the TCR has a high affinity, there is a negative selection that leads to apoptosis of the T cell. After these processes, only low-affinity autoreactive T cells enter the peripheral blood [Citation38].
The second mechanism is peripheral. It consists of the inactivation of circulating autoreactive lymphocytes that have escaped the central process. The Treg cells (previously selected in the thymus), when detecting naive (virgin) T cells that recognize self-antigens, block their differentiation and proliferation by contact-dependent inhibition, whereas when detecting autoreactive effector T cells, Treg cells block their function by secreting IL-10 and TGF-β [Citation37,Citation38].
Finally, there is an additional control mechanism mediated by B cells. Many antibodies are autoreactive, but most of them do not reach circulation thanks to the clonal selection that occurs in the bone marrow [Citation39]. This selection is only effective when self-proteins are present at high concentrations. If the antigens are present at low concentrations, the specific B cells can survive, and therefore an additional peripheral mechanism is required [Citation39]. Since B cell activation depends on Th cells, the removal or anergy of T cells with TCR for the same antigens can, by itself, prevent production of autoantibodies.
Immune system alterations in ITP
Approximately 25% of patients with ITP do not have detectable platelet autoantibodies [Citation40], on the one hand due to the lack of standardization of detection methods and on the other hand due to the existence of additional pathophysiological mechanisms. Besides anti-platelet autoantibodies, the alterations include abnormalities in T cells, such as platelet destruction mediated by Th1 and CTL and alterations in the function of Th1 and Treg cells [Citation27], which in turn is associated with clonal activation of autoreactive B cells [Citation41] (Fig. and Table ).
Figure 2 Immunological alterations in ITP: (A) the presence of an inflammatory stimulus or a stressful situation (associated with an infection, drugs, etc.) transforms certain platelet GPs into autoantigens that trigger an immune response; (B) antigen-presenting cells (activated macrophages) process the platelet autoantigens and present them on MHC class I and II molecules; (C) circulating Th cells with TCR specific for platelet antigens activate and become autoreactive, secreting cytokines which act as co-stimulatory signals of B lymphocytes; (D) activated autoreactive B cells recognize platelet GPs and initiate mass production of autoantibodies (IgG) that destroy platelets and megakaryocytes through the humoral response; (E) in parallel, activated macrophages present platelet autoantigens to cytotoxic T cells and activate them, inducing platelet destruction mediated by the cellular response. Adapted from John W. Semple's Laboratory Homepage, by M. Perera. Retrieved from http://www.angelfire.com/ut/johnsnotes/index.html. Copyright 2009 by John W. Semple. Adapted with permission.
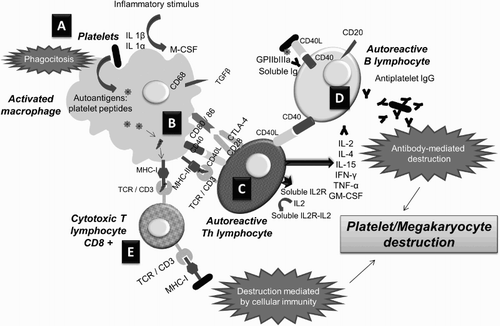
Table 1 Main alterations of the immune system described in patients with ITP and the possible effects achievable using different available therapies
Alterations in B lymphocytes
Development of anti-platelet autoantibodies
Although their existence has been known since 1951 [Citation9], in 1971 there was the discovery of IgG autoantibodies directed against the glycoprotein (GP) of platelet membranes (mainly GPIIb/IIIa, GPIb/IX, and others like GPIa/IIa, GPIV, and GPV) [Citation42]. These opsonic antibodies facilitate binding of phagocytes that destroy platelets. To this end, they bind to the receptor of the constant portion of the IgG (Fc-R) present on the surface of macrophages of the reticuloendothelial system (RES). These GPs are also found in megakaryocytes [Citation43].
The origin of autoreactive B cells may be due to several factors. For example, there are environmental factors, prompted by molecular mimicry between foreign and self-molecules. Some infectious agents with structures (usually of protein nature) similar to those of the host may develop autoreactivity. These include several viruses (human immunodeficiency virus, Epstein–Barr virus, hepatitis C virus) and bacteria (Helicobacter pylori), and even some drugs [Citation1]. Evidence regarding the cross-reactivity of virus-specific antibodies with normal platelet antigens was firstly demonstrated in patients developing ITP after human immunodeficiency virus [Citation44] or varicella zoster virus infection [Citation45]. Molecules that are highly conserved in the phylogeny are the best candidates since they contain common epitopes. Heat shock proteins (HSP) are expressed in all prokaryotic and eukaryotic cells under stress conditions (increased temperature, lack of water or glucose, radiation, etc.) [Citation46]. It is hypothesized that an immune response against a microorganism HSP could cross-react with a self HSP. Anti-HSP antibodies have been found in several autoimmune diseases such as type I diabetes, Crohn's disease, rheumatoid arthritis, and lupus erythematosus [Citation46]. In ITP, the conversion of HSP60 or HSP70 in autoantigens could lead to the loss of tolerance.
Additionally, genetic susceptibility of the individual is probably involved. For example, ITP has been found to be significantly associated with several polymorphisms: MHC HLAB8DR3 [Citation47], polymorphisms in the genes encoding TNF-β [Citation48], R-Fc [Citation49], DNA methyltransferase 3B (DNMT3B), and IL-1 receptor antagonist [Citation50].
Synergy between autoantibodies and platelet toll-like receptors
Recently, it has been shown that platelets express Toll-like receptors (TLRs), particularly TLR4, TLR-7, and BAFF1, so they can present antigens through MHC-II [Citation51]. TLRs are transmembrane proteins that act as pattern recognition receptors [Citation52]. They have affinity for different groups of pathogen-associated molecular patterns (PAMP) [Citation53], such as lipopolysaccharides or nucleic acids of viruses and bacteria. The TLR send intracellular signals that activate different pathways, including mitogen-activated protein (MAP) kinases, signal transducers and activators of transcription, and the pathway of the nuclear factor kappa B [Citation54]. Then, the affected cells produce inflammatory cytokines [ILs, interferons, and tumour necrosis factor alpha (TNF-α)] that send ‘danger signals’ to the immune system [Citation55], triggering a rapid recruitment of effector cells and stimulation of the APC [Citation56].
Recent studies show that TLR can recognize, in addition to PAMP, molecules from injured tissues [Citation53]. The presence of TLRs in platelets is probably because these cells can act as circulating sentinels for infection [Citation51]. This would explain the worsening of the disease observed during infections in some patients with ITP. It is postulated that infections caused by pathogens recognized by TLR increase platelet destruction, multiplied in the presence of anti-platelet antibodies due to the synergy with TLR, which increases R-Fc-mediated phagocytosis [Citation57] (Fig. ). In turn, the TLR can increase the production of antibodies, since they activate the secretion of factor B lymphocyte stimulator by dendritic cells, which has been correlated with the level of autoantibodies [Citation58].
Figure 3 Possible role of platelet TLRs in the pathophysiology of ITP: platelets act as circulating sentinels and, in the event of detecting pathogens or molecules derived from injured tissues, the TLRs that recognize them send ‘danger signals’ to the immune system by stimulating the effector cells and the APCs of the RES; if anti-platelet antibodies are also present, the TLRs stimulate R-Fc-mediated phagocytosis. Adapted from John W. Semple's Laboratory Homepage, by M. Perera. Retrieved from http://www.angelfire.com/ut/johnsnotes/index.html. Copyright 2009 by John W. Semple. Adapted with permission.
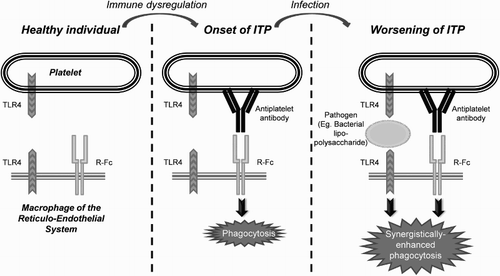
These mechanisms could also explain why, in some patients, there is spontaneous ITP resolution after treatment with antibacterial agents, since they would inactivate the mechanisms of TLR-mediated destruction.
Autoantibodies with different isotypes
It has been hypothesized that not all anti-platelet antibodies are equal, but there are several isotypes associated with different alterations in platelet function. An in vitro study showed that there are differences in SRE-mediated phagocytosis between platelets opsonized by different antibody isotypes [Citation59]. It is further postulated that some isotypes could inhibit platelet activation instead of inducing phagocytosis, which may explain why, at a given platelet count, some patients bleed and others do not. The subtype of antibody could be related also to the severity of the disease, and to different responses to the administration of IVIG.
Increase in splenic B lymphocytes
A large number of studies suggest the presence of abnormalities in B lymphocytes. Olsson et al. [Citation60] found an increase in the number of B lymphocytes in the spleens of 29 ITP patients compared to healthy controls. Audia et al. [Citation61] observed a decrease in circulating and splenic B lymphocytes in 18 splenectomized patients with or without rituximab, independently of treatment response. They also observed an increase in the ratio of Th1/Treg lymphocytes, suggesting that rituximab, besides inhibiting B cells, might modulate the T lymphocyte response.
Decrease in Breg lymphocytes
In untreated patients with ITP, a decrease in the population of Breg was observed, phenotypically characterized as CD19+CD24hiCD38hi cells [Citation62,Citation63]. Although this interaction has not yet been demonstrated in humans, a plausible hypothesis in ITP would be that the alteration of Breg cells contributes to the already known disruption in Treg cells, as will be explained below [Citation64].
Two subtypes of B-regulatory lymphocytes were identified in patients with ITP: CD19+ CD24+ CD38+ and CD19+ CD24+ FOXP3+. A prior report indicated no difference in splenic B-lymphocytes and between ITP patients and controls, but lower splenic Treg cells in ITP [Citation61]. Altered Breg function has also been described in ITP, with decreased IL-10 secretion and less inhibitory capacity [Citation62]. In mouse models, the IL-10-secreting Breg cells promote the differentiation or recruitment of Treg cells [Citation65,Citation66].
The CD19+CD24+CD38 B-regulatory cells from ITP patients were not significantly different from controls, but CD19+CD24+FOXP3+ Bregs were markedly increased in the spleen. Glucocorticoids and other lympholytic agents in the treatment of ITP before splenic removal are less likely to explain the finding.
This CD19+CD24+FOXP3+ B-regulatory population has been reported in normal human blood [Citation67], and is diminished in the blood of patients with autoimmune disease [Citation68]. Increased splenic CD19+CD24+FOXP3+ Bregs may be due to sequestration, comparable to the sequestration of T-regulatory cells noted in the thymuses of experimental ITP mice [Citation51]. These findings raise the question of the role of B-regulatory sub-types in the alteration of T and B lymphocyte interaction, and immune modulation that results in the antibody and T-lymphocyte destruction of platelets in chronic ITP [Citation69].
Alterations in T lymphocytes
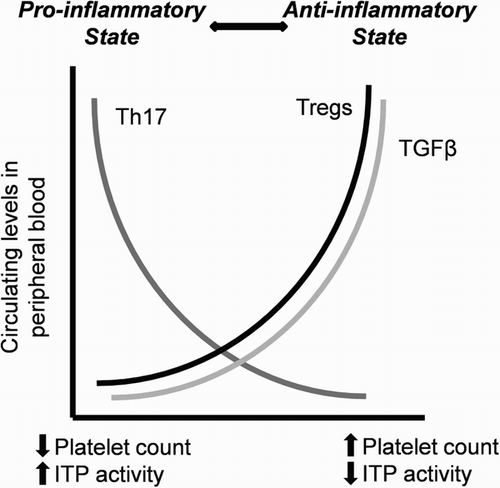
To date, there have been three types of T cell alterations in ITP described: an increase in Th1 over Th2 lymphocytes; an increase in Th17 cells; and a decrease in Treg lymphocytes (Fig. ) [Citation64].
Figure 4 Alterations in T lymphocytes in active ITP (pro-inflammatory state): there is an increase in Th17 cells and a decrease in Treg levels, which release less amount of the anti-inflammatory cytokine TGF-β to the blood. These changes are reversed after the response to several treatments, such as A-TPOs. Adapted by M. Perera with permission from John W. Semple. Copyright 2009 by John W. Semple.
Increase in Th1 lymphocytes
Patients with active ITP have an imbalance in the Th1/Th2 response when compared with healthy controls and patients with ITP who have responded to treatment [Citation24]. The pro-inflammatory CD16+ monocytes, which are increased in ITP in detriment of CD14hiCD16− monocytes [Citation70], release TNF and promote the development of Th1 and the release of IL-17, downregulating Tregs [Citation70]. Th1 release cytokines that activate the cellular response: IL-2, a factor stimulating Th1, Th2, and CTLs; IFN-γ, which induces conversion of macrophages to APC; and TNF-β, which recruits more macrophages [Citation71]. Higher IL-23 levels and increased expression of TLR4 are also observed in monocytes [Citation72]. On the other hand, there is a decrease in Th2 cells and in the inhibitory cytokines IL-10 and TGF-β. In chronic ITP, the levels of TGF-β are inversely related to disease activity [Citation71]. (Fig. ).
Increase in Th17 cells
In ITP there is an increase in pro-inflammatory Th17 cells that secrete IL-17 and IL-22 [Citation72,Citation73], although not all studies have replicated these findings [Citation74,Citation75]. In most autoimmune diseases, there is an imbalance between pro-inflammatory and anti-inflammatory mechanisms. The Th17 cells develop from Th0 through a different pathway than Th1 or Th2. The IL17 plays an important role in many autoimmune diseases [Citation34]. In ITP, several studies have shown increased IL-17 during active disease in both children and adults [Citation73]. IL-22 is also elevated in active phases of disease, and decreases in patients responsive to dexamethasone [Citation76].
Decrease in Treg lymphocytes
Phenotypically, Treg cells are CD4+CD25+FoxP3+ [Citation77]. They represent 5–10% of all lymphocytes, and suppress humoral and cellular response of autoreactive lymphocytes by secreting IL-10 and TGF-β. Their effect is contrary to that of Th17 cells [Citation78]. ITP patients have decreased Treg levels, which normalize after a response to different therapies [Citation72,Citation77]. The Treg/Th17 ratio correlates with disease activity [Citation79], and lower levels of Treg, IL-10, and TGF-β are associated with lower platelet counts [Citation75,Citation80].
In murine ITP models, it has been observed that Treg reduction occurs only in the spleen, whereas there is an increase in Tregs in the thymus. Treatment of these mice with IVIG increased platelet counts, decreased autoantibody production, and normalized the levels of Tregs in both the thymus and spleen. Therefore, these models suggest that ITP could be associated with a peripheral Tregs deficiency due to their retention in the thymus, which normalizes after treatment [Citation81].
Lymphoid organs
Studies of peripheral blood APCs, lymphocyte subpopulations, and cytokines in the peripheral blood might not mirror the organ-specific environments, such as in the spleen, which is a known site of anti-platelet antibody synthesis. Follicular T-helper cells and Tregs within the proliferative lymphoid nodules (PLNs) and germinal centres (GCs) of spleens from patients with ITP were reduced compared with spleens removed for other reasons [Citation82]. The density of follicular T-helper cells and Tregs was lower in the PLNs, but not GCs, which also contain GP IIb/IIIa within IgM-containing immune complexes tightly bound to follicular dendritic cells, closely approximated to proliferating B-cells. These studies suggest that PLNs are the sites of auto-antigen stimulation related to a lack of T-cell control [Citation82]. The ratio of Th1 cells to Tregs was reported to be increased in the spleens of ITP patients who failed rituximab therapy despite depletion of peripheral blood B-cells [Citation61]. It has also been suggested that rituximab may induce the differentiation of autoimmune plasma cells into long-lived resident cells in the spleen of patients who failed treatment [Citation83]. These studies serve to highlight potential differences between findings in specialized lymphoid organs and those made on components of peripheral blood.
Relationship between pathophysiology and disease course
According to its clinical evolution, ITP is classified as ‘newly diagnosed,’ ‘persistent’ (3–12 months) or ‘chronic’ (>12 months) [Citation1]. More aggressive treatment options, such as splenectomy, are usually reserved for patients with chronic ITP [Citation30].
As mentioned previously, autoreactivity against HSP60 may lead to the lack of anti-inflammatory response due to a decreased activation of Tregs, which can lead to the chronic form of the disease. HSP60 serum levels in newly diagnosed ITP are lower than those in patients with chronic ITP or healthy controls [Citation84]. They are also higher in patients with counts >30 × 109/l and are positively correlated with platelet counts [Citation84]. Therefore, in the initial stages, it is possible that the autoantibodies generated by molecular mimicry with HSP are the only mechanism causing thrombocytopenia. Since these autoantibodies induce changes in Treg and Breg cells, the disease may become chronic if the inhibited regulatory mechanisms fail to completely eliminate autoreactive cells. In this case, despite the temporary removal of autoantibodies, anti-platelet autoreactivity would manifest again sooner or later.
The natural course of the disease differs between children and adults. In the paediatric population, most cases resolve spontaneously and do not require treatment. Pathophysiologically, this course of ITP would be explained by a transient autoantibody production during a viral infection. Spontaneous remission is associated with the age of onset, being most frequent between 2 and 12 months of age (90%), compared to 1–8 years (71%) or 9–18 years (49%) [Citation85]. Moreover, in children over 1 year of age, there are more remissions if the initial count is very low (<10 × 109/l), and if there is a history of other diseases. The remission rate at 6 months is independent of treatment with corticosteroids or IVIG [Citation86]. If the disease does become chronic, spontaneous remissions in children are less common, about 33%, of which 45% occur in the short term. After splenectomy, 80% achieve remission, but some cases may relapse after several years [Citation87]. Late relapses could be explained by patients continuing to produce low amounts of autoantibodies that cause sustained platelet destruction and inhibited formation; in the long term, destruction is greater than formation [Citation87].
Severe bleeding occurs rarely in children (0.1% developed an intracranial haemorrhage in the IPARC registry[Citation86]), although it is unknown whether this is due to the presence of a certain isotype of autoantibodies.
In adults, spontaneous remission rates are traditionally lower, ranging from 5 to 11% [Citation88], although a recent study in patients treated with romiplostim found an increased rate of up to 32% [Citation89]. At 12 months, approximately 70% develop chronic ITP [Citation90]. The elderly are more likely to suffer chronic disease [Citation87]. At a pathophysiological level, the positivity for autoantibodies at diagnosis is associated with more severe bleeding and a chronic course [Citation90]. It seems plausible that changes in the Th and Treg lymphocytes induced by autoreactive B cells (Fig. ) alter the inhibitory mechanisms of the immune system, thereby perpetuating the disease.
Relationship between pathophysiology and treatment
Treatment of ITP has traditionally been based on corticosteroids, IVIG, and splenectomy [Citation1]. However, the authorization of the A-TPO romiplostim and eltrombopag within Europe in 2009 and 2010, that lead to increased platelet production, resulted in higher response rates of up to 70–80% being achieved even in patients refractory to other treatments [Citation91].
Several recent studies have examined the immunological changes induced by some treatments (Table ).
Corticosteroids and other immunosuppressive agents
Following response to corticosteroids, associated or not with other immunosuppressive agents such as rituximab or rapamycin, a recovery of the Treg cells population has been observed.
A study of high-dose dexamethasone was found to result in the normalization of the Th1/Th2 ratio, Treg and Th17 cells levels and, by inhibition of RORγt and GATA3, an increased expression of Foxp3 [Citation92]. In patients with rapamycin and low-dose prednisone, there was an increase in Treg and a strong correlation between the levels of TGF-β and Treg after treatment. Furthermore, the upregulation was maintained after discontinuation [Citation93].
A study of rituximab and corticosteroids confirmed the improvement in Treg cells, which was greater and more prolonged than in patients treated with corticosteroids alone [Citation94]. Stasi et al. [Citation95] also observed a rituximab-mediated modulation of Treg cells.
The number of circulating plasmacytoid dendritic cells (PDC) is low in patients with ITP, both with primary or with Helicobacter (H.) pylori-associated disease. However, no reduction is observed in the levels of myeloid dendritic cells (MDC) or CTLs. Interestingly, an increase in PDC was observed in responders but not in non-responders after eradicating H. pylori with an antibiotic, while PDC and MDCs decreased in both responders and in non-responders after administration of prednisolone. In untreated patients, the low number of PDCs persisted [Citation74]. Another study of high-dose dexamethasone found an increase in MDCs, a decrease in PDCs, and a lower expression of CD11c in MDCs after four days of treatment [Citation96].
IVIG
Multiple immunological changes have been described during IVIG therapy. They include inhibition of the production of autoantibodies, their neutralization by anti-idiotype antibodies, inhibition of complement-mediated damage; modulation of cytokine production, induction of apoptosis of lymphocytes and monocytes, and modulation of the functions of B and T lymphocytes [Citation97]. They also appear to block the activating R-Fc and favour the expression of inhibiting R-Fc [Citation97]. Therefore, they are a well-established therapeutic option, and have been associated with long-term responses [Citation30].
A-TPO
The A-TPOs are indicated in patients who do not respond to splenectomy or in whom splenectomy is contraindicated. Due to their mechanism of action, treatment should be administered on a continuous basis and, once it is suspended, the platelet count is expected to decrease [Citation30]. However, there are an increasing number of published cases of maintenance of response after cessation of romiplostim [Citation98–Citation101], and also some cases published with eltrombopag [Citation100,Citation102]. During the clinical development of romiplostim, 7% of patients had remission, which, at that time, was regarded as spontaneous [Citation91]. A later study designed to assess remission prospectively found a rate of 32% [Citation89].
Two recent studies suggest that A-TPO could induce immunomodulatory changes beyond its stimulatory effect on platelet production. Bao et al. [Citation103] demonstrated that A-TPOs increased Treg activity and decreased levels of IL-2-releasing Th1 cells (CD4+), consistent with an immune response dampening effect by the A-TPO. An increase in circulating TGF-β was also observed, which was correlated with the recovery of platelet counts. This suggests that the platelets could mediate the recovery of Treg cells during treatment with A-TPO, either directly or indirectly by an increased release of TGF-β, as a result of the increased platelet renewal rate. Another study by the same group observed a Breg increase in non-splenectomized patients after a response to A-TPOs [Citation70]. Moreover, in the patients with higher platelet counts, there was an enhancement of the monocyte-suppressor activity of B lymphocytes. They also observed a normalization of pro-inflammatory CD16+ monocytes in responders, while the non-responders continued with increased levels [Citation70].
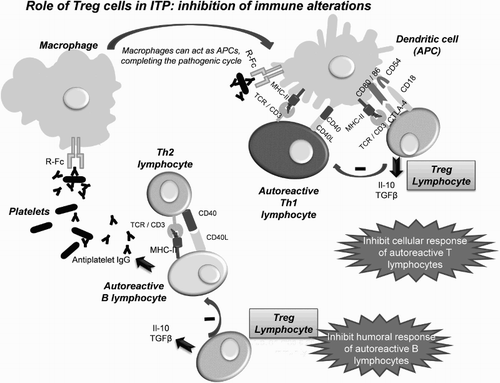
Both studies suggest that treatment with A-TPO in ITP patients could not only recover platelet counts, but also modulate the immune response or even restore the immune tolerance. Since immune cells do not express TPO-R, the effect should be mediated by other mechanisms. One of the most plausible hypotheses is direct stimulation of Breg cells by the newly generated platelets [Citation64], which would express CD40L due to the activation by the autoantibodies still present [Citation104]. The Breg cells are activated through CD40 [Citation63]. In turn, the Bregs would induce a recovery in the Treg levels, which would inhibit the activity of effector T cells (CTL and Th) and autoreactive B cells (Fig. ), leading to the complete remission of ITP. Further research is needed to corroborate these findings.
Figure 5 Effects of recovery in the level of Treg cells in ITP patients: the Tregs inhibit the activity of autoreactive effector T cells (CTL and Th) in the presence of APCs with autoantigens by releasing soluble IL-10 and TGF-β; they also inhibit, by direct contact, autoreactive B lymphocytes, which fail to produce antibodies against platelets.
Conclusions
Patients with ITP have multiple immune system alterations. At the onset of disease, there is a loss of tolerance to platelet GP antigens, caused by molecular mimicry phenomena with infectious agents or drugs, which may be modulated by genetic factors and by the presence of TLR on the platelet surface. As a result, autoreactive effector B and T lymphocytes are activated, and Breg and Treg subpopulations are inhibited, perpetuating the disease in some patients. Activated B lymphocytes release autoantibodies that destroy opsonized platelets, inducing their phagocytosis and inhibiting their maturation from megakaryocytes. In some patients with certain autoantibody isotypes, instead of an increased phagocytosis, there is an inhibition of platelet activation, which increases the probability of bleeding. In T lymphocytes there is an increase in pro-inflammatory Th1 and Th17 subtypes, and a reduction in Th2 and in the cytokines IL-10 and TGF-β that suppress the immune response. Several treatments, such as steroids, rituximab, and A-TPOs could present further benefits to the recovery of platelet counts, thanks to the restoration of subpopulations of T lymphocytes and levels TGF-β. The A-TPOs have also demonstrated a recovery in the levels of Breg lymphocytes, probably by direct stimulation by platelets through CD40. Further studies are needed to verify whether the recovery of Breg and Treg is the underlying mechanism that explains complete remission. Such knowledge would allow tailoring of the treatment choice, not only with the aim of recovering platelet levels, but also aiming to completely restore the immune system of the patient.
Disclaimer statements
Contributors María Perera wrote the article and approved the final version. Teresa Garrido reviewed it and approved the final version.
Funding The project and the writing of this document have been funded by an unrestricted educational grant provided by Amgen S.A.
Conflicts of interest The medical writing support has been provided by Dr Neus Valveny from TFS Develop. Teresa Garrido is an employee of Amgen S.A.
Ethics approval None.
References
- Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–93.
- Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. 2012;11(10):754–65.
- Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999;94(3):909–13.
- Neylon AJ, Saunders PWG, Howard MR, Proctor SJ, Taylor PRA. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122(6):966–74.
- Anon. Ferri consultor clinico, 2006–2007: claves diagnosticas y tratamiento. Available from: http://www.agapea.com/libros/Ferri-consultor-clinico-2006-2007-claves-diagnosticas-y-tratamiento-9788481749144-i.htm [Accessed March 13, 2014].
- Segal JB, Powe NR. Prevalence of immune thrombocytopenia: analyses of administrative data. J Thromb Haemost JTH. 2006;4(11):2377–83.
- Hedman A, Henter JI, Hedlund I, Elinder G. Prevalence and treatment of chronic idiopathic thrombocytopenic purpura of childhood in Sweden. Acta Paediatr Oslo Nor 1992. 1997;86(2):226–7.
- Shulman NR, Weinrach RS, Libre EP, Andrews HL. The role of the reticuloendothelial system in the pathogenesis of idiopathic thrombocytopenic purpura. Trans Assoc Am Physicians. 1965;78:374–90.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med. 1951;38(1):1–10.
- Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann N Y Acad Sci. 1965;124(2):499–542.
- Bartley TD, Bogenberger J, Hunt P, Li YS, Lu HS, Martin F, et al. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell 1994;77(7):1117–24.
- Kuter DJ, Beeler DL, Rosenberg RD. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Natl Acad Sci USA. 1994;91(23):11104–8.
- de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 1994;369(6481):533–8.
- Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature 1994;369(6481):565–8.
- Kaushansky K, Drachman JG. The molecular and cellular biology of thrombopoietin: the primary regulator of platelet production. Oncogene 2002;21(21):3359–67.
- Wolber E-M, Jelkmann W. Thrombopoietin: the novel hepatic hormone. News Physiol Sci. 2002;17:6–10.
- Li J, Xia Y, Kuter DJ. Interaction of thrombopoietin with the platelet c-mpl receptor in plasma: binding, internalization, stability and pharmacokinetics. Br J Haematol. 1999;106(2):345–56.
- Kappers-Klunne MC, de Haan M, Struijk PC, van Vliet HHDM. Serum thrombopoietin levels in relation to disease status in patients with immune thrombocytopenic purpura. Br J Haematol. 2001;115(4):1004–6.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest. 1987;80(1):33–40.
- Emmons RV, Reid DM, Cohen RL, Meng G, Young NS, Dunbar CE, et al. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood 1996;87(10):4068–71.
- Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115(12):3339–47.
- Houwerzijl EJ, Blom NR, van der Want JJL, Esselink MT, Koornstra JJ, Smit JW, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004;103(2):500–6.
- Semple JW, Freedman J. Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia. Blood 1991;78(10):2619–25.
- Semple JW, Milev Y, Cosgrave D, Mody M, Hornstein A, Blanchette V, et al. Differences in serum cytokine levels in acute and chronic autoimmune thrombocytopenic purpura: relationship to platelet phenotype and antiplatelet T-cell reactivity. Blood 1996;87(10):4245–54.
- Ogawara H, Handa H, Morita K, Hayakawa M, Kojima J, Amagai H, et al. High Th1/Th2 ratio in patients with chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 2003;71(4):283–8.
- Kuwana M, Kaburaki J, Ikeda Y. Autoreactive T cells to platelet GPIIb-IIIa in immune thrombocytopenic purpura. Role in production of anti-platelet autoantibody. J Clin Invest. 1998;102(7):1393–402.
- Olsson B, Andersson P-O, Jernås M, Jacobsson S, Carlsson B, Carlsson LMS, et al. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med. 2003;9(9):1123–4.
- Zhang F, Chu X, Wang L, Zhu Y, Li L, Ma D, et al. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 2006;76(5):427–31.
- Stasi R. Immune thrombocytopenia: pathophysiologic and clinical update. Semin Thromb Hemost. 2012;38(5):454–62.
- Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115(2):168–86.
- Sanz MÁ, Vicente García V, Fernández A, López MF, Grande C, Jarque I, et al. [Guidelines for diagnosis, treatment and monitoring of primary immune thrombocytopenia]. Med Clínica. 2012;138(6):261.e1–e17.
- Neunert CE, Buchanan GR, Imbach P, Bolton-Maggs PHB, Bennett CM, Neufeld E, et al. Bleeding manifestations and management of children with persistent and chronic immune thrombocytopenia: data from the Intercontinental Cooperative ITP Study Group (ICIS). Blood 2013;121(22):4457–62.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004;104(4):956–60.
- Roitt I, Delves P. Roitt's essential immunology. 10th ed. Oxford: Wiley; 2001.
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–50.
- Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M. B lymphocytes regulate dendritic cell (DC) function in vivo: increased interleukin 12 production by DCs from B cell-deficient mice results in T helper cell type 1 deviation. J Exp Med. 2000;192(4):475–82.
- Coutinho A, Caramalho I, Seixas E, Demengeot J. Thymic commitment of regulatory T cells is a pathway of TCR-dependent selection that isolates repertoires undergoing positive or negative selection. Curr Top Microbiol Immunol. 2005;293:43–71.
- Hsieh C-S, Lee H-M, Lio C-WJ. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12(3):157–67.
- Salinas GF, Braza F, Brouard S, Tak P-P, Baeten D. The role of B lymphocytes in the progression from autoimmunity to autoimmune disease. Clin Immunol Orlando Fla. 2013;146(1):34–45.
- Zhang H-Y, Hou M, Zhang X-H, Guan X-H, Sun G-Z. The diagnostic value of platelet glycoprotein-specific autoantibody detection in idiopathic thrombocytopenic purpura. [Zhongguo Shi Yan Xue Ye Xue Za Zhi Zhongguo Bing Li Sheng Li Xue Hui] J Exp Hematol Chin Assoc Pathophysiol. 2004;12(2):204–6.
- Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood 2002;100(4):1388–98.
- McMillan R, Smith RS, Longmire RL, Yelenosky R, Reid RT, Craddock CG. Immunoglobulins associated with human platelets. Blood 1971;37(3):316–22.
- Stahl CP, Zucker-Franklin D, McDonald TP. Incomplete antigenic cross-reactivity between platelets and megakaryocytes: relevance to ITP. Blood 1986;67(2):421–8.
- Bettaieb A, Fromont P, Louache F, Oksenhendler E, Vainchenker W, Duédari N, et al. Presence of cross-reactive antibody between human immunodeficiency virus (HIV) and platelet glycoproteins in HIV-related immune thrombocytopenic purpura. Blood 1992;80(1):162–9.
- Wright JF, Blanchette VS, Wang H, Arya N, Petric M, Semple JW, et al. Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br J Haematol. 1996;95(1):145–52.
- Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32(2):89–95.
- Porges A, Bussel J, Kimberly R, Schulman I, Pollack M, Pandey J, et al. Elevation of platelet associated antibody levels in patients with chronic idiopathic thrombocytopenic purpura expressing the B8 and/or DR3 allotypes. Tissue Antigens. 1985;26(2):132–7.
- Satoh T, Pandey JP, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y, et al. Single nucleotide polymorphisms of the inflammatory cytokine genes in adults with chronic immune thrombocytopenic purpura. Br J Haematol. 2004;124(6):796–801.
- Satoh T, Miyazaki K, Shimohira A, Amano N, Okazaki Y, Nishimoto T, et al. Fcγ receptor IIB gene polymorphism in adult Japanese patients with primary immune thrombocytopenia. Blood 2013;122(11):1991–2.
- Pesmatzoglou M, Lourou M, Goulielmos GN, Stiakaki E. DNA methyltransferase 3B gene promoter and interleukin-1 receptor antagonist polymorphisms in childhood immune thrombocytopenia. Clin Dev Immunol. 2012;2012:352059.
- Aslam R, Speck ER, Kim M, Crow AR, Bang KWA, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006;107(2):637–41.
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–45.
- Xu D, Liu H, Komai-Koma M. Direct and indirect role of Toll-like receptors in T cell mediated immunity. Cell Mol Immunol. 2004;1(4):239–46.
- Akira S. Mammalian Toll-like receptors. Curr Opin Immunol. 2003;15(1):5–11.
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
- Matzinger P. The danger model: a renewed sense of self. Science 2002;296(5566):301–5.
- Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood 2007;109(11):4803–5.
- Yu H, Liu Y, Han J, Yang Z, Sheng W, Dai H, et al. TLR7 regulates dendritic cell-dependent B-cell responses through BlyS in immune thrombocytopenic purpura. Eur J Haematol. 2011;86(1):67–74.
- Hoemberg M, Stahl D, Schlenke P, Sibrowski W, Pachmann U, Cassens U. The isotype of autoantibodies influences the phagocytosis of antibody-coated platelets in autoimmune thrombocytopenic purpura. Scand J Immunol. 2011;74(5):489–95.
- Olsson B, Ridell B, Jernås M, Wadenvik H. Increased number of B-cells in the red pulp of the spleen in ITP. Ann Hematol. 2012;91(2):271–7.
- Audia S, Samson M, Guy J, Janikashvili N, Fraszczak J, Trad M, et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood 2011;118(16):4394–400.
- Li X, Zhong H, Bao W, Boulad N, Evangelista J, Haider MA, et al. Defective regulatory B-cell compartment in patients with immune thrombocytopenia. Blood 2012;120(16):3318–25.
- Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010;32(1):129–40.
- Yazdanbakhsh K, Zhong H, Bao W. Immune dysregulation in immune thrombocytopenia (ITP). Semin Hematol. 2013;50(1):S63–7.
- Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol. 2010;125(5):1114–24.e8.
- Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol Baltim Md 1950. 2011;186(10):5569–79.
- Noh J, Choi WS, Noh G, Lee JH. Presence of Foxp3-expressing CD19(+)CD5(+) B cells in human peripheral blood mononuclear cells: human CD19(+)CD5(+)Foxp3(+) regulatory B cell (Breg). Immune Netw. 2010;10(6):247–9.
- Guo Y, Zhang X, Qin M, Wang X. Changes in peripheral CD19(+)Foxp3(+) and CD19(+)TGFβ(+) regulatory B cell populations in rheumatoid arthritis patients with interstitial lung disease. J Thorac Dis. 2015;7(3):471–7.
- Aslam R, Segel GB, Burack R, Spence SA, Speck ER, Guo L, et al. Splenic lymphocyte subtypes in immune thrombocytopenia: increased presence of a subtype of B-regulatory cells. Br J Haematol. 2016;173(1):159–60.
- Zhong H, Bao W, Li X, Miller A, Seery C, Haq N, et al. CD16+ monocytes control T-cell subset development in immune thrombocytopenia. Blood 2012;120(16):3326–35.
- Andersson P-O, Olsson A, Wadenvik H. Reduced transforming growth factor-beta1 production by mononuclear cells from patients with active chronic idiopathic thrombocytopenic purpura. Br J Haematol. 2002;116(4):862–7.
- Liu H, Ouyang X, Li Y, Zeng H, Wang X, Xie S, et al. Involvement of levels of Toll like receptor-4 in monocytes, CD4+ T-lymphocyte subsets, and cytokines in patients with immune thrombocytopenic purpura. Thromb Res. 2013;132(2):196–201.
- Rocha AMC, Souza C, Rocha GA, de Melo FF, Clementino NCD, Marino MCA, et al. The levels of IL-17A and of the cytokines involved in Th17 cell commitment are increased in patients with chronic immune thrombocytopenia. Haematologica 2011;96(10):1560–4.
- Saito A, Yokohama A, Osaki Y, Ogawa Y, Nakahashi H, Toyama K, et al. Circulating plasmacytoid dendritic cells in patients with primary and helicobacter pylori-associated immune thrombocytopenia. Eur J Haematol. 2012;88(4):340–9.
- Chang D, Ouyang J, Zhou R, Xu J, Chen B, Yang Y, et al. [Profiles of different subsets of CD(4)(+) T cells in chronic idiopathic thrombocytopenic purpura]. Zhonghua Nei Ke Za Zhi. 2010;49(3):213–6.
- Cao J, Chen C, Zeng L, Li L, Li X, Li Z, et al. Elevated plasma IL-22 levels correlated with Th1 and Th22 cells in patients with immune thrombocytopenia. Clin Immunol Orlando Fla. 2011;141(1):121–3.
- Nishimoto T, Kuwana M. CD4+CD25+Foxp3+ regulatory T cells in the pathophysiology of immune thrombocytopenia. Semin Hematol. 2013;50(Suppl 1):S43–49.
- Cao J, Li X-Q, Chen C, Zeng L-Y, Cheng H, Li Z-Y, et al. [Imbalance of Th17/Treg cells ratio in peripheral blood of patients with immune thrombocytopenia]. [Zhongguo Shi Yan Xue Ye Xue Za Zhi Zhongguo Bing Li Sheng Li Xue Hui] J Exp Hematol Chin Assoc Pathophysiol. 2011;19(3):730–3.
- Ji L, Zhan Y, Hua F, Li F, Zou S, Wang W, et al. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PloS One. 2012;7(12):e50909.
- Jia R-P, Zhao X-Y. [Expression and clinical significance of CD4(+)CD25(+)Treg cells, sFas and sFasL in peripheral blood of patients with autoimmune thrombocytopenic purpura]. [Zhongguo Shi Yan Xue Ye Xue Za Zhi Zhongguo Bing Li Sheng Li Xue Hui] J Exp Hematol Chin Assoc Pathophysiol. 2011;19(5):1264–7.
- Aslam R, Hu Y, Gebremeskel S, Segel GB, Speck ER, Guo L, et al. Thymic retention of CD4+CD25+FoxP3+ T regulatory cells is associated with their peripheral deficiency and thrombocytopenia in a murine model of immune thrombocytopenia. Blood 2012;120(10):2127–32.
- Daridon C, Loddenkemper C, Spieckermann S, Kuhl AA, Salama A, Burmester GR, et al. Splenic proliferative lymphoid nodules distinct from germinal centers are sites of autoantigen stimulation in immune thrombocytopenia. Blood 2012;120(25):5021–31.
- Mahévas M, Patin P, Huetz F, Descatoire M, Cagnard N, Bole-Feysot C, et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J Clin Invest. 2013;123(1):432–42.
- Dolasik I, Birtas Atesoglu E, Tarkun P, Mehtap O, Keski H, Dogru A, et al. Decreased serum heat shock protein 60 levels in newly diagnosed immune thrombocytopenia patients. Platelets 2015;26(3):220–3.
- Donato H, Picón A, Martinez M, Rapetti MC, Rosso A, Gomez S, et al. Demographic data, natural history, and prognostic factors of idiopathic thrombocytopenic purpura in children: a multicentered study from Argentina. Pediatr Blood Cancer. 2009;52(4):491–6.
- Kühne T. Update on the Intercontinental Cooperative ITP Study Group (ICIS) and on the Pediatric and Adult Registry on Chronic ITP (PARC ITP). Pediatr Blood Cancer. 2013;60(Suppl 1):S15–18.
- Warrier R, Chauhan A. Management of immune thrombocytopenic purpura: an update. Ochsner J. 2012;12(3):221–7.
- Fogarty PF, Segal JB. The epidemiology of immune thrombocytopenic purpura. Curr Opin Hematol. 2007;14(5):515–9.
- Newland A, Godeau B, Priego V, Viallard J-F, Lopez Fernandez MF, Orejudos A, et al. A final analysis of a phase 2, single-arm study of platelet (Plt) responses and remission rates in patients with Immune Thrombocytopenia (ITP) receiving romiplostim. Abstract # 2775. Presented at the 56th ASH Annual Meeting, San Francisco, December, 6–9 2014. Available from: https://ash.confex.com/ash/2014/webprogram/Paper67031.html. [Accessed February 5, 2015].
- Grimaldi D, Canouï-Poitrine F, Croisille L, Lee K, Roudot-Thoraval F, Languille L, et al. Antiplatelet antibodies detected by the MAIPA assay in newly diagnosed immune thrombocytopenia are associated with chronic outcome and higher risk of bleeding. Ann Hematol. 2014;93(2):309–15.
- Kuter DJ, Bussel JB, Lyons RM, Pullarkat V, Gernsheimer TB, Senecal FM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008;371(9610):395–403.
- Li J, Wang Z, Hu S, Zhao X, Cao L. Correction of abnormal T cell subsets by high-dose dexamethasone in patients with chronic idiopathic thrombocytopenic purpura. Immunol Lett. 2013;154(1–2):42–8.
- Li J, Wang Z, Dai L, Cao L, Su J, Zhu M, et al. Effects of rapamycin combined with low dose prednisone in patients with chronic immune thrombocytopenia. Clin Dev Immunol. 2013;2013:548085.
- Li Z, Mou W, Lu G, Cao J, He X, Pan X, et al. Low-dose rituximab combined with short-term glucocorticoids up-regulates Treg cell levels in patients with immune thrombocytopenia. Int J Hematol. 2011;93(1):91–8.
- Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 2008;112(4):1147–50.
- Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur J Haematol. 2007;79(4):310–6.
- Clynes R. Immune complexes as therapy for autoimmunity. J Clin Invest. 2005;115(1):25–7.
- Mahévas M, Fain O, Ebbo M, Roudot-Thoraval F, Limal N, Khellaf M, et al. The temporary use of thrombopoietin-receptor agonists may induce a prolonged remission in adult chronic immune thrombocytopenia. Results of a French observational study. Br J Haematol. 2014;165(6):865–9.
- Vlachaki E, Papageorgiou V, Klonizakis F, Spandonidou M, Chisan S, Vetsiou E, et al. Total remission of severe immune thrombocytopenia after short term treatment with romiplostim. Hematol Rep. 2011;3(3):e20.
- Ghadaki B, Nazi I, Kelton JG, Arnold DM. Sustained remissions of immune thrombocytopenia associated with the use of thrombopoietin receptor agonists. Transfusion (Paris) 2013;53(11):2807–12.
- Bussel JB, Wang X, Lopez A, Eisen M. Case study of remission in adults with immune thrombocytopenia following cessation of treatment with the thrombopoietin mimetic romiplostim. Hematology. 2016;21(4):257–62.
- José González-López T, Pascual C, Álvarez-Román MT, Fernández-Fuertes F, Sánchez-González B, Caparrós I, et al. Successful discontinuation of eltrombopag after complete remission in patients with primary immune thrombocytopenia. Am J Hematol. 2015;90(3):E40–3.
- Bao W, Bussel JB, Heck S, He W, Karpoff M, Boulad N, et al. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood 2010;116(22):4639–45.
- May AE, Kälsch T, Massberg S, Herouy Y, Schmidt R, Gawaz M. Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation 2002;106(16):2111–7.